
A Pervasive History of Gene Flow in Madagascar’s True Lemurs (Genus Eulemur)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
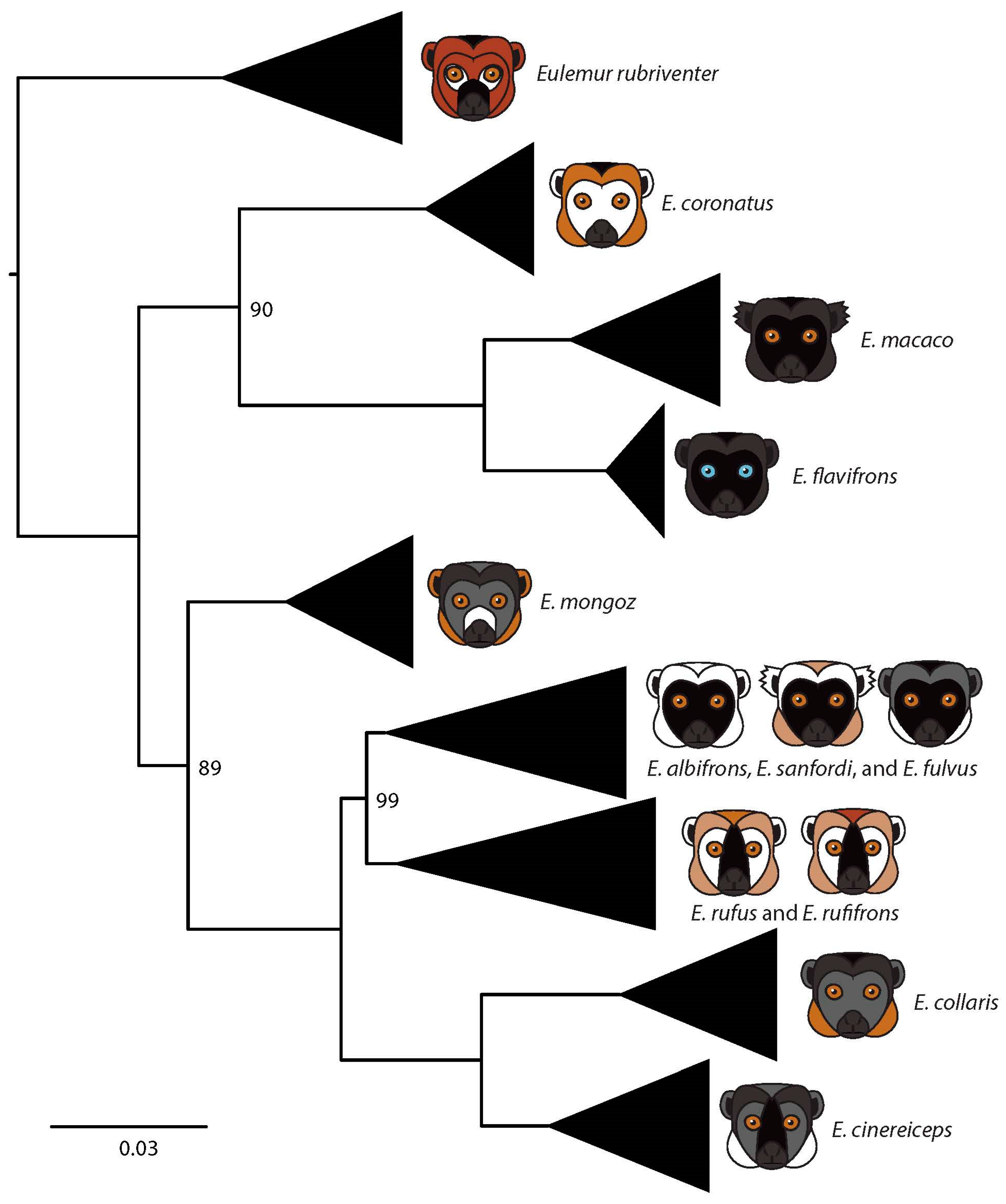
3.1. Mitochondrial Phylogeny
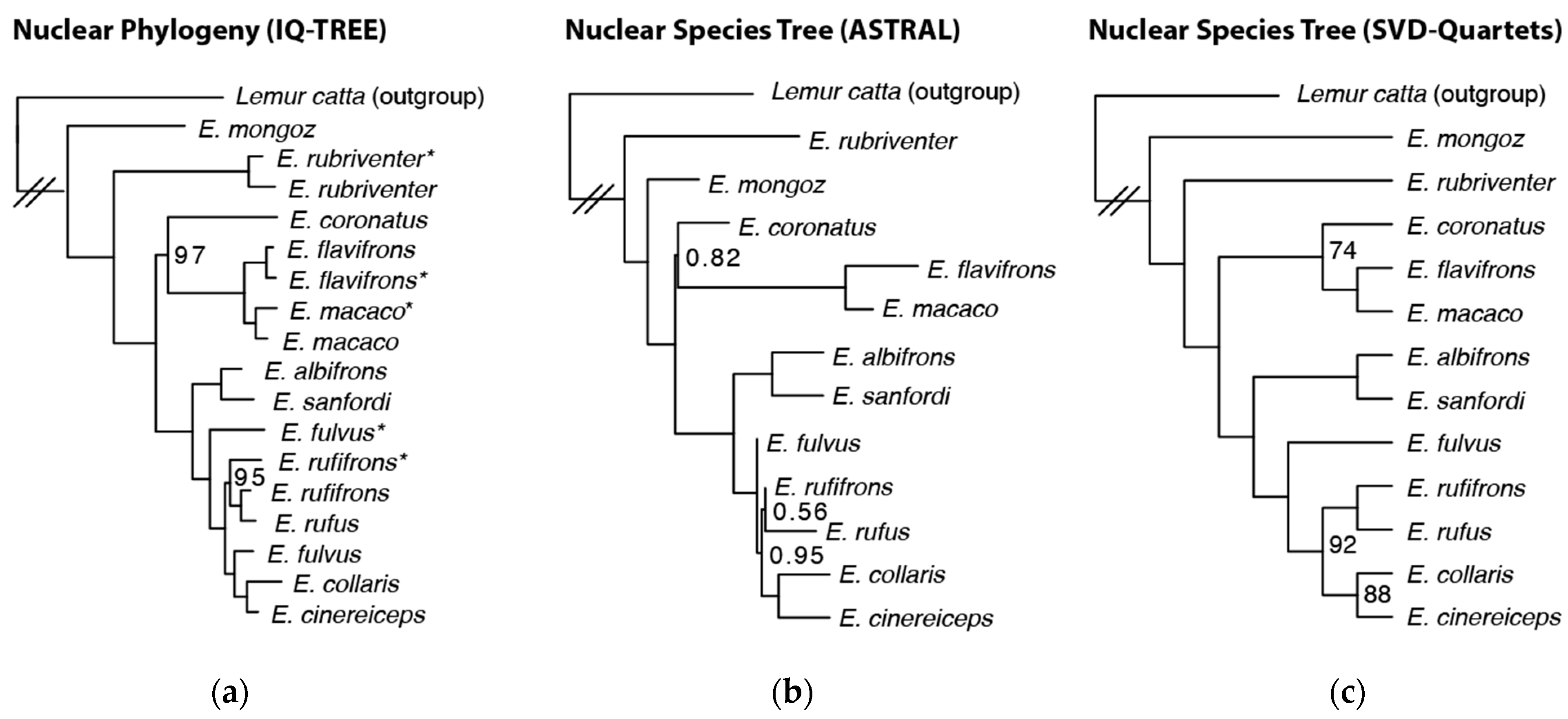
3.2. Nuclear Species Tree
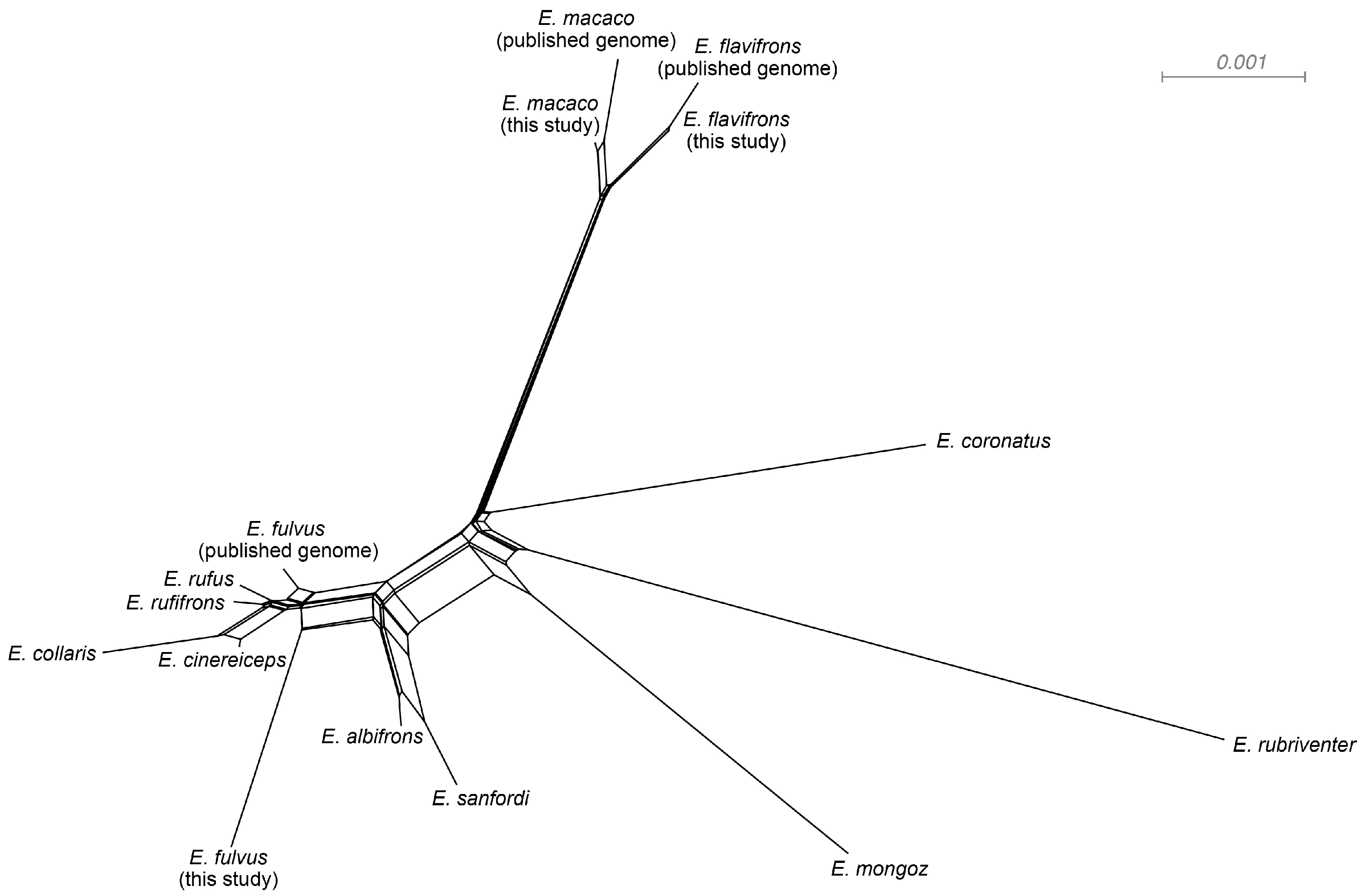
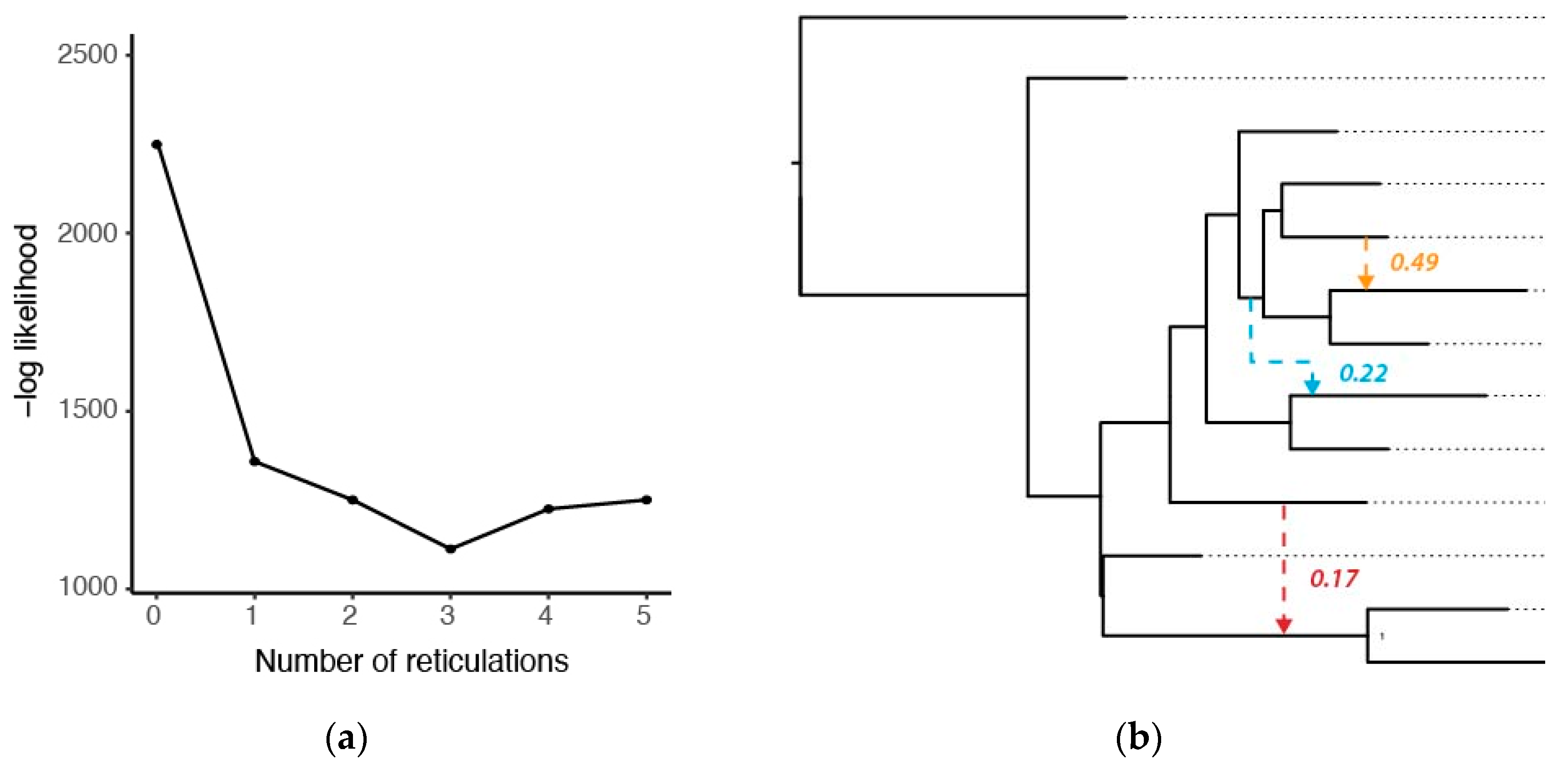
3.3. Phylogenetic Networks
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barth, J.M.I.; Gubili, C.; Matschiner, M.; Tørresen, O.K.; Watanabe, S.; Egger, B.; Han, Y.-S.; Feunteun, E.; Sommaruga, R.; Jehle, R.; et al. Stable Species Boundaries despite Ten Million Years of Hybridization in Tropical Eels. Nat. Commun. 2020, 11, 1433. [Google Scholar] [CrossRef] [PubMed]
- Nieto Feliner, G.; Álvarez, I.; Fuertes-Aguilar, J.; Heuertz, M.; Marques, I.; Moharrek, F.; Piñeiro, R.; Riina, R.; Rosselló, J.A.; Soltis, P.S.; et al. Is Homoploid Hybrid Speciation That Rare? An Empiricist’s View. Heredity 2017, 118, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Folk, R.A.; Soltis, P.S.; Soltis, D.E.; Guralnick, R. New Prospects in the Detection and Comparative Analysis of Hybridization in the Tree of Life. Am. J. Bot. 2018, 105, 364–375. [Google Scholar] [CrossRef]
- Mallet, J.; Besansky, N.; Hahn, M.W. How Reticulated Are Species? BioEssays 2016, 38, 140–149. [Google Scholar] [CrossRef]
- Sites, J.W.; Marshall, J.C. Operational Criteria for Delimiting Species. Annu. Rev. Ecol. Evol. Syst. 2004, 35, 199–227. [Google Scholar] [CrossRef]
- Harrison, R.G.; Larson, E.L. Hybridization, Introgression, and the Nature of Species Boundaries. J. Hered. 2014, 105, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E. Animal Species and Evolution; Harvard University Press: Cambridge, MA, USA, 1963. [Google Scholar]
- Mallet, J. Hybridization as an Invasion of the Genome. Trends Ecol. Evol. 2005, 20, 229–237. [Google Scholar] [CrossRef]
- Abbott, R.J.; Barton, N.H.; Good, J.M. Genomics of Hybridization and Its Evolutionary Consequences. Mol. Ecol. 2016, 25, 2325–2332. [Google Scholar] [CrossRef]
- Taylor, S.A.; Larson, E.L. Insights from Genomes into the Evolutionary Importance and Prevalence of Hybridization in Nature. Nat. Ecol. Evol. 2019, 3, 170–177. [Google Scholar] [CrossRef]
- Mallet, J. Hybrid Speciation. Nature 2007, 446, 279–283. [Google Scholar] [CrossRef]
- Yu, Y.; Barnett, R.; Nakhleh, L. Parsimonious Inference of Hybridization in the Presence of Incomplete Lineage Sorting. Syst. Biol. 2013, 62, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Kubatko, L.S. Detecting Hybrid Speciation in the Presence of Incomplete Lineage Sorting Using Gene Tree Incongruence: A Model. Theor. Popul. Biol. 2009, 75, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.; McLenachan, P.; Lockhart, P. A Statistical Approach for Distinguishing Hybridization and Incomplete Lineage Sorting. Am. Nat. 2009, 174, E54–E70. [Google Scholar] [CrossRef] [PubMed]
- Holland, B.; Benthin, S.; Lockhart, P.; Moulton, V.; Huber, K. Using Supernetworks to Distinguish Hybridization from Lineage-Sorting. BMC Evol. Biol. 2008, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Bloomquist, E.W.; Suchard, M.A. Unifying Vertical and Nonvertical Evolution: A Stochastic ARG-Based Framework. Syst. Biol. 2010, 59, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Leaché, A.D.; Harris, R.B.; Rannala, B.; Yang, Z. The Influence of Gene Flow on Species Tree Estimation: A Simulation Study. Syst. Biol. 2014, 63, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.J.; Carstens, B.C. Does Gene Flow Destroy Phylogenetic Signal? The Performance of Three Methods for Estimating Species Phylogenies in the Presence of Gene Flow. Mol. Phylogenet Evol. 2008, 49, 832–842. [Google Scholar] [CrossRef]
- Morales, A.E.; Jackson, N.D.; Dewey, T.A.; O’Meara, B.C.; Carstens, B.C. Speciation with Gene Flow in North American Myotis Bats. Syst. Biol. 2016, 66, 440–452. [Google Scholar] [CrossRef]
- Evans, P.D.; Mekel-Bobrov, N.; Vallender, E.J.; Hudson, R.R.; Lahn, B.T. Evidence That the Adaptive Allele of the Brain Size Gene Microcephalin Introgressed into Homo Sapiens from an Archaic Homo Lineage. Proc. Natl. Acad. Sci. USA 2006, 103, 18178–18183. [Google Scholar] [CrossRef]
- Hawks, J.; Cochran, G.; Harpending, H.C.; Lahn, B.T. A Genetic Legacy from Archaic Homo. Trends Genet. 2008, 24, 19–23. [Google Scholar] [CrossRef]
- Racimo, F.; Sankararaman, S.; Nielsen, R.; Huerta-Sánchez, E. Evidence for Archaic Adaptive Introgression in Humans. Nat. Rev. Genet. 2015, 16, 359–371. [Google Scholar] [CrossRef]
- Rinker, D.C.; Simonti, C.N.; McArthur, E.; Shaw, D.; Hodges, E.; Capra, J.A. Neanderthal Introgression Reintroduced Functional Ancestral Alleles Lost in Eurasian Populations. Nat. Ecol. Evol. 2020, 4, 1332–1341. [Google Scholar] [CrossRef]
- Racimo, F.; Marnetto, D.; Huerta-Sánchez, E. Signatures of Archaic Adaptive Introgression in Present-Day Human Populations. Mol. Biol. Evol. 2017, 34, 296–317. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Ortiz, L.; Duda, T.F., Jr.; Canales-Espinosa, D.; García-Orduña, F.; Rodríguez-Luna, E.; Bermingham, E. Hybridization in Large-Bodied New World Primates. Genetics 2007, 176, 2421. [Google Scholar] [CrossRef] [PubMed]
- Tung, J.; Charpentier, M.J.E.; Garfield, D.A.; Altmann, J.; Alberts, S.C. Genetic Evidence Reveals Temporal Change in Hybridization Patterns in a Wild Baboon Population. Mol. Ecol. 2008, 17, 1998–2011. [Google Scholar] [CrossRef]
- Zinner, D.; Chuma, I.; Knauf, S.; Roos, C. Inverted Intergeneric Introgression between Critically Endangered Kipunjis and Yellow Baboons in Two Disjunct Populations. Biol. Lett. 2018, 14, 20170729. [Google Scholar] [CrossRef] [PubMed]
- Malukiewicz, J.; Boere, V.; Fuzessy, L.; Grativol, A.; de Oliveira e Silva, I.; Pereira, L.; Ruiz-Miranda, C.; Valença, Y.; Stone, A. Natural and Anthropogenic Hybridization in Two Species of Eastern Brazilian Marmosets (Callithrix Jacchus and C. Penicillata). PLoS ONE 2015, 10, e0127268. [Google Scholar] [CrossRef]
- Ackermann, R.R.; Bishop, J.M. Morphological and Molecular Evidence Reveals Recent Hybridization between Gorilla Taxa. Evolution 2010, 64, 271–290. [Google Scholar] [CrossRef]
- Johnson, S.E. Ecology and Speciation in Brown Lemurs: White-Collared Lemurs (Eulemur Albocollaris) and Hybrids (Eulemur Albocollaris x Eulemur Fulvus Fulvus) in Southeastern Madagascar. Ph.D. Thesis, University of Texas at Austin, Austin, TX, USA, 2002. [Google Scholar]
- Wyner, Y.M.; Johnson, S.E.; Stumpf, R.M.; Desalle, R. Genetic Assessment of a White-Collared×red-Fronted Lemur Hybrid Zone at Andringitra, Madagascar. Am. J. Primatol. 2002, 57, 51–66. [Google Scholar] [CrossRef]
- Coimbra-Filho, A.F.; Mittermeier, R.A.; Rylands, A.B.; Mendes, S.L.; Kierulff, M.C.M.; Pinto, L.P.D.S. The Taxonomic Status of Wied’s Black-Tufted-Ear Marmoset, Callithrix Kuhlii (Callitrichidae, Primates). Primate Conserv. 2006, 21, 1–24. [Google Scholar] [CrossRef]
- Tosi, A.; Morales, J.; Melnick, D. Comparison of Y Chromosome and MtDNA Phylogenies Leads to Unique Inferences of Macaque Evolutionary History. Mol. Phylogenet. Evol. 2000, 17, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Ramakrishnan, U.; Panor, J.; Mishra, C.; Sinha, A. Phylogenetic Relationships and Morphometric Affinities of the Arunachal Macaque Macaca Munzala, a Newly Described Primate from Arunachal Pradesh, Northeastern India. Mol. Phylogenet. Evol. 2007, 44, 838–849. [Google Scholar] [CrossRef]
- Roos, C.; Zinner, D.; Kubatko, L.S.; Schwarz, C.; Yang, M.; Meyer, D.; Nash, S.D.; Xing, J.; Batzer, M.A.; Brameier, M.; et al. Nuclear versus Mitochondrial DNA: Evidence for Hybridization in Colobine Monkeys. BMC Evol. Biol. 2011, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Osterholz, M.; Walter, L.; Roos, C. Phylogenetic Position of the Langur Genera Semnopithecus and Trachypithecus among Asian Colobines, and Genus Affiliations of Their Species Groups. BMC Evol. Biol. 2008, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Yoder, A.D.; Irwin, J.A. Phylogeny of the Lemuridae: Effects of Character and Taxon Sampling on Resolution of Species Relationships within Eulemur. Cladistics 1999, 15, 351–361. [Google Scholar] [CrossRef]
- Markolf, M.; Kappeler, P.M. Phylogeographic Analysis of the True Lemurs (Genus Eulemur) Underlines the Role of River Catchments for the Evolution of Micro-Endemism in Madagascar. Front. Zool. 2013, 10, 70. [Google Scholar] [CrossRef]
- Ralainasolo, F.; Ratsimbazafy, J.; Stevens, N. Behavior and Diet of the Critically Endangered Eulemur Cinereiceps in Manombo Forest, Southeast Madagascar. Madag. Conserv. Dev. 2009, 3, 38–43. [Google Scholar] [CrossRef]
- Donati, G.; Campera, M.; Balestri, M.; Barresi, M.; Kesch, K.; Ndremifidy, K.; Rabenantoandro, J.; Racevska, E.; Randriatafika, F.; Ravaolahy, M.; et al. Life in a Fragment: Evolution of Foraging Strategies of Translocated Collared Brown Lemurs, Eulemur Collaris, over an 18-Year Period. Am. J. Primatol. 2020, 82, e23106. [Google Scholar] [CrossRef]
- Ossi, K.; Kamilar, J.M. Environmental and Phylogenetic Correlates of Eulemur Behavior and Ecology (Primates: Lemuridae). Behav. Ecol. Sociobiol. 2006, 61, 53–64. [Google Scholar] [CrossRef]
- Chen, D.; Narváez-Torres, P.; Tiafinjaka, O.; Farris, Z.; Rasoloharijaona, S.; Louis, E.; Johnson, S. Lemur Paparazzi: Arboreal Camera Trapping and Occupancy Modeling as Conservation Tools for Monitoring Threatened Lemur Species. Am. J. Primatol. 2021, 83, e23270. [Google Scholar] [CrossRef]
- Tinsman, J.C.; Eschmann, C.L.; Solofondranohatra, J.S.; Ralainirina, J.R.; Holderied, M.; McCabe, G. Range and Conservation Updates for the Critically Endangered Blue-Eyed Black Lemur Eulemur Flavifrons and the Vulnerable Black Lemur Eulemur Macaco. Oryx 2019, 54, 819–827. [Google Scholar] [CrossRef]
- Scheffrahn, W.; Rabarivola, C.; Rumpler, Y. Field Studies of Population Genetics in Eulemur: A Discussion of Their Potential Importance in Conservation. Folia Primatol. 1998, 69, 147–151. [Google Scholar] [CrossRef]
- Tattersall, I. Madagascar’s Lemurs: Cryptic Diversity or Taxonomic Inflation? Evol. Anthropol. 2007, 16, 12–23. [Google Scholar] [CrossRef]
- Tattersall, I.; Sussman, R.W. Little Brown Lemurs Come of Age: Summary and Perspective. Int. J. Primatol. 2016, 37, 3–9. [Google Scholar] [CrossRef]
- Pastorini, J.; Zaramody, A.; Curtis, D.J.; Nievergelt, C.M.; Mundy, N.I. Genetic Analysis of Hybridization and Introgression between Wild Mongoose and Brown Lemurs. BMC Evol. Biol. 2009, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.; Sterling, E.; Dusch, M.; Raxworthy, C.; Pearson, R. Ecological Divergence and Speciation between Lemur (Eulemur) Sister Species in Madagascar. J. Evol. Biol. 2013, 26, 1790–1801. [Google Scholar] [CrossRef]
- Rabarivola, C.; Meyers, D.; Rumpler, Y. Distribution and Morphological Characters of Intermediate Forms between the Black Lemur (Eulemur Macaco Macaco) and the Sclater’s Lemur (E. m. Flavifrons). Primates 1991, 32, 269–273. [Google Scholar] [CrossRef]
- Pinkus, S.; Smith, J.N.M.; Jolly, A. Feeding Competition between Introduced Eulemur Fulvus and Native Lemur Catta during the Birth Season at Berenty Reserve, Southern Madagascar. In Ringtailed Lemur Biology; Springer: Boston, MA, USA, 2006; pp. 119–140. [Google Scholar]
- Tanaka, M. Habitat Use and Social Structure of a Brown Lemur Hybrid Population in the Berenty Reserve, Madagascar. Am. J. Primatol. 2007, 69, 1189–1194. [Google Scholar] [CrossRef]
- Delmore, K.E.; Louis, E.E.; Johnson, S.E. Morphological Characterization of a Brown Lemur Hybrid Zone (Eulemur Rufifrons × E. Cinereiceps). Am. J. Phys. Anthropol. 2011, 145, 55–66. [Google Scholar] [CrossRef]
- Delmore, K.E.; Brenneman, R.A.; Lei, R.; Bailey, C.A.; Brelsford, A.; Louis, E.E.; Johnson, S.E. Clinal Variation in a Brown Lemur (Eulemur Spp.) Hybrid Zone: Combining Morphological, Genetic and Climatic Data to Examine Stability. J. Evol. Biol. 2013, 26, 1677–1690. [Google Scholar] [CrossRef]
- Johnson, S.E.; Delmore, K.E.; Brown, K.A.; Wyman, T.M.; Louis, E.E. Niche Divergence in a Brown Lemur (Eulemur Spp.) Hybrid Zone: Using Ecological Niche Models to Test Models of Stability. Int. J. Primatol. 2016, 37, 69–88. [Google Scholar] [CrossRef]
- Markolf, M.; Rakotonirina, H.; Fichtel, C.; von Grumbkow, P.; Brameier, M.; Kappeler, P. True Lemurs…true Species—Species Delimitation Using Multiple Data Sources in the Brown Lemur Complex. BMC Evol. Biol. 2013, 13, 233. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, K. Species Concepts and Species Delimitation. Syst. Biol. 2007, 56, 879–886. [Google Scholar] [CrossRef]
- Everson, K.M.; Pozzi, L.; Barrett, M.A.; Blair, M.E.; Donohue, M.E.; Kappeler, P.M.; Kitchener, A.C.; Lemmon, A.R.; Lemmon, E.M.; Pavón-Vázquez, C.J.; et al. Not One, but Multiple Radiations Underlie the Biodiversity of Madagascar’s Endangered Lemurs. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lemmon, A.; Emme, S.; Lemmon, E. Anchored Hybrid Enrichment for Massively High-Throughput Phylogenomics. Syst. Biol. 2012, 61, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lemmon, A.R.; Lemmon, E.M.; Pyron, R.A.; Burbrink, F.T. Using Phylogenomics to Understand the Link between Biogeographic Origins and Regional Diversification in Ratsnakes. Mol. Phylogenet Evol. 2017, 111, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Lemmon, A.R.; Margres, M.J.; Aronow, K. The Venom-Gland Transcriptome of the Eastern Diamondback Rattlesnake (Crotalus Adamanteus). BMC Genomics 2012, 13, 312. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Mittermeier, R.A.; Konstant, W.R.; Hawkins, F.; Louis, E.E.; Langrand, O.; Ratsimbazafy, J.; Rasoloarison, R.; Ganzhorn, J.U.; Rajaoblina, S.; Tattersall, I.; et al. Lemurs of Madagascar, 2nd ed.; Conservation International: Washington, DC, USA, 2006. [Google Scholar]
- Mittermeier, R.A.; Ganzhorn, J.U.; Konstant, W.R.; Glander, K.; Tattersall, I.; Groves, C.P.; Rylands, A.B.; Hapke, A.; Ratsimbazafy, J.; Mayor, M.I.; et al. Lemur Diversity in Madagascar. Int. J. Primatol. 2008, 29, 1607–1656. [Google Scholar] [CrossRef]
- Groves, C.P. Red-Fronted Lemurs Are Not the Same as Red Lemurs. Australas. Primatol. 2007, 18, 23. [Google Scholar]
- Johnson, S.E.; Lei, R.; Martin, S.K.; Irwin, M.T.; Louis, E.E. Does Eulemur Cinereiceps Exist? Preliminary Evidence from Genetics and Ground Surveys in Southeastern Madagascar. Am. J. Primatol. 2008, 70, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Chifman, J.; Kubatko, L. Quartet Inference from SNP Data under the Coalescent Model. Bioinformatics 2014, 30, 3317–3324. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using. Parsimony (* and Other Methods); Version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-Scale Coalescent-Based Species Tree Estimation. Bioinformatics 2014, 30, 541–548. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Huson, D.H. SplitsTree: Analyzing and Visualizing Evolutionary Data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef]
- Bryant, D.; Moulton, V. Neighbor-Net: An Agglomerative Method for the Construction of Phylogenetic Networks. Mol. Biol. Evol. 2003, 21, 255–265. [Google Scholar] [CrossRef]
- Solís-Lemus, C.; Bastide, P.; Ané, C. PhyloNetworks: A Package for Phylogenetic Networks. Mol. Biol. Evol. 2017, 34, 3292–3298. [Google Scholar] [CrossRef]
- Solís-Lemus, C.; Ané, C. Inferring Phylogenetic Networks with Maximum Pseudolikelihood under Incomplete Lineage Sorting. PLoS Genet. 2016, 12, e1005896. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, T.; Leblois, R.; Rousset, F.; Crochet, P.A. A Reassessment of Explanations for Discordant Introgressions of Mitochondrial and Nuclear Genomes. Evolution 2017, 71, 2140–2158. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.E. Evolutionary Divergence in the Brown Lemur Species Complex. In Lemurs; Springer: Boston, MA, USA, 2006; pp. 187–210. [Google Scholar]
- Overdorff, D.J. Ecological and Reproductive Correlates to Range Use in Red-Bellied Lemurs (Eulemur Rubriventer) and Rufous Lemurs (Eulemur Fulvus Rufus). In Lemur Social. Systems and Their Ecological Basis; Springer: Boston, MA, USA, 1993; pp. 167–178. [Google Scholar]
- Gross, B.L.; Rieseberg, L.H. The Ecological Genetics of Homoploid Hybrid Speciation. J. Hered. 2005, 96, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Buerkle, C.A.; Morris, R.J.; Asmussen, M.A.; Rieseberg, L.H. The Likelihood of Homoploid Hybrid Speciation. Heredity 2000, 84, 441–451. [Google Scholar] [CrossRef]
- Schwitzer, C.; Mittermeier, R.A.; Johnson, S.E.; Donati, G.; Irwin, M.; Peacock, H.; Ratsimbazafy, J.; Razafindramanana, J.; Louis, E.E.; Chikhi, L.; et al. Averting Lemur Extinctions amid Madagascar’s Political Crisis. Science 2014, 343, 842–843. [Google Scholar] [CrossRef]
- Michielsen, N.M.; Goodman, S.M.; Soarimalala, V.; van der Geer, A.A.E.; Dávalos, L.M.; Saville, G.I.; Upham, N.; Valente, L. The Macroevolutionary Impact of Recent and Imminent Mammal Extinctions on Madagascar. Nat. Commun. 2023, 14, 14. [Google Scholar] [CrossRef]
- Whiteley, A.R.; Fitzpatrick, S.W.; Funk, W.C.; Tallmon, D.A. Genetic Rescue to the Rescue. Trends Ecol. Evol. 2015, 30, 42–49. [Google Scholar] [CrossRef]
- Johnson, W.E.; Onorato, D.P.; Roelke, M.E.; Land, E.D.; Cunningham, M.; Belden, R.C.; McBride, R.; Jansen, D.; Lotz, M.; Shindle, D.; et al. Genetic Restoration of the Florida Panther. Science 2010, 329, 1641–1645. [Google Scholar] [CrossRef]
- Westemeier, R.L.; Brawn, J.D.; Simpson, S.A.; Esker, T.L.; Jansen, R.W.; Walk, J.W.; Kershner, E.L.; Bouzat, J.L.; Paige, K.N. Tracking the Long-Term Decline and Recovery of an Isolated Population. Science 1998, 282, 1695–1698. [Google Scholar] [CrossRef]
- Edmands, S. Between a Rock and a Hard Place: Evaluating the Relative Risks of Inbreeding and Outbreeding for Conservation and Management. Mol. Ecol. 2007, 16, 463–475. [Google Scholar] [CrossRef]
- Meyer, W.K.; Venkat, A.; Kermany, A.R.; van de Geijn, B.; Sidi Zhang, S.; Przeworski, M. Evolutionary history inferred from the de novo assembly of a nonmodel organism, the blue-eyed black lemur. Mol. Ecol. 2015, 24, 4392–4405. [Google Scholar] [CrossRef] [PubMed]
- Shearn, R.; Lecompte, E.; Régis, C.; Mousset, S.; Penel, S.; Douay, G.; Crouau-Roy, B.; Marais, G.A. Contrasted sex chromosome evolution in primates with and without sexual dimorphism. eLife 2020, 9, e63650. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.; Mughal, M.R.; Godfrey, L.R.; Bankoff, R.J.; Randrianatoandro, H.; Crowley, B.E.; Bergey, C.M.; Muldoon, K.M.; Randrianasy, J.; Raharivololona, B.M.; et al. Evolutionary and phylogenetic insights from a nuclear genome sequence of the extinct, giant, “subfossil” koala lemur Megaladapis edwardsi. Proc. Natl. Acad. Sci. USA 2021, 118, e2022117118. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Everson, K.M.; Donohue, M.E.; Weisrock, D.W. A Pervasive History of Gene Flow in Madagascar’s True Lemurs (Genus Eulemur). Genes 2023, 14, 1130. https://doi.org/10.3390/genes14061130
Everson KM, Donohue ME, Weisrock DW. A Pervasive History of Gene Flow in Madagascar’s True Lemurs (Genus Eulemur). Genes. 2023; 14(6):1130. https://doi.org/10.3390/genes14061130
Chicago/Turabian StyleEverson, Kathryn M., Mariah E. Donohue, and David W. Weisrock. 2023. "A Pervasive History of Gene Flow in Madagascar’s True Lemurs (Genus Eulemur)" Genes 14, no. 6: 1130. https://doi.org/10.3390/genes14061130