

A Review of the Epigenetic Clock: Emerging Biomarkers for Asthma and Allergic Disease

Abstract
:1. Introduction
2. Purpose
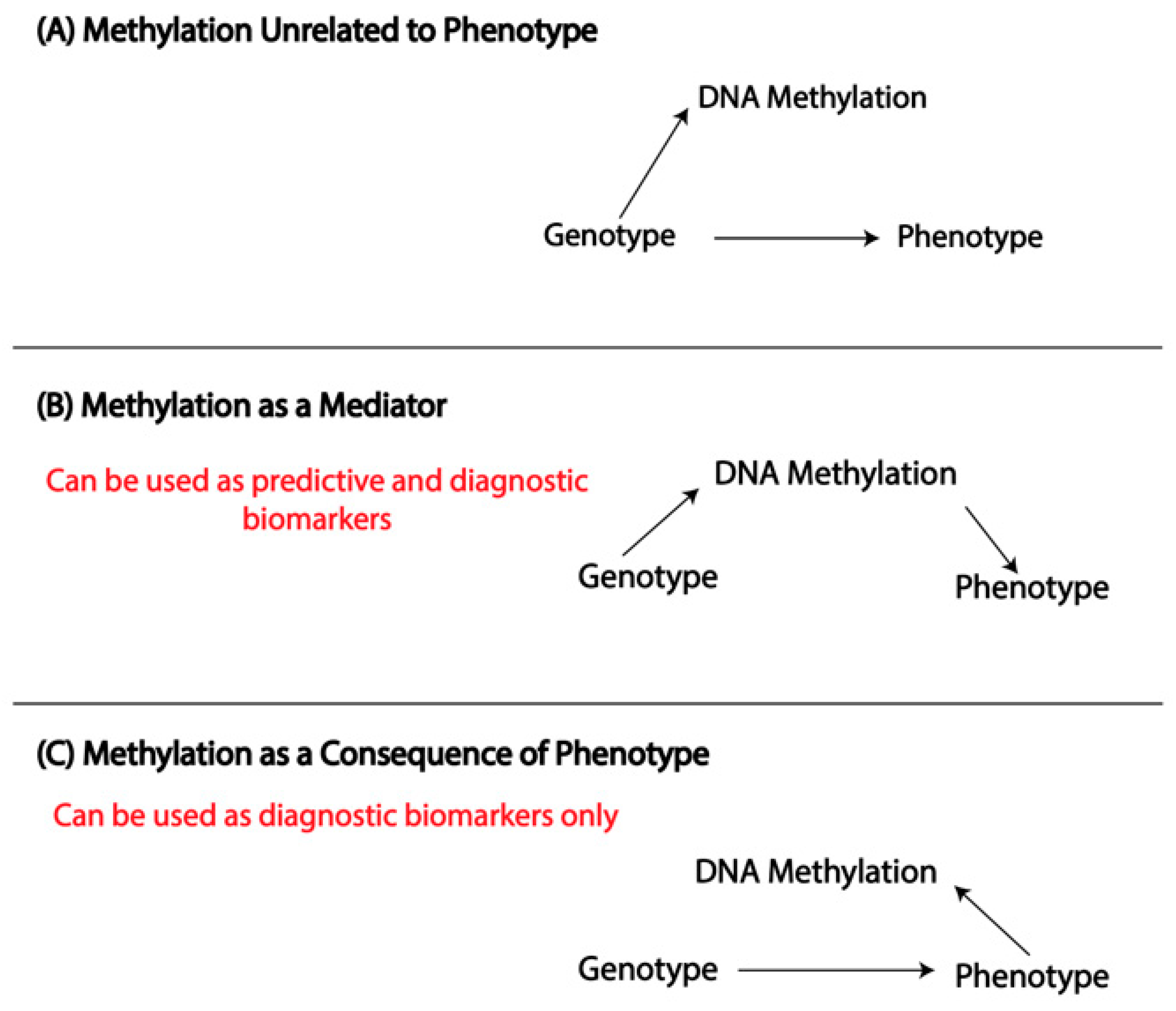
3. Overview of DNA Methylation
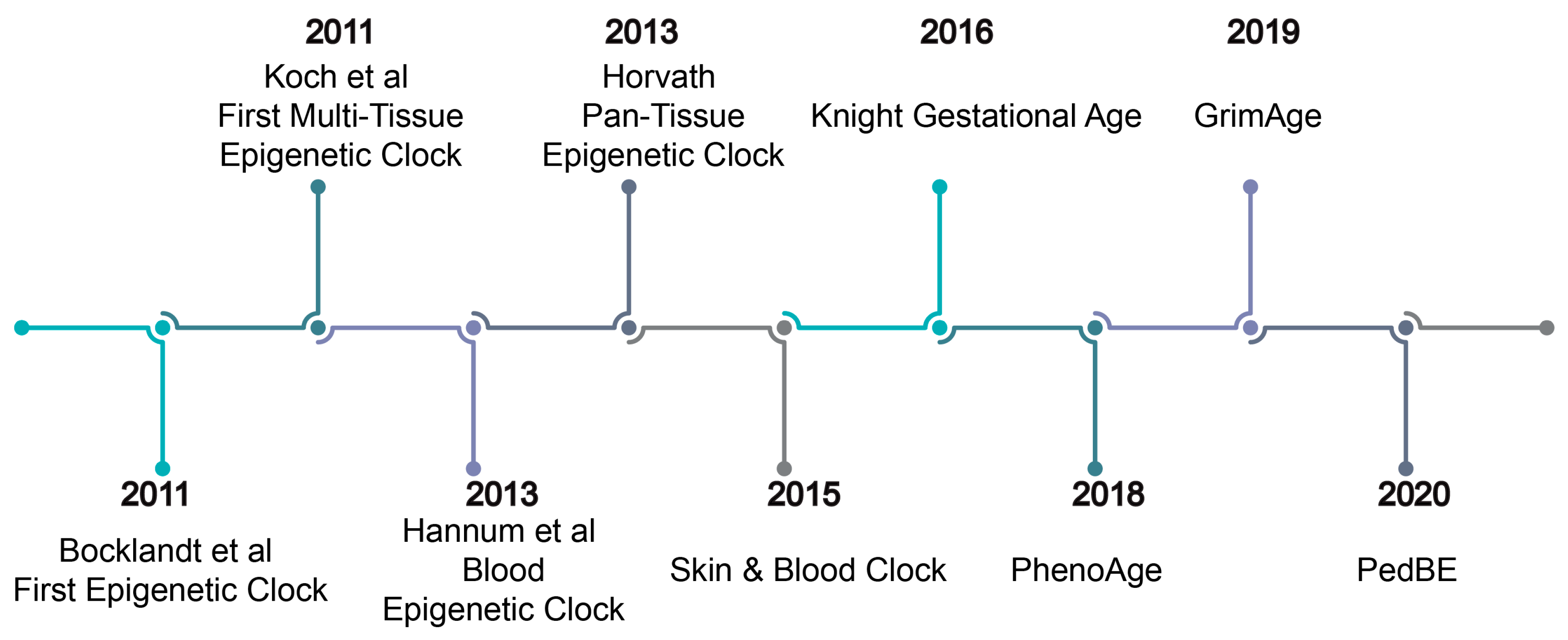
4. Epigenetic Clocks
5. Epigenetic Clock Training Metrics
6. First-Generation Epigenetic Clocks
7. First-Generation Multi-Tissue Epigenetic Clocks
8. Pediatric Epigenetic Clock
9. Gestational Age Clocks
10. Second-Generation Epigenetic Clocks
11. Metrics of Epigenetic Age Acceleration
- Intrinsic Epigenetic Age Acceleration (IEAA)—epigenetic age acceleration independent of cell type composition. This captures the “intrinsic” process of aging and should be universal, regardless of cell and tissue type. This metric is calculated by extracting the residuals of the linear regression:
- 2.
- Extrinsic Epigenetic Age Acceleration (EEAA)—a measure of age acceleration including both intrinsic age-related processes and changes in cell type, calculated in two steps:
- Enhanced Hannum epigenetic age is the weighted average of epigenetic age predicted by the Hannum clock and a combination of cell types. The weights are determined using a correlation between cell type and chronological age.
- The second step is a regression of enhanced Hannum age on chronological age
- 3.
- Age Acceleration (AgeAccel) [96]—a measure obtained by extracting the residuals of the linear regression of epigenetics on chronological age without accounting for cell type: Epigenetic Age ~ Chronological Age.
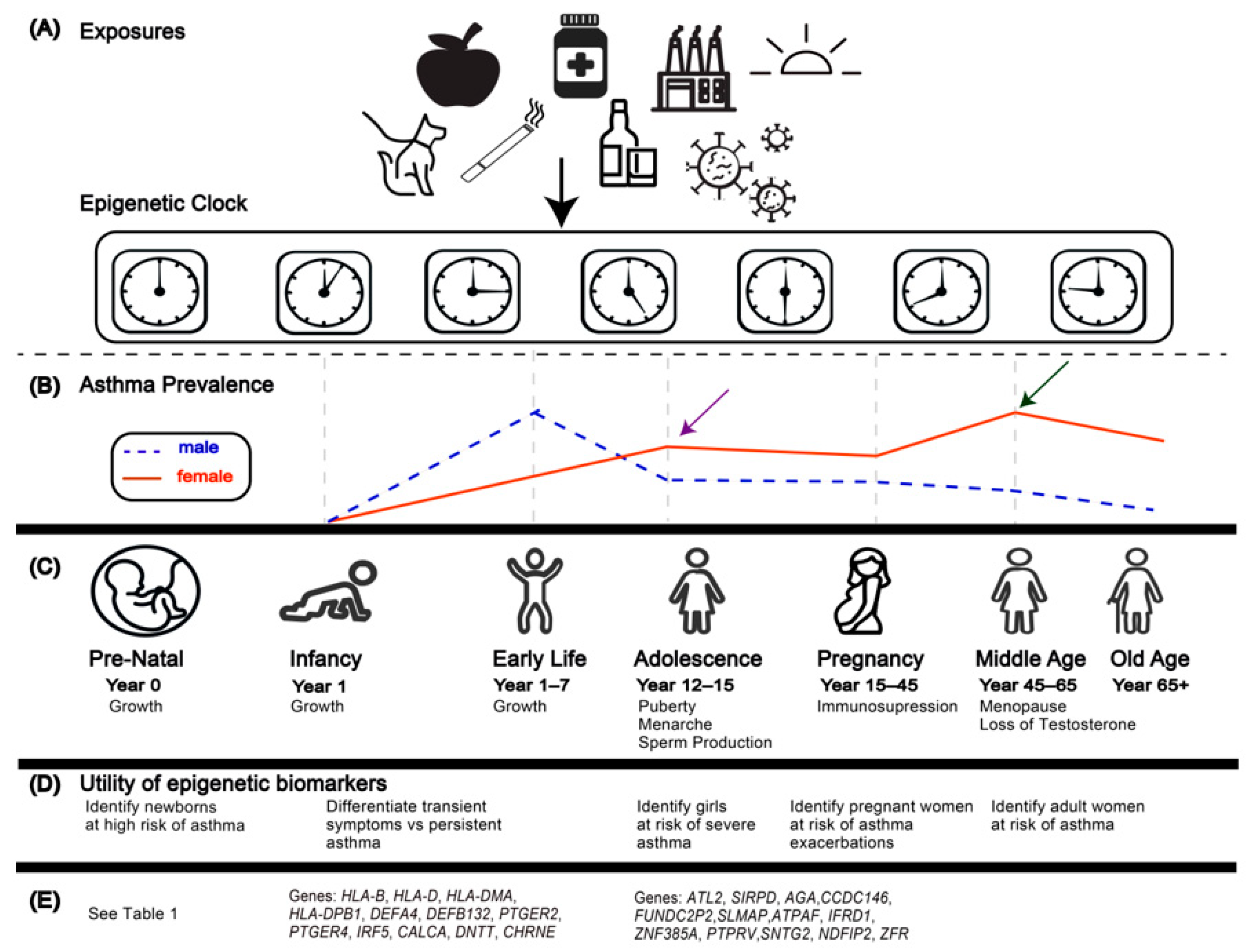
12. Applications of the First-Generation Epigenetic Clocks to Asthma and Allergic Disease
13. Epigenetic Age Acceleration and the Developmental Origins of Health and Disease
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev Genet 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef]
- Zeilinger, S.; Kühnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Hoang, T.T.; Qi, C.; Paul, K.C.; Lee, M.; White, J.D.; Richards, M.; Auerbach, S.S.; Long, S.; Shrestha, S.; Wang, T.; et al. Epigenome-Wide DNA Methylation and Pesticide Use in the Agricultural Lung Health Study. Environ. Health Perspect. 2021, 129, 97008. [Google Scholar] [CrossRef]
- Legaki, E.; Arsenis, C.; Taka, S.; Papadopoulos, N.G. DNA methylation biomarkers in asthma and rhinitis: Are we there yet? Clin. Transl. Allergy 2022, 12, e12131. [Google Scholar] [CrossRef]
- Titus, A.J.; Gallimore, R.M.; Salas, L.A.; Christensen, B.C. Cell-type deconvolution from DNA methylation: A review of recent applications. Hum. Mol. Genet. 2017, 26, R216–R224. [Google Scholar] [CrossRef] [PubMed]
- Grant, O.A.; Wang, Y.; Kumari, M.; Zabet, N.R.; Schalkwyk, L. Characterising sex differences of autosomal DNA methylation in whole blood using the Illumina EPIC array. Clin. Epigenet. 2022, 14, 62. [Google Scholar] [CrossRef]
- Bjornsson, H.T.; Sigurdsson, M.I.; Fallin, M.D.; Irizarry, R.A.; Aspelund, T.; Cui, H.; Yu, W.; Rongione, M.A.; Ekström, T.J.; Harris, T.B.; et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA 2008, 299, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Florath, I.; Butterbach, K.; Müller, H.; Bewerunge-Hudler, M.; Brenner, H. Cross-sectional and longitudinal changes in DNA methylation with age: An epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum. Mol. Genet. 2014, 23, 1186–1201. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.-P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Grodstein, F.; Lemos, B.; Yu, L.; Klein, H.U.; Iatrou, A.; Buchman, A.S.; Shireby, G.L.; Mill, J.; Schneider, J.A.; De Jager, P.L.; et al. The association of epigenetic clocks in brain tissue with brain pathologies and common aging phenotypes. Neurobiol. Dis. 2021, 157, 105428. [Google Scholar] [CrossRef]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef]
- Dugué, P.A.; Bassett, J.K.; Joo, J.E.; Jung, C.H.; Ming Wong, E.; Moreno-Betancur, M.; Schmidt, D.; Makalic, E.; Li, S.; Severi, G.; et al. DNA methylation-based biological aging and cancer risk and survival: Pooled analysis of seven prospective studies. Int. J. Cancer 2018, 142, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Cardenas, A.; Rifas-Shiman, S.L.; Hivert, M.-F.; Gold, D.R.; Platts-Mills, T.A.; Lin, X.; Oken, E.; Avila, L.; Celedón, J.C.; et al. Epigenetic age acceleration is associated with allergy and asthma in children in Project Viva. J. Allergy Clin. Immunol. 2019, 143, 2263–2270.e14. [Google Scholar] [CrossRef]
- Cardenas, A.; Sordillo, J.E.; Rifas-Shiman, S.L.; Chung, W.; Liang, L.; Coull, B.A.; Hivert, M.-F.; Lai, P.S.; Forno, E.; Celedón, J.C.; et al. The nasal methylome as a biomarker of asthma and airway inflammation in children. Nat. Commun. 2019, 10, 3095. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Ghildayal, N.; Fore, R.; Lutz, S.M.; Cardenas, A.; Perron, P.; Bouchard, L.; Hivert, M.F. Early-pregnancy maternal body mass index is associated with common DNA methylation markers in cord blood and placenta: A paired-tissue epigenome-wide association study. Epigenetics 2022, 17, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, W.J.; Kim, J.; Jeong, C.Y.; Park, H.; Hong, Y.C.; Ha, M.; Kim, Y.; Won, S.; Ha, E. Prenatal Exposure to Traffic-Related Air Pollution and the DNA Methylation in Cord Blood Cells: MOCEH Study. Int. J. Environ. Res. Public Health 2022, 19, 3292. [Google Scholar] [CrossRef]
- Akhabir, L.; Stringer, R.; Desai, D.; Mandhane, P.J.; Azad, M.B.; Moraes, T.J.; Subbarao, P.; Turvey, S.E.; Paré, G.; Anand, S.S. DNA methylation changes in cord blood and the developmental origins of health and disease—A systematic review and replication study. BMC Genom. 2022, 23, 221. [Google Scholar] [CrossRef]
- Hannon, E.; Schendel, D.; Ladd-Acosta, C.; Grove, J.; Hansen, C.S.; Hougaard, D.M.; Bresnahan, M.; Mors, O.; Hollegaard, M.V.; Bækvad-Hansen, M.; et al. Variable DNA methylation in neonates mediates the association between prenatal smoking and birth weight. Philos. Trans. R Soc. Lond. B Biol. Sci. 2019, 374, 20180120. [Google Scholar] [CrossRef]
- Küpers, L.K.; Xu, X.; Jankipersadsing, S.A.; Vaez, A.; la Bastide-van Gemert, S.; Scholtens, S.; Nolte, I.M.; Richmond, R.C.; Relton, C.L.; Felix, J.F.; et al. DNA methylation mediates the effect of maternal smoking during pregnancy on birthweight of the offspring. Int. J. Epidemiol. 2015, 44, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Frank, J.; Gilles, M.; Lang, M.; Treutlein, J.; Streit, F.; Wolf, I.A.C.; Peus, V.; Scharnholz, B.; Send, T.S.; et al. Impact on birth weight of maternal smoking throughout pregnancy mediated by DNA methylation. BMC Genom. 2018, 19, 290. [Google Scholar] [CrossRef] [PubMed]
- Küpers, L.K.; Monnereau, C.; Sharp, G.C.; Yousefi, P.; Salas, L.A.; Ghantous, A.; Page, C.M.; Reese, S.E.; Wilcox, A.J.; Czamara, D.; et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat. Commun. 2019, 10, 1893. [Google Scholar] [CrossRef]
- Bohlin, J.; Håberg, S.E.; Magnus, P.; Reese, S.E.; Gjessing, H.K.; Magnus, M.C.; Parr, C.L.; Page, C.M.; London, S.J.; Nystad, W. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 2016, 17, 207. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef]
- Joubert, B.R.; Håberg, S.E.; Nilsen, R.M.; Wang, X.; Vollset, S.E.; Murphy, S.K.; Huang, Z.; Hoyo, C.; Midttun, Ø.; Cupul-Uicab, L.A.; et al. 450 K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012, 120, 1425–1431. [Google Scholar] [CrossRef]
- Zhang, B.; Hong, X.; Ji, H.; Tang, W.Y.; Kimmel, M.; Ji, Y.; Pearson, C.; Zuckerman, B.; Surkan, P.J.; Wang, X. Maternal smoking during pregnancy and cord blood DNA methylation: New insight on sex differences and effect modification by maternal folate levels. Epigenetics 2018, 13, 505–518. [Google Scholar] [CrossRef]
- Fuseini, H.; Newcomb, D.C. Mechanisms Driving Gender Differences in Asthma. Curr. Allergy Asthma Rep. 2017, 17, 19. [Google Scholar] [CrossRef]
- Chowdhury, N.U.; Guntur, V.P.; Newcomb, D.C.; Wechsler, M.E. Sex and gender in asthma. Eur. Respir. Rev. 2021, 30, 210067. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Newcomb, D.C. Sex Bias in Asthma Prevalence and Pathogenesis. Front. Immunol. 2018, 9, 2997. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, N.; Reinius, L.E.; Vitezic, M.; Fortino, V.; Söderhäll, C.; Honkanen, H.; Veijola, R.; Simell, O.; Toppari, J.; Ilonen, J.; et al. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clin. Epigenet. 2015, 7, 34. [Google Scholar] [CrossRef]
- Martino, D.; Loke, Y.J.; Gordon, L.; Ollikainen, M.; Cruickshank, M.N.; Saffery, R.; Craig, J.M. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013, 14, R42. [Google Scholar] [CrossRef]
- Patel, R.; Solatikia, F.; Zhang, H.; Wolde, A.; Kadalayil, L.; Karmaus, W.; Ewart, S.; Arathimos, R.; Relton, C.; Ring, S.; et al. Sex-specific associations of asthma acquisition with changes in DNA methylation during adolescence. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2021, 51, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Palmer, E.; Sanver, D.; Kirtland, M.; Shamji, M.H. Molecular allergology approach to allergic asthma. Mol. Asp. Med. 2022, 85, 101027. [Google Scholar] [CrossRef] [PubMed]
- Abellan, A.; Mensink-Bout, S.M.; Garcia-Esteban, R.; Beneito, A.; Chatzi, L.; Duarte-Salles, T.; Fernandez, M.F.; Garcia-Aymerich, J.; Granum, B.; Iñiguez, C.; et al. In utero exposure to bisphenols and asthma, wheeze, and lung function in school-age children: A prospective meta-analysis of 8 European birth cohorts. Environ. Int. 2022, 162, 107178. [Google Scholar] [CrossRef]
- Lee, A.; Leon Hsu, H.H.; Mathilda Chiu, Y.H.; Bose, S.; Rosa, M.J.; Kloog, I.; Wilson, A.; Schwartz, J.; Cohen, S.; Coull, B.A.; et al. Prenatal fine particulate exposure and early childhood asthma: Effect of maternal stress and fetal sex. J. Allergy Clin. Immunol. 2018, 141, 1880–1886. [Google Scholar] [CrossRef]
- Burke, H.; Leonardi-Bee, J.; Hashim, A.; Pine-Abata, H.; Chen, Y.; Cook, D.G.; Britton, J.R.; McKeever, T.M. Prenatal and passive smoke exposure and incidence of asthma and wheeze: Systematic review and meta-analysis. Pediatrics 2012, 129, 735–744. [Google Scholar] [CrossRef]
- Sheikhpour, M.; Maleki, M.; Ebrahimi Vargoorani, M.; Amiri, V. A review of epigenetic changes in asthma: Methylation and acetylation. Clin. Epigenetics 2021, 13, 65. [Google Scholar] [CrossRef]
- Cardenas, A.; Fadadu, R.P.; Koppelman, G.H. Epigenome-wide association studies of allergic disease and the environment. J. Allergy Clin. Immunol. 2023, 7, S0091. [Google Scholar] [CrossRef]
- de Prado-Bert, P.; Ruiz-Arenas, C.; Vives-Usano, M.; Andrusaityte, S.; Cadiou, S.; Carracedo, Á.; Casas, M.; Chatzi, L.; Dadvand, P.; González, J.R.; et al. The early-life exposome and epigenetic age acceleration in children. Environ. Int. 2021, 155, 106683. [Google Scholar] [CrossRef]
- Strichman-Almashanu, L.Z.; Lee, R.S.; Onyango, P.O.; Perlman, E.; Flam, F.; Frieman, M.B.; Feinberg, A.P. A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 2002, 12, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Le, J.; Barnes, B.; Saedinia-Melnyk, S.; Zhou, L.; Shen, R.; Gunderson, K.L. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 2009, 1, 177–200. [Google Scholar] [CrossRef]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef]
- Talens, R.P.; Christensen, K.; Putter, H.; Willemsen, G.; Christiansen, L.; Kremer, D.; Suchiman, H.E.; Slagboom, P.E.; Boomsma, D.I.; Heijmans, B.T. Epigenetic variation during the adult lifespan: Cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell 2012, 11, 694–703. [Google Scholar] [CrossRef]
- Day, K.; Waite, L.L.; Thalacker-Mercer, A.; West, A.; Bamman, M.M.; Brooks, J.D.; Myers, R.M.; Absher, D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013, 14, R102. [Google Scholar] [CrossRef]
- Bell, J.T.; Tsai, P.C.; Yang, T.P.; Pidsley, R.; Nisbet, J.; Glass, D.; Mangino, M.; Zhai, G.; Zhang, F.; Valdes, A.; et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012, 8, e1002629. [Google Scholar] [CrossRef]
- Martino, D.J.; Tulic, M.K.; Gordon, L.; Hodder, M.; Richman, T.R.; Metcalfe, J.; Prescott, S.L.; Saffery, R. Evidence for age-related and individual-specific changes in DNA methylation profile of mononuclear cells during early immune development in humans. Epigenetics 2011, 6, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Herbstman, J.B.; Wang, S.; Perera, F.P.; Lederman, S.A.; Vishnevetsky, J.; Rundle, A.G.; Hoepner, L.A.; Qu, L.; Tang, D. Predictors and consequences of global DNA methylation in cord blood and at three years. PLoS ONE 2013, 8, e72824. [Google Scholar] [CrossRef] [PubMed]
- Urdinguio, R.G.; Torró, M.I.; Bayón, G.F.; Álvarez-Pitti, J.; Fernández, A.F.; Redon, P.; Fraga, M.F.; Lurbe, E. Longitudinal study of DNA methylation during the first 5 years of life. J. Transl. Med. 2016, 14, 160. [Google Scholar] [CrossRef] [PubMed]
- Daya, M.; Cox, C.; Acevedo, N.; Boorgula, M.P.; Campbell, M.; Chavan, S.; Cho, M.H.; David, G.L.; Kachroo, P.; Lasky-Su, J.; et al. Multiethnic genome-wide and HLA association study of total serum IgE level. J. Allergy Clin. Immunol. 2021, 148, 1589–1595. [Google Scholar] [CrossRef]
- Madore, A.M.; Vaillancourt, V.T.; Asai, Y.; Alizadehfar, R.; Ben-Shoshan, M.; Michel, D.L.; Kozyrskyj, A.L.; Becker, A.; Chan-Yeung, M.; Clarke, A.E.; et al. HLA-DQB1*02 and DQB1*06:03P are associated with peanut allergy. Eur. J. Hum. Genet. 2013, 21, 1181–1184. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the immune system in humans from infancy to old age. Proc. Biol. Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Berger, A. Th1 and Th2 responses: What are they? BMJ 2000, 321, 424. [Google Scholar] [CrossRef]
- Holgate, S.T. Innate and adaptive immune responses in asthma. Nat. Med. 2012, 18, 673–683. [Google Scholar] [CrossRef]
- Vijeyakumaran, M.; Jawhri, M.A.; Fortunato, J.; Solomon, L.; Shrestha Palikhe, N.; Vliagoftis, H.; Cameron, L. Dual activation of estrogen receptor alpha and glucocorticoid receptor upregulate CRTh2-mediated type 2 inflammation; mechanism driving asthma severity in women? Allergy 2023, 78, 767–779. [Google Scholar] [CrossRef]
- Zhang, H.; Tong, X.; Holloway, J.W.; Rezwan, F.I.; Lockett, G.A.; Patil, V.; Ray, M.; Everson, T.M.; Soto-Ramirez, N.; Arshad, S.H.; et al. The interplay of DNA methylation over time with Th2 pathway genetic variants on asthma risk and temporal asthma transition. Clin. Epigenet. 2014, 6, 8. [Google Scholar] [CrossRef]
- Alisch, R.S.; Barwick, B.G.; Chopra, P.; Myrick, L.K.; Satten, G.A.; Conneely, K.N.; Warren, S.T. Age-associated DNA methylation in pediatric populations. Genome Res. 2012, 22, 623–632. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Snir, S.; Farrell, C.; Pellegrini, M. Human epigenetic ageing is logarithmic with time across the entire lifespan. Epigenetics 2019, 14, 912–926. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef]
- Vaiserman, A. Developmental Tuning of Epigenetic Clock. Front. Genet. 2018, 9, 584. [Google Scholar] [CrossRef] [PubMed]
- Cephus, J.Y.; Stier, M.T.; Fuseini, H.; Yung, J.A.; Toki, S.; Bloodworth, M.H.; Zhou, W.; Goleniewska, K.; Zhang, J.; Garon, S.L.; et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep. 2017, 21, 2487–2499. [Google Scholar] [CrossRef]
- Murakami, Y.; Fahmy, S.; Goldblum, R.M.; Watson, C.S.; Midoro- Horiuti, T. Environmental estrogen exposures alter molecular signaling in immune cells that promote the development of childhood asthma. Mol. Immunol. 2023, 157, 142–145. [Google Scholar] [CrossRef]
- Sowers, M.L.; Tang, H.; Tian, B.; Goldblum, R.; Midoro-Horiuti, T.; Zhang, K. Bisphenol A Activates an Innate Viral Immune Response Pathway. J. Proteome Res. 2020, 19, 644–654. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Society. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Cardenas, A.; Ecker, S.; Fadadu, R.P.; Huen, K.; Orozco, A.; McEwen, L.M.; Engelbrecht, H.-R.; Gladish, N.; Kobor, M.S.; Rosero-Bixby, L.; et al. Epigenome-wide association study and epigenetic age acceleration associated with cigarette smoking among Costa Rican adults. Sci. Rep. 2022, 12, 4277. [Google Scholar] [CrossRef] [PubMed]
- Perna, L.; Zhang, Y.; Mons, U.; Holleczek, B.; Saum, K.-U.; Brenner, H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin. Epigenet. 2016, 8, 64. [Google Scholar] [CrossRef]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017, 9, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Fransquet, P.D.; Wrigglesworth, J.; Woods, R.L.; Ernst, M.E.; Ryan, J. The epigenetic clock as a predictor of disease and mortality risk: A systematic review and meta-analysis. Clin. Epigenet. 2019, 11, 62. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- McEwen, L.M.; O’Donnell, K.J.; McGill, M.G.; Edgar, R.D.; Jones, M.J.; MacIsaac, J.L.; Lin, D.T.S.; Ramadori, K.; Morin, A.; Gladish, N.; et al. The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells. Proc. Natl. Acad. Sci. USA 2020, 117, 23329–23335. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.R.; Lin, N.; Liu, J.; Storniolo, A.M.V.; Shendre, A.; Hou, L.; Horvath, S.; Liu, Y.; Wang, C.; He, C. Estimating breast tissue-specific DNA methylation age using next-generation sequencing data. Clin. Epigenet. 2020, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.M.; Wagner, W. Epigenetic-aging-signature to determine age in different tissues. Aging 2011, 3, 1018–1027. [Google Scholar] [CrossRef]
- Knight, A.K.; Craig, J.M.; Theda, C.; Bækvad-Hansen, M.; Bybjerg-Grauholm, J.; Hansen, C.S.; Hollegaard, M.V.; Hougaard, D.M.; Mortensen, P.B.; Weinsheimer, S.M.; et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 2016, 17, 206. [Google Scholar] [CrossRef]
- Haftorn, K.L.; Lee, Y.; Denault, W.R.P.; Page, C.M.; Nustad, H.E.; Lyle, R.; Gjessing, H.K.; Malmberg, A.; Magnus, M.C.; Næss, Ø.; et al. An EPIC predictor of gestational age and its application to newborns conceived by assisted reproductive technologies. Clin. Epigenet. 2021, 13, 82. [Google Scholar] [CrossRef]
- Simpkin, A.J.; Hemani, G.; Suderman, M.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Sharp, G.C.; Tilling, K.; Horvath, S.; et al. Prenatal and early life influences on epigenetic age in children: A study of mother-offspring pairs from two cohort studies. Hum. Mol. Genet. 2016, 25, 191–201. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- McEwen, L.M.; Jones, M.J.; Lin, D.T.S.; Edgar, R.D.; Husquin, L.T.; MacIsaac, J.L.; Ramadori, K.E.; Morin, A.M.; Rider, C.F.; Carlsten, C.; et al. Systematic evaluation of DNA methylation age estimation with common preprocessing methods and the Infinium MethylationEPIC BeadChip array. Clin. Epigenet. 2018, 10, 123. [Google Scholar] [CrossRef]
- El Khoury, L.Y.; Gorrie-Stone, T.; Smart, M.; Hughes, A.; Bao, Y.; Andrayas, A.; Burrage, J.; Hannon, E.; Kumari, M.; Mill, J.; et al. Systematic underestimation of the epigenetic clock and age acceleration in older subjects. Genome Biol. 2019, 20, 283. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Gurven, M.; Levine, M.E.; Trumble, B.C.; Kaplan, H.; Allayee, H.; Ritz, B.R.; Chen, B.; Lu, A.T.; Rickabaugh, T.M.; et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016, 17, 171. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Xue, L.; Salfati, E.L.; Chen, B.H.; Ferrucci, L.; Levy, D.; Joehanes, R.; Murabito, J.M.; Kiel, D.P.; Tsai, P.-C.; et al. GWAS of epigenetic aging rates in blood reveals a critical role for TERT. Nat. Commun. 2018, 9, 387. [Google Scholar] [CrossRef]
- Smith, J.A.; Raisky, J.; Ratliff, S.M.; Liu, J.; Kardia, S.L.R.; Turner, S.T.; Mosley, T.H.; Zhao, W. Intrinsic and extrinsic epigenetic age acceleration are associated with hypertensive target organ damage in older African Americans. BMC Med. Genom. 2019, 12, 141. [Google Scholar] [CrossRef]
- Bantz, S.K.; Zhu, Z.; Zheng, T. The Atopic March: Progression from Atopic Dermatitis to Allergic Rhinitis and Asthma. J. Clin. Cell Immunol. 2014, 5, 202. [Google Scholar] [CrossRef]
- Chan, E.S.; Abrams, E.M.; Hildebrand, K.J.; Watson, W. Early introduction of foods to prevent food allergy. Allergy Asthma Clin. Immunol. 2018, 14, 57. [Google Scholar] [CrossRef]
- McGeachie, M.J.; Stahl, E.A.; Himes, B.E.; Pendergrass, S.A.; Lima, J.J.; Irvin, C.G.; Peters, S.P.; Ritchie, M.D.; Plenge, R.M.; Tantisira, K.G. Polygenic heritability estimates in pharmacogenetics: Focus on asthma and related phenotypes. Pharmacogenetics Genom. 2013, 23, 324–328. [Google Scholar] [CrossRef]
- Thomsen, S.F. Genetics of asthma: An introduction for the clinician. Eur. Clin. Respir. J. 2015, 2, 24643. [Google Scholar] [CrossRef] [PubMed]
- Kontakioti, E.; Domvri, K.; Papakosta, D.; Daniilidis, M. HLA and asthma phenotypes/endotypes: A review. Hum. Immunol. 2014, 75, 930–939. [Google Scholar] [CrossRef]
- Asai, Y.; Eslami, A.; van Ginkel, C.D.; Akhabir, L.; Wan, M.; Yin, D.; Ellis, G.; Ben-Shoshan, M.; Marenholz, I.; Martino, D.; et al. A Canadian genome-wide association study and meta-analysis confirm HLA as a risk factor for peanut allergy independent of asthma. J. Allergy Clin. Immunol. 2018, 141, 1513–1516. [Google Scholar] [CrossRef]
- Everson, T.M.; Zhang, H.; Lockett, G.A.; Kaushal, A.; Forthofer, M.; Ewart, S.L.; Burrows, K.; Relton, C.L.; Sharp, G.C.; Henderson, A.J.; et al. Epigenome-wide association study of asthma and wheeze characterizes loci within HK1. Allergy Asthma Clin. Immunol. 2019, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Forno, E.; Wang, T.; Qi, C.; Yan, Q.; Xu, C.J.; Boutaoui, N.; Han, Y.Y.; Weeks, D.E.; Jiang, Y.; Rosser, F.; et al. DNA methylation in nasal epithelium, atopy, and atopic asthma in children: A genome-wide study. Lancet Respir. Med. 2019, 7, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Park, J.E.; He, J.Q.; Yan, J.; Akhabir, L.; Stefanowicz, D.; Becker, A.B.; Chan-Yeung, M.; Bossé, Y.; Kozyrskyj, A.L.; et al. Associations and interactions of genetic polymorphisms in innate immunity genes with early viral infections and susceptibility to asthma and asthma-related phenotypes. J. Allergy Clin. Immunol. 2012, 130, 1284–1293. [Google Scholar] [CrossRef]
- Paller, A.S.; Spergel, J.M.; Mina-Osorio, P.; Irvine, A.D. The atopic march and atopic multimorbidity: Many trajectories, many pathways. J. Allergy Clin. Immunol. 2019, 143, 46–55. [Google Scholar] [CrossRef]
- Daley, D. The evolution of the hygiene hypothesis: The role of early-life exposures to viruses and microbes and their relationship to asthma and allergic diseases. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, S.; Rivero-Calle, I.; Gómez-Carballa, A.; Cebey-López, M.; Barral-Arca, R.; Gómez-Rial, J.; Pardo-Seco, J.; Curras-Tuala, M.J.; Viz-Lasheras, S.; Bello, X.; et al. Role and Diagnostic Performance of Host Epigenome in Respiratory Morbidity after RSV Infection: The EPIRESVi Study. Front. Immunol. 2022, 13, 875691. [Google Scholar] [CrossRef]
- Joyce, B.T.; Gao, T.; Zheng, Y.; Ma, J.; Hwang, S.J.; Liu, L.; Nannini, D.; Horvath, S.; Lu, A.T.; Bai Allen, N.; et al. Epigenetic Age Acceleration Reflects Long-Term Cardiovascular Health. Circ. Res. 2021, 129, 770–781. [Google Scholar] [CrossRef]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Ritchie, S.J.; Muniz-Terrera, G.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396. [Google Scholar] [CrossRef]
- Breen, M.; Nwanaji-Enwerem, J.C.; Karrasch, S.; Flexeder, C.; Schulz, H.; Waldenberger, M.; Kunze, S.; Ollert, M.; Weidinger, S.; Colicino, E.; et al. Accelerated epigenetic aging as a risk factor for chronic obstructive pulmonary disease and decreased lung function in two prospective cohort studies. Aging 2020, 12, 16539–16554. [Google Scholar] [CrossRef]
- Arefeen, M.A.; Nimi, S.T.; Rahman, M.S.; Arshad, S.H.; Holloway, J.W.; Rezwan, F.I. Prediction of Lung Function in Adolescence Using Epigenetic Aging: A Machine Learning Approach. Methods Protoc. 2020, 3, 77. [Google Scholar] [CrossRef]
- Chlamydas, S.; Markouli, M.; Strepkos, D.; Piperi, C. Epigenetic mechanisms regulate sex-specific bias in disease manifestations. J. Mol. Med. 2022, 100, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.; Harden, K.; Forsythe, A.; Chilingar, L.; Hoffman, C.; Sperling, W.; Zeiger, R.S. The course of asthma during pregnancy, post partum, and with successive pregnancies: A prospective analysis. J. Allergy Clin. Immunol. 1988, 81, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Murphy, V.E.; Gibson, P.G.; Smith, R.; Clifton, V.L. Asthma during pregnancy: Mechanisms and treatment implications. Eur. Respir. J. 2005, 25, 731–750. [Google Scholar] [CrossRef]
- Taneja, V. Sex Hormones Determine Immune Response. Front Immunol 2018, 9, 1931. [Google Scholar] [CrossRef]
- Yung, J.A.; Fuseini, H.; Newcomb, D.C. Hormones, sex, and asthma. Ann. Allergy Asthma Immunol. 2018, 120, 488–494. [Google Scholar] [CrossRef]
- Lieberoth, S.; Gade, E.J.; Brok, J.; Backer, V.; Thomsen, S.F. Age at menarche and risk of asthma: Systematic review and meta-analysis. J. Asthma 2014, 51, 559–565. [Google Scholar] [CrossRef] [PubMed]
- McCleary, N.; Nwaru, B.I.; Nurmatov, U.B.; Critchley, H.; Sheikh, A. Endogenous and exogenous sex steroid hormones in asthma and allergy in females: A systematic review and meta-analysis. J. Allergy Clin. Immunol. 2018, 141, 1510–1513.e8. [Google Scholar] [CrossRef]
- Balzano, G.; Fuschillo, S.; Melillo, G.; Bonini, S. Asthma and sex hormones. Allergy 2001, 56, 13–20. [Google Scholar] [CrossRef]
- Han, Y.Y.; Forno, E.; Celedón, J.C. Sex Steroid Hormones and Asthma in a Nationwide Study of U.S. Adults. Am. J. Respir. Crit. Care Med. 2020, 201, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Julia, D. Use of oestrogen only hormone replacement therapy associated with increased risk of asthma onset in postmenopausal women. Evid. Based Med. 2010, 15, 190. [Google Scholar] [CrossRef]
- Midoro-Horiuti, T.; Tiwari, R.; Watson, C.S.; Goldblum, R.M. Maternal bisphenol a exposure promotes the development of experimental asthma in mouse pups. Environ. Health Perspect. 2010, 118, 273–277. [Google Scholar] [CrossRef]
- Nwaru, B.I.; Sheikh, A. Hormonal contraceptives and asthma in women of reproductive age: Analysis of data from serial national Scottish Health Surveys. J. R Soc. Med. 2015, 108, 358–371. [Google Scholar] [CrossRef]
- Macsali, F.; Real, F.G.; Omenaas, E.R.; Bjorge, L.; Janson, C.; Franklin, K.; Svanes, C. Oral contraception, body mass index, and asthma: A cross-sectional Nordic-Baltic population survey. J. Allergy Clin. Immunol. 2009, 123, 391–397. [Google Scholar] [CrossRef]
- Kankaanpää, A.; Tolvanen, A.; Saikkonen, P.; Heikkinen, A.; Laakkonen, E.K.; Kaprio, J.; Ollikainen, M.; Sillanpää, E. Do Epigenetic Clocks Provide Explanations for Sex Differences in Life Span? A Cross-Sectional Twin Study. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Yusipov, I.; Bacalini, M.G.; Kalyakulina, A.; Krivonosov, M.; Pirazzini, C.; Gensous, N.; Ravaioli, F.; Milazzo, M.; Giuliani, C.; Vedunova, M.; et al. Age-related DNA methylation changes are sex-specific: A comprehensive assessment. Aging 2020, 12, 24057–24080. [Google Scholar] [CrossRef] [PubMed]
- Binder, A.M.; Corvalan, C.; Mericq, V.; Pereira, A.; Santos, J.L.; Horvath, S.; Shepherd, J.; Michels, K.B. Faster ticking rate of the epigenetic clock is associated with faster pubertal development in girls. Epigenetics 2018, 13, 85–94. [Google Scholar] [CrossRef]
- Sugrue, V.J.; Zoller, J.A.; Narayan, P.; Lu, A.T.; Ortega-Recalde, O.J.; Grant, M.J.; Bawden, C.S.; Rudiger, S.R.; Haghani, A.; Bond, D.M.; et al. Castration delays epigenetic aging and feminizes DNA methylation at androgen-regulated loci. eLife 2021, 10, e64932. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Chen, B.H.; Hernandez, D.G.; Singleton, A.B.; Ferrucci, L.; Bandinelli, S.; Salfati, E.; Manson, J.E.; Quach, A.; et al. Menopause accelerates biological aging. Proc. Natl. Acad. Sci. USA 2016, 113, 9327–9332. [Google Scholar] [CrossRef]
- Stubbs, T.M.; Bonder, M.J.; Stark, A.-K.; Krueger, F.; Bolland, D.; Butcher, G.; Chandra, T.; Clark, S.J.; Corcoran, A.; Eckersley-Maslin, M.; et al. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, H.; Kaushal, A.; Rezwan, F.I.; Kadalayil, L.; Karmaus, W.; Henderson, A.J.; Relton, C.L.; Ring, S.; Arshad, S.H.; et al. Changes in DNA methylation from pre- to post-adolescence are associated with pubertal exposures. Clin. Epigenetics 2019, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Kananen, L.; Marttila, S.; Nevalainen, T.; Kummola, L.; Junttila, I.; Mononen, N.; Kähönen, M.; Raitakari, O.T.; Hervonen, A.; Jylhä, M.; et al. The trajectory of the blood DNA methylome ageing rate is largely set before adulthood: Evidence from two longitudinal studies. AGE 2016, 38, 65. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Publication | CpG Site | CHR | Position (hg38) | Gene | Associated Exposures or Phenotypes |
|---|---|---|---|---|---|
| [20] | cg14547404 | 10 | 48653753 | ARHGAP22 | Air Pollution |
| [20] | cg06517429 | 10 | 113679876 | CASP7 | Air Pollution |
| [21,25] | cg26995690 | 13 | 35772239 | DCLK1 | Birthweight |
| [21,25] | cg00637745 | 2 | 120739758 | Birthweight | |
| [21,25] | cg07133097 | 2 | 120739962 | Birthweight | |
| [19] | cg10593758 | 5 | 76952917 | CRHBP | Elevated Maternal BMI |
| [19] | cg07621682 | 19 | 41321853 | CCDC97 | Elevated Maternal BMI |
| [21,22,26] | cg11932158 | 3 | 155704340 | PLCH1 | Gestational Age |
| [21,26] | cg18623216 | 3 | 155704181 | PLCH1 | Gestational Age |
| [21,26] | cg16103712 | 8 | 98011641 | MATN2 | Gestational Age |
| [21,26] | cg17133774 | 1 | 6138607 | CHD5 | Gestational Age |
| [21,26] | cg12713583 | 19 | 940724 | ARID3A | Gestational Age |
| [21,26] | cg04347477 | 12 | 124517461 | NCOR2 | Gestational Age |
| [21,26] | cg08817867 | 17 | 19753241 | Gestational Age | |
| [21,26] | cg02001279 | 19 | 940967 | ARID3A | Gestational Age |
| [21,26] | cg08412913 | 16 | 85395916 | DOCK6 | Gestational Age |
| [21,26] | cg06870470 | 19 | 11205091 | Gestational Age | |
| [21,24,27] | cg05549655 | 15 | 74726802 | CYP1A1 | Maternal Smoking |
| [21,23,27,28] | cg11924019 | 15 | 74726942 | CYP1A1 | Maternal Smoking |
| [21,23,27,28] | cg22549041 | 15 | 74726910 | CYP1A1 | Maternal Smoking |
| [21,23,24,27,28,29] | cg23067299 | 5 | 323791 | AHRR | Maternal Smoking |
| [21,23,24,27] | cg22132788 | 7 | 44962886 | MYO1G | Maternal Smoking |
| [21,23,27,28] | cg18092474 | 15 | 74726961 | CYP1A1 | Maternal Smoking |
| [21,23,24,27,28] | cg12803068 | 7 | 44963320 | MYO1G | Maternal Smoking |
| [21,23,27] | cg12101586 | 15 | 74726862 | CYP1A1 | Maternal Smoking |
| Type | Epigenetic Clock | Tissue Type | Methodology Used | Methylation Technology | Strengths | Limitations |
|---|---|---|---|---|---|---|
| Single Tissue | [83] | Single Tissue: Saliva | Association Analysis | Illumina 27 K Array | First epigenetic clock | Low accuracy (mean AE: 5.2 years) |
| [79] | Single Tissue: Whole Blood | Elastic Net with bootstrapping | Illumina 450 K Array | Accurate in blood. Extensively used | Limited age range of training samples: 19–101 years | |
| [84] | Single Tissue: Whole Blood | Multivariate Linear Regression | Pyrosequencing | Consists of only three CpG sites | Low accuracy (mean AE: 5.4 years) | |
| [85] | Single Tissue: Breast Tissue | Elastic Net Regression with cross-validation | TruSeq Methyl Capture EPIC library | Improved accuracy in breast tissue | TruSeq Methyl Capture not yet widely used | |
| Multi-Tissue | [86] | Multi-Tissue: Epidermis, dermis, T-cells, cervical smear, and monocytes | Pearson Correlation | Illumina 27 K Array | First multi-tissue clock | Relative low accuracy (mean AE: 11 years) |
| [65] | Multi-Tissue: 51 tissue and cell types | Elastic Net with ten-fold cross-validation | Illumina 27 K Array and Illumina 450 K Array | Accurate across tissues; extensively used | Mostly adult samples Age of neonate samples set at “0” | |
| [80] | Multi-Tissue including blood | Elastic Net with ten-fold cross-validation | Illumina 450 K and Illumina EPIC Array | Accurate (mean AE: 2.5 years) | Not widely used yet | |
| Pediatric Single Tissue | [81] | Single Tissue: Buccal Cells | Elastic Net with cross-validation | Illumina EPIC Array | Pediatric-only clock | Low accuracy in blood |
| Gestational Age | [87] | Cord Blood | Elastic Net Regression with cross-validation | Illumina 27 K array and Illumina 450 K Array | Median error: 1.24 weeks | Gestational Age Only |
| [88] | Cord Blood | Lasso Regression with cross-validation | Illumina EPIC Array | Uses the EPIC Array | Gestational Age Only | |
| [26] | Cord Blood | Elastic Net Regression | Illumina 450 K Array | Correlation with Gestational Age | Gestational Age Only |
| Epigenetic Clock Citation | Tissue Type | Methodology Used | Platform | Strengths | Limitations |
|---|---|---|---|---|---|
| [77] | Elastic Net with Cross Validation | Phenotypic Age | Illumina EPIC Array | Composite of aging; well correlated with morbidity | Utility of childhood samples is unknown |
| [90] | Elastic Net with Cross Validation | Time-to-Death | Illumina 450 K and EPIC Arrays | Well correlated with mortality | Utility of childhood samples is unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasileva, D.; Greenwood, C.M.T.; Daley, D. A Review of the Epigenetic Clock: Emerging Biomarkers for Asthma and Allergic Disease. Genes 2023, 14, 1724. https://doi.org/10.3390/genes14091724
Vasileva D, Greenwood CMT, Daley D. A Review of the Epigenetic Clock: Emerging Biomarkers for Asthma and Allergic Disease. Genes. 2023; 14(9):1724. https://doi.org/10.3390/genes14091724
Chicago/Turabian StyleVasileva, Denitsa, Celia M. T. Greenwood, and Denise Daley. 2023. "A Review of the Epigenetic Clock: Emerging Biomarkers for Asthma and Allergic Disease" Genes 14, no. 9: 1724. https://doi.org/10.3390/genes14091724