
Comprehensive Analysis of Clinically Relevant Copy Number Alterations (CNAs) Using a 523-Gene Next-Generation Sequencing Panel and NxClinical Software in Solid Tumors

 , , ,
, , ,
Abstract
:1. Introduction
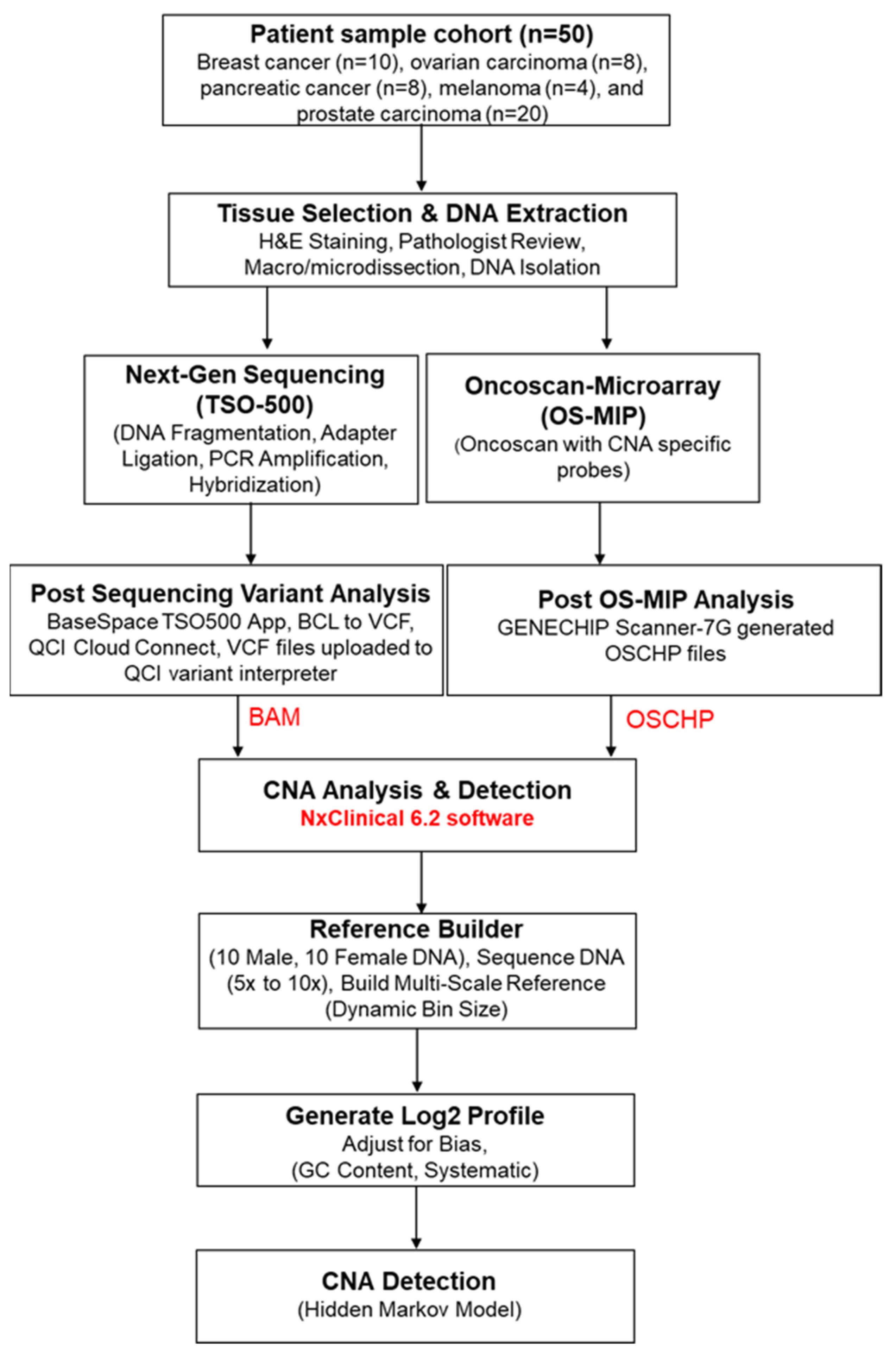
2. Materials and Methods
2.1. Patient Samples
2.2. Tissue Selection and DNA Extraction
2.3. OncoScan Microarray Inversion Probe (OS-MIP)
2.4. Library Preparation for Next-Generation Sequencing
2.5. Post-Sequencing Variant Analysis
2.6. CNA Analysis
3. Results
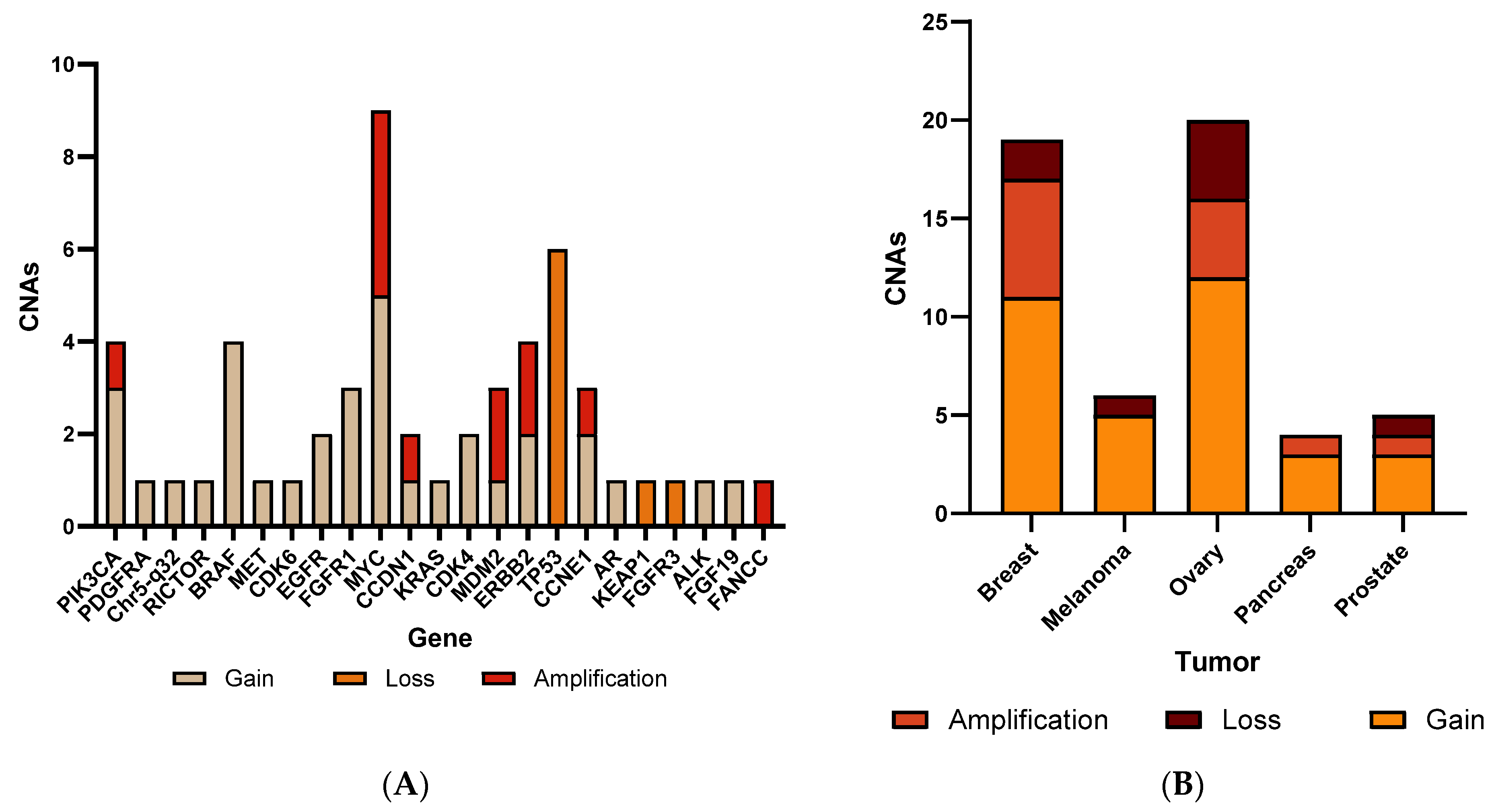
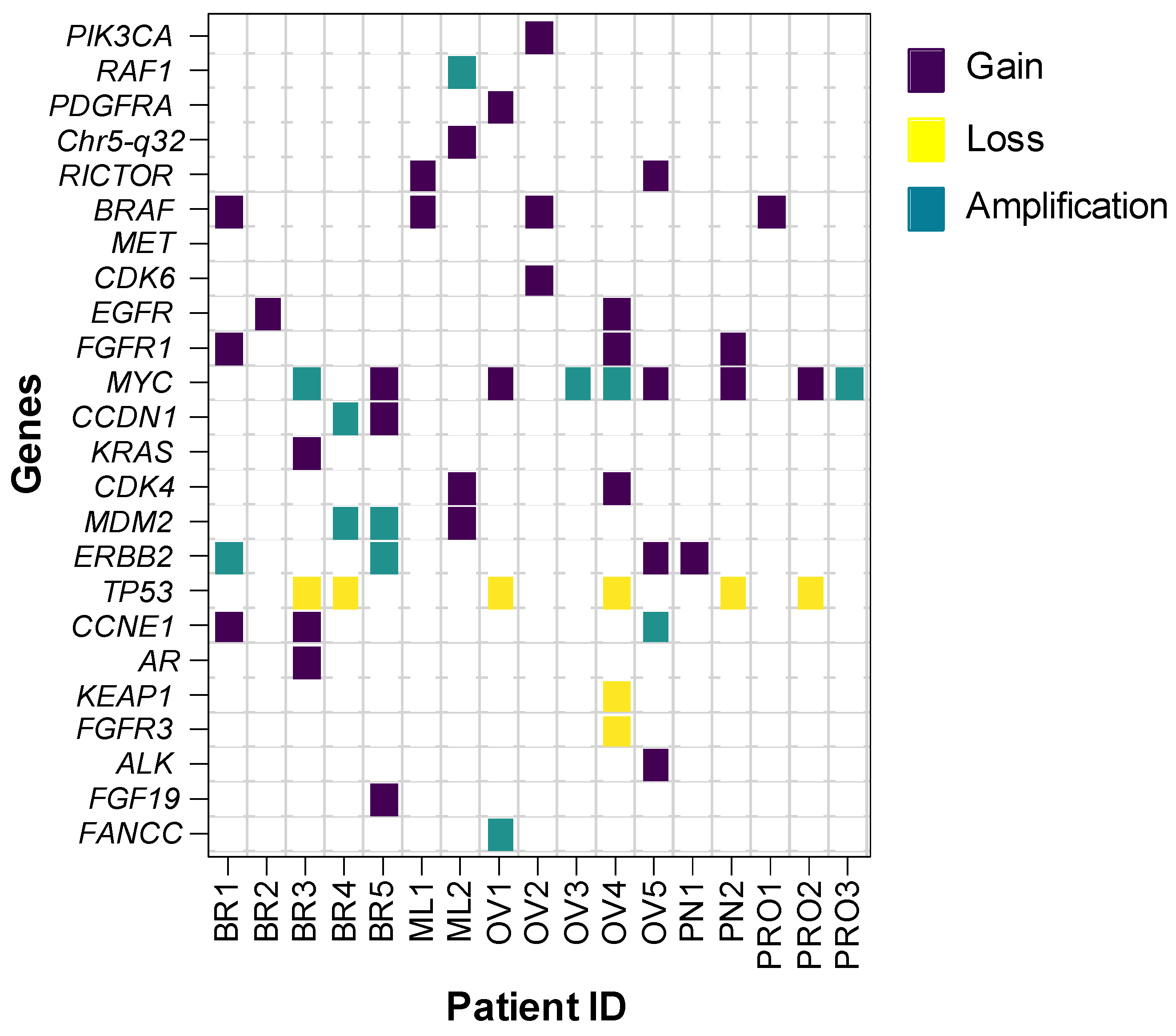
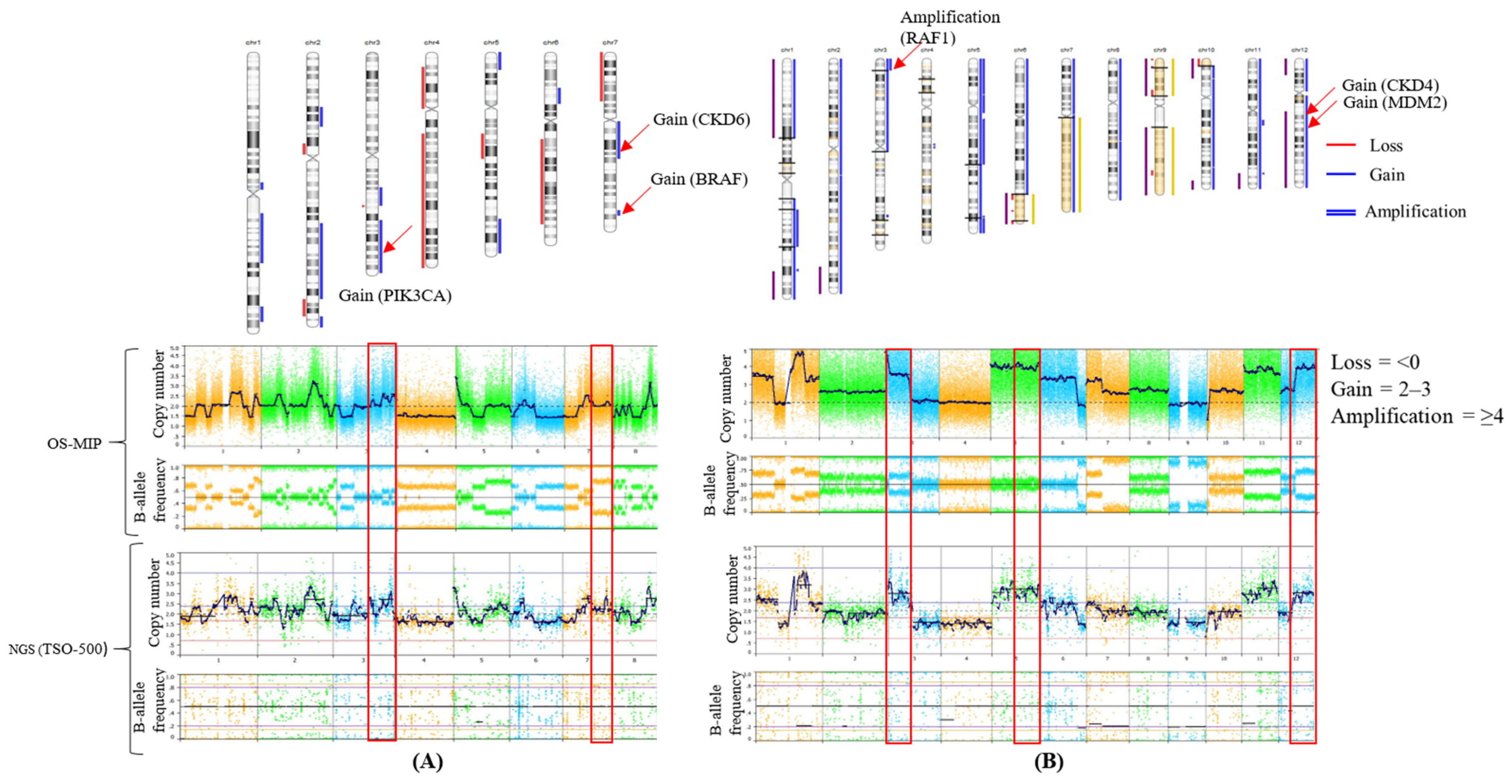
3.1. Concordance of CNA Detection
3.2. CNA Patterns According to Tumor Type
3.3. Sensitivity, Specificity, and Accuracy
4. Discussion
5. Strengths and Limitations
5.1. Strengths
- Comprehensive Analysis: NGS platforms offer extensive genomic data, encompassing CNAs, SNVs, and other genetic aberrations. By leveraging NxClinical for CNA analysis within NGS data, the study consolidates multiple facets of genetic testing into a unified workflow. This integration facilitates a holistic understanding of the genomic landscape, enabling clinicians to gain insights into both structural and sequence-level alterations from a single analysis.
- Streamlined Workflow: Integration of CNA analysis into NGS data processing through NxClinical enhances laboratory efficiency by streamlining workflow processes. This integration reduces the need for disparate tests, such as chromosomal microarrays, thereby optimizing sample processing time. The resultant expedited turnaround times contribute to timely generation of comprehensive genomic profiles, facilitating swift clinical decision making.
- Cost Efficiency: Despite the inherent costs associated with NGS technology, the adoption of a unified NGS test for both CNA and sequence variant analysis can yield significant cost efficiency.
5.2. Limitations
- Scope Limitation: The study’s focus on 24 predetermined genomic regions of clinical relevance within the NxClinical platform excludes other potential areas analyzed by the software. This selective approach may restrict the breadth of comprehensive CNA detection and interpretation. Clinicians interpreting study findings should acknowledge this scope limitation, which could impact the generalizability of the results to broader genomic landscapes.
- Sample Size Inadequacy: With a sample size of 50, the study may lack the statistical power necessary to establish a coherent approach for a streamlined workflow utilizing NGS and NxClinical for CNA detection. A larger sample size is imperative to ensure robustness and generalizability of findings, particularly in understanding the nuances and complexities of genomic alterations.
- Limited Cancer Type Validation: The validation of the NxClinical workflow was conducted with a restricted range of cancer types. Diverse categories of cancers should be included in future assessments to comprehensively evaluate the efficacy and accuracy of NxClinical in detecting CNAs across various malignancies.
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, R.R.; Mehrotra, M.; Chen, H.; Almohammedsalim, A.A.; Sahin, A.; Bosamra, A.; Patel, K.P.; Routbort, M.J.; Lu, X.; Ronald, A.; et al. Comprehensive Screening of Gene Copy Number Aberrations in Formalin-Fixed, Paraf-fin-Embedded Solid Tumors Using Molecular Inversion Probe–Based Single-Nucleotide Polymorphism Array. J. Mol. Diagn. 2016, 18, 676–687. [Google Scholar] [CrossRef]
- Garcia, E.P.; Minkovsky, A.; Jia, Y.; Ducar, M.D.; Shivdasani, P.; Gong, X.; Ligon, A.H.; Sholl, L.M.; Kuo, F.C.; MacConaill, L.E.; et al. Validation of OncoPanel: A Targeted Next-Generation Sequencing Assay for the Detection of So-matic Variants in Cancer. Arch. Pathol. Lab. Med. 2017, 141, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cabrera, J.M.; Del Valle, J.; Feliubadaló, L.; Pineda, M.; González, S.; Campos, O.; Cuesta, R.; Brunet, J.; Serra, E.; Capellà, G.; et al. Screening of CNVs Using NGS Data Improves Mutation Detection Yield and Decreases Costs in Ge-netic Testing for Hereditary Cancer. J. Med. Genet. 2022, 59, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimizadeh, W.; Guérard, K.P.; Rouzbeh, S.; Scarlata, E.; Brimo, F.; Patel, P.G.; Jamaspishvili, T.; Hamel, L.; Aprikian, A.G.; Lee, A.Y.; et al. A DNA Copy Number Alteration Classifier as a Prognostic Tool for Prostate Cancer Patients. Br. J. Cancer 2023, 128, 2165–2174. [Google Scholar]
- Li, J.; Greytak, S.R.; Guan, P.; Engel, K.B.; Goerlitz, D.S.; Islam, M.; Varghese, R.S.; Moore, H.M.; Ressom, H.W. Formalin Fixation, Delay to Fixation, and Time in Fixative Adversely Impact Copy Number Variation Analysis by aCGH. Biopreserv. Biobank. 2023, 21, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Xi, H.; Chen, J.; Peng, Y.; Jia, Z.; Yang, S.; Hu, J.; Pang, J.; Zhang, Y.; Hu, R.; et al. Integrated CNV-Seq, Kary-otyping and SNP-Array Analyses for Effective Prenatal Diagnosis of Chromosomal Mosaicism. BMC Med. Genomics 2021, 14, 1–12. [Google Scholar]
- Serin Harmanci, A.; Harmanci, A.O.; Zhou, X. CaSpER Identifies and Visualizes CNV Events by Integrative Analysis of Single-Cell or Bulk RNA-Sequencing Data. Nat. Commun. 2020, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Zito Marino, F.; Buono, S.; Montella, M.; Giannatiempo, R.; Messina, F.; Casaretta, G.; Arpino, G.; Vita, G.; Fiorentino, F.; Insabato, L.; et al. NTRK Gene Aberrations in Triple-Negative Breast Cancer: Detection Challenges Using IHC, FISH, RT-PCR, and NGS. J. Pathol. Clin. Res. 2023. [CrossRef] [PubMed]
- Ito, T.; Kawashima, Y.; Fujikawa, T.; Honda, K.; Makabe, A.; Kitamura, K.; Tsutsumi, T. Rapid Screening of Copy Num-ber Variations in STR C by Droplet Digital PCR in Patients with Mild-to-Moderate Hearing Loss. Hum. Genome Var. 2019, 6, 41. [Google Scholar] [CrossRef]
- Vives-Usano, M.; García Pelaez, B.; Román Lladó, R.; Garzón Ibañez, M.; Aldeguer, E.; Rodriguez, S.; Aguilar, A.; Pons, F.; Viteri, S.; Cabrera, C.; et al. Analysis of Copy Number Variations in Solid Tumors Using a Next Genera-tion Sequencing Custom Panel. J. Mol. Pathol. 2021, 2, 123–134. [Google Scholar] [CrossRef]
- Mu, W.; Li, B.; Wu, S.; Chen, J.; Sain, D.; Xu, D.; Black, M.H.; Karman, R.; Gillespie, K.; Hagman, K.D.F.; et al. Detection of Structural Variation Using Target Captured Next-Generation Sequenc-ing Data for Genetic Diagnostic Testing. Genes 2019, 21, 1603–1610. [Google Scholar]
- Zhang, Z.; Hao, K. SAAS-CNV: A Joint Segmentation Approach on Aggregated and Allele Specific Signals for the Iden-tification of Somatic Copy Number Alterations with Next-Generation Sequencing Data. PLoS Comput. Biol. 2015, 11, e1004618. [Google Scholar] [CrossRef] [PubMed]
- Abel, H.J.; Duncavage, E.J. Detection of Structural DNA Variation from Next Generation Sequencing Data: A Review of Informatic Approaches. Cancer Genet. 2013, 206, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Truty, R.; Paul, J.; Kennemer, M.; Lincoln, S.E.; Olivares, E.; Nussbaum, R.L.; Aradhya, S. Prevalence and Properties of Intragenic Copy-Number Variation in Mendelian Disease Genes. Genet. Med. 2019, 21, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Eric, T.; Hunter, S.A.; Thomas, B.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar]
- Klambauer, G.; Schwarzbauer, K.; Mayr, A.; Clever, D.A.; Mitterecker, H.; Bodenhofer, U.; Hochreiter, S. cn.MOPS: Mixture of Poissons for Discovering Copy Number Variations in Next-Generation Sequencing Data with a Low False Discovery Rate. Nucleic Acids Res. 2012, 40, e69. [Google Scholar] [CrossRef]
- Johansson, L.F.; van Dijk, F.; de Boer, E.N.; van Dijk-Bos, K.K.; Jongbloed, J.D.; van der Hout, A.H.; Westers, H.; Sinke, R.J.; Swertz, M.A.; Sijmons, R.H.; et al. CoNVaDING: Single Exon Variation Detection in Targeted NGS Data. Hum. Mutat. 2016, 37, 457–464. [Google Scholar] [CrossRef]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate Clinical Detection of Exon Copy Number Variants in a Targeted NGS Panel Using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef]
- Roca, I.; González-Castro, L.; Fernández, H.; Couce, M.L.; Fernández-Marmiesse, A. Free-Access Copy-Number Variant Detection Tools for Targeted Next-Generation Sequencing Data. Mutat. Res. Rev. Mutat. Res. 2019, 779, 114–125. [Google Scholar] [CrossRef]
- Illumina. TruSight Oncology 500 High Throughput: Reference Guide. Document #1000000094853 v00. 2022. [Google Scholar]
- Iafrate, A.J.; Feuk, L.; Rivera, M.N.; Listewnik, M.L.; Donahoe, P.K.; Qi, Y.; Scherer, S.W.; Lee, C. Detection of Large-Scale Variation in the Human Genome. Nat. Genet. 2004, 36, 949–951. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Manér, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-Scale Copy Number Polymorphism in the Human Genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef]
- Lupski, J.R. Genomic Rearrangements and Sporadic Disease. Nat. Genet. 2007, 39 (Suppl 7), S43–S47. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Altshuler, D.M. Copy-Number Variation and Association Studies of Human Disease. Nat. Genet. 2007, 39 (Suppl. 7), S37–S42. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration Across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-Cancer Patterns of Somatic Copy Number Alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, M.; Luthra, R.; Abraham, R.; Mishra, B.M.; Virani, S.; Chen, H.; Routbort, M.J.; Patel, K.P.; Medeiros, L.J.; Singh, R.R. Validation of Quantitative PCR-Based Assays for Detection of Gene Copy Number Aberrations in Forma-lin-Fixed, Paraffin-Embedded Solid Tumor Samples. Cancer Genet. 2017, 212, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, R.; Reuther, J.; Gandhi, I.; Voicu, H.; Alvarez, K.R.; Plon, S.E.; Lopez-Terrada, D.H.; Fisher, K.E.; Parsons, D.W.; Roy, A. A validation framework for somatic copy number detection in targeted sequencing panels. J. Mol. Diagn. 2022, 24, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Kerkhof, J.; Schenkel, L.C.; Reilly, J.; McRobbie, S.; Aref-Eshghi, E.; Stuart, A.; Rupar, C.A.; Adams, P.; Hegele, R.A.; Lin, H.; et al. Clinical validation of copy number variant detection from targeted next-generation sequencing panels. J. Mol. Diagn. 2017, 19, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Borge, K.S.; Nord, S.; Van Loo, P.; Lingjærde, O.C.; Gunnes, G.; Alnæs, G.I.; Solvang, H.K.; Lüders, T.; Kristensen, V.N.; Børresen-Dale, A.L.; et al. Canine Mammary Tumours Are Affected by Frequent Copy Number Aberrations, In-cluding Amplification of MYC and Loss of PTEN. PLoS ONE 2015, 10, e0126371. [Google Scholar] [CrossRef] [PubMed]
- Koçak, A.; Heselmeyer-Haddad, K.; Lischka, A.; Hirsch, D.; Fiedler, D.; Hu, Y.; Doberstein, N.; Torres, I.; Chen, W.D.; Gertz, E.M.; et al. High Levels of Chromosomal Copy Number Alterations and TP53 Mutations Correlate with Poor Outcome in Younger Breast Cancer Patients. Am. J. Pathol. 2020, 190, 1643–1656. [Google Scholar] [CrossRef]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/Akt/mTOR Pathway in Ovarian Cancer: Therapeutic Opportunities and Challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef]
- Ko, J.; Meyer, A.N.; Haas, M.; Donoghue, D.J. Characterization of FGFR Signaling in Prostate Cancer Stem Cells and In-hibition via TKI Treatment. Oncotarget 2021, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR Gene Copy Number Predicts Poor Outcome in Triple-Negative Breast Cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, K.L.; Martin, A.M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.T.; et al. Tumor Genetic Analyses of Patients with Metastatic Melanoma Treated with the BRAF Inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef] [PubMed]
- Varella-Garcia, M.; Aisner, D.L. ALK Rearrangement in Non Small Cell Lung Carcinoma. Assoc. Mol. Path. Mol. Rev. Solid Tumors 2010. [Google Scholar]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Amin, H.M.; McDonnell, T.J.; Ma, Y.; Lin, Q.; Fujio, Y.; Kunisada, K.; Leventaki, V.; Das, P.; Rassidakis, G.Z.; Cutler, C.; et al. Selective inhibition of STAT3 induces apoptosis and G1 cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene 2004, 23, 5426–5434. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Todd, W. Oncogenic mutations of PIK3CA in human cancers. Curr. Top. Microbiol. Immunol. 2010, 347, 21–41. [Google Scholar]
- Kawano, O.; Sasaki, H.; Okuda, K.; Yukiue, H.; Yokoyama, T.; Yano, M.; Fujii, Y. PIK3CA gene amplification in Japanese non-small cell lung cancer. Lung Cancer 2007, 58, 159–160. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Bian, D.; Su, S.; Mahanivong, C.; Cheng, R.K.; Han, Q.; Pan, Z.K.; Sun, P.; Huang, S. Lysophosphatidic acid stimulates ovarian cancer cell migration via a Ras-MEK kinase 1 pathway. Cancer Res. 2004, 64, 4209–4217. [Google Scholar] [CrossRef]
- Graziano, S.L.; Pfeifer, A.M.; Testa, J.R.; Mark, G.E.; Johnson, B.E.; Hallinan, E.J.; Pettengill, O.S.; Sorenson, G.D.; Tatum, A.H.; Brauch, H.; et al. Involvement of the RAFI locus, at band 3p25, in the 3p deletion of small-cell lung cancer. Genes Chromosomes Cancer 1991, 3, 283–293. [Google Scholar] [CrossRef]
- Jansson, S.; Bendahl, P.-O.; Grabau, D.A.; Falck, A.-K.; Fernö, M.; Aaltonen, K.; Rydén, L. The three receptor tyrosine kinases c-KIT, VEGFR2 and PDGFRalpha, closely spaced at 4q12, show increased protein expression in triple-negative breast cancer. PLoS ONE 2014, 9, e102176. [Google Scholar] [CrossRef]
- Gorringe, K.L.; Jacobs, S.; Thompson, E.R.; Sridhar, A.; Qiu, W.; Choong, D.Y.; Campbell, I.G. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin. Cancer Res. 2007, 13, 4731–4739. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Krishna, N.S.; Witton, C.J.; Bartlett, J.M.S. Gene amplifications associated with the development of hormone-resistant prostate cancer. Clin. Cancer Res. 2003, 9, 5271–5281. [Google Scholar] [PubMed]
- Sapi, E. The role of CSF-1 in normal physiology of mammary gland and breast cancer: An update. Exp. Biol. Med. 2004, 229, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, W.; Simeone, I.; Anjum, S.; Mokrab, Y.; Bertucci, F.; Finetti, P.; Curigliano, G.; Seliger, B.; Cerulo, L.; Tomei, S.; et al. Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis. Oncoimmunology 2017, 6, e1253654. [Google Scholar] [CrossRef] [PubMed]
- Joly, M.M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Estrada, M.V.; Young, C.; Williams, M.; Rexer, B.N.; Sarbassov, D.D.; Muller, W.J.; et al. Rictor/mTORC2 drives progression and therapeutic resistance of HER2-amplified breast cancers. Cancer Res. 2016, 76, 4752–4764. [Google Scholar] [CrossRef] [PubMed]
- Jebali, A.; Battistella, M.; Lebbé, C.; Dumaz, N. RICTOR affects melanoma tumorigenesis and its resistance to targeted therapy. Biomedicines 2021, 9, 1498. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Varkaris, A.; Corn, P.G.; Gaur, S.; Dayyani, F.; Logothetis, C.J.; E Gallick, G. The role of HGF/c-Met signaling in prostate cancer progression and c-Met inhibitors in clinical trials. Expert Opin. Investig. Drugs 2011, 20, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, K.Y.; Giubellino, A. The role of MET in melanoma and melanocytic lesions. Am. J. Pathol. 2019, 189, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Garutti, M.; Targato, G.; Buriolla, S.; Palmero, L.; Minisini, A.M.; Puglisi, F. CDK4/6 Inhibitors in Melanoma: A Comprehensive Review. Cells 2021, 10, 1334. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, A.; Sonego, M.; Pellizzari, I.; Pellarin, I.; Canzonieri, V.; D’Andrea, S.; Benevol, S.; Sorio, R.; Giorda, G.; Califano, D.; et al. CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol. Med. 2017, 9, 1415–1433. [Google Scholar] [CrossRef] [PubMed]
- Ala, M. Target c-Myc to treat pancreatic cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Boufaied, N.; Hallal, T.; Feit, A.; de Polo, A.; Luoma, A.M.; Alahmadi, W.; Larocque, J.; Zadra, G.; Xie, Y.; et al. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat. Commun. 2022, 13, 2559. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.-L.; Liu, J.-Y.; Qu, L.-X.; La, H.; Wang, H.-L.; Chen, X.-X.; Wang, N.; Wei, Z.-Z. Expression of Cyclin D1 gene in ovarian cancer and effect of silencing its expression on ovarian cancer cells based on the Oncomine database. Bioengineered 2021, 12, 9290–9300. [Google Scholar] [CrossRef]
- Valla, M.; Klæstad, E.; Ytterhus, B.; Bofin, A.M. CCND1 amplification in breast cancer-associations with proliferation, histopathological grade, molecular subtype and prognosis. J. Mammary Gland. Biol. Neoplasia 2022, 27, 67–77. [Google Scholar] [CrossRef]
- Francavilla, C.; O’Brien, C.S. Fibroblast growth factor receptor signalling dysregulation and targeting in breast cancer. Open Biol. 2022, 12, 210373. [Google Scholar] [CrossRef]
- Chia, L.; Wang, B.; Kim, J.-H.; Luo, L.Z.; Shuai, S.; Herrera, I.; Chen, S.Y.; Li, L.; Xian, L.; Huso, T.; et al. HMGA1 induces FGF19 to drive pancreatic carcinogenesis and stroma formation. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Ratner, E.; Keane, F.; Lindner, R.; A Tassi, R.; Paranjape, T.; Glasgow, M.; Nallur, S.; Deng, Y.; Lu, L.; Steele, L.; et al. A KRAS variant is a biomarker of poor outcome, platinum chemotherapy resistance and a potential target for therapy in ovarian cancer. Oncogene 2012, 31, 4559–4566. [Google Scholar] [CrossRef]
- Mayr, D.; Hirschmann, A.; Löhrs, U.; Diebold, J. KRAS and BRAF mutations in ovarian tumors: A comprehensive study of invasive carcinomas, borderline tumors and extraovarian implants. Gynecol. Oncol. 2006, 103, 883–887. [Google Scholar] [CrossRef]
- Wu, C.; Peng, S.; Pilié, P.G.; Geng, C.; Park, S.; Manyam, G.C.; Lu, Y.; Yang, G.; Tang, Z.; Kondraganti, S.; et al. PARP and CDK4/6 inhibitor combination therapy induces apoptosis and suppresses neuroendocrine differentiation in prostate cancer. Mol. Cancer Ther. 2021, 20, 1680–1691. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qin, J.-J.; Voruganti, S.; Nijampatnam, B.; Velu, S.E.; Ruan, K.-H.; Hu, M.; Zhou, J.; Zhang, R. Discovery and characterization of dual inhibitors of MDM2 and NFAT1 for pancreatic cancer therapy. Cancer Res. 2018, 78, 5656–5667. [Google Scholar] [CrossRef]
- Makii, C.; Oda, K.; Ikeda, Y.; Sone, K.; Hasegawa, K.; Uehara, Y.; Nishijima, A.; Asada, K.; Koso, T.; Fukuda, T.; et al. MDM2 is a potential therapeutic target and prognostic factor for ovarian clear cell carcinomas with wild type TP53. Oncotarget 2016, 7, 75328–75338. [Google Scholar] [CrossRef] [PubMed]
- Dörk, T.; Peterlongo, P.; Mannermaa, A.; Bolla, M.K.; Wang, Q.; Dennis, J.; Ahearn, T.; Andrulis, I.L.; Anton-Culver, H.; Arndt, V.; et al. Two truncating variants in FANCC and breast cancer risk. Sci. Rep. 2019, 9, 12524. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.W.; Wang, X.J.; Chen, T.; Ding, X.W.; Jiang, X.; Gao, Y.; Mo, W.-J.; Huang, Y.; Lou, C.-J.; Cao, W.-M. Deleterious mutations in DNA repair gene FANCC exist in BRCA1/2-negative Chinese familial breast and/or ovarian cancer patients. Front. Oncol. 2019, 9, 169. [Google Scholar] [CrossRef]
- Révillion, F.; Bonneterre, J.; Peyrat, J.P. ERBB2 oncogene in human breast cancer and its clinical significance. Eur. J. Cancer 1998, 34, 791–808. [Google Scholar] [CrossRef]
- Wu, X.; Yan, J.; Yu, J.; Cheng, Z.; Guo, Q.; Kong, Y.; Guo, J. Clinical significance of CCNE1 copy number gain in acral melanoma patients. Melanoma Res. 2021, 31, 352–357. [Google Scholar] [CrossRef]
- Fox, D.B.; Ebright, R.Y.; Hong, X.; Russell, H.C.; Guo, H.; LaSalle, T.J.; Wittner, B.S.; Poux, N.; Vuille, J.A.; Toner, M.; et al. Downregulation of KEAP1 in melanoma promotes resistance to immune checkpoint blockade. NPJ Precis. Oncol. 2023, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, G.S.; Biswal, S. Loss of Keap1 function in prostate cancer cells causes chemo-and radio-resistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NxClinical (BAM Files TSO500) | NxClinical (OSCHP Files OncoScan) | ||
|---|---|---|---|
| POSITIVE | NEGATIVE | TOTAL | |
| POSITIVE | 54 (TP) | 0 (FP) | 54 |
| NEGATIVE | 0 (FN) | 1146 (TN) | 1146 |
| TOTAL | 54 | 1146 | 1200 |
| Sensitivity TP/(TP + FN) = 100% | PPV TP/(TP + FP) = 100% | ||
| Specificity TN/(FP + TN) = 100% | NPV TN/(FN + TN) = 100% | ||
| Diagnostic accuracy (TP + TN)/TP + TN + FP + FN) = 100% | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, V.; Vashisht, V.; Vashisht, A.; Mondal, A.K.; Alptekin, A.; Singh, H.; Kolhe, R. Comprehensive Analysis of Clinically Relevant Copy Number Alterations (CNAs) Using a 523-Gene Next-Generation Sequencing Panel and NxClinical Software in Solid Tumors. Genes 2024, 15, 396. https://doi.org/10.3390/genes15040396
Gupta V, Vashisht V, Vashisht A, Mondal AK, Alptekin A, Singh H, Kolhe R. Comprehensive Analysis of Clinically Relevant Copy Number Alterations (CNAs) Using a 523-Gene Next-Generation Sequencing Panel and NxClinical Software in Solid Tumors. Genes. 2024; 15(4):396. https://doi.org/10.3390/genes15040396
Chicago/Turabian StyleGupta, Vivek, Vishakha Vashisht, Ashutosh Vashisht, Ashis K. Mondal, Ahmet Alptekin, Harmanpreet Singh, and Ravindra Kolhe. 2024. "Comprehensive Analysis of Clinically Relevant Copy Number Alterations (CNAs) Using a 523-Gene Next-Generation Sequencing Panel and NxClinical Software in Solid Tumors" Genes 15, no. 4: 396. https://doi.org/10.3390/genes15040396