Epigenetic Alterations of Brain Non-Neuronal Cells in Major Mental Diseases


Abstract
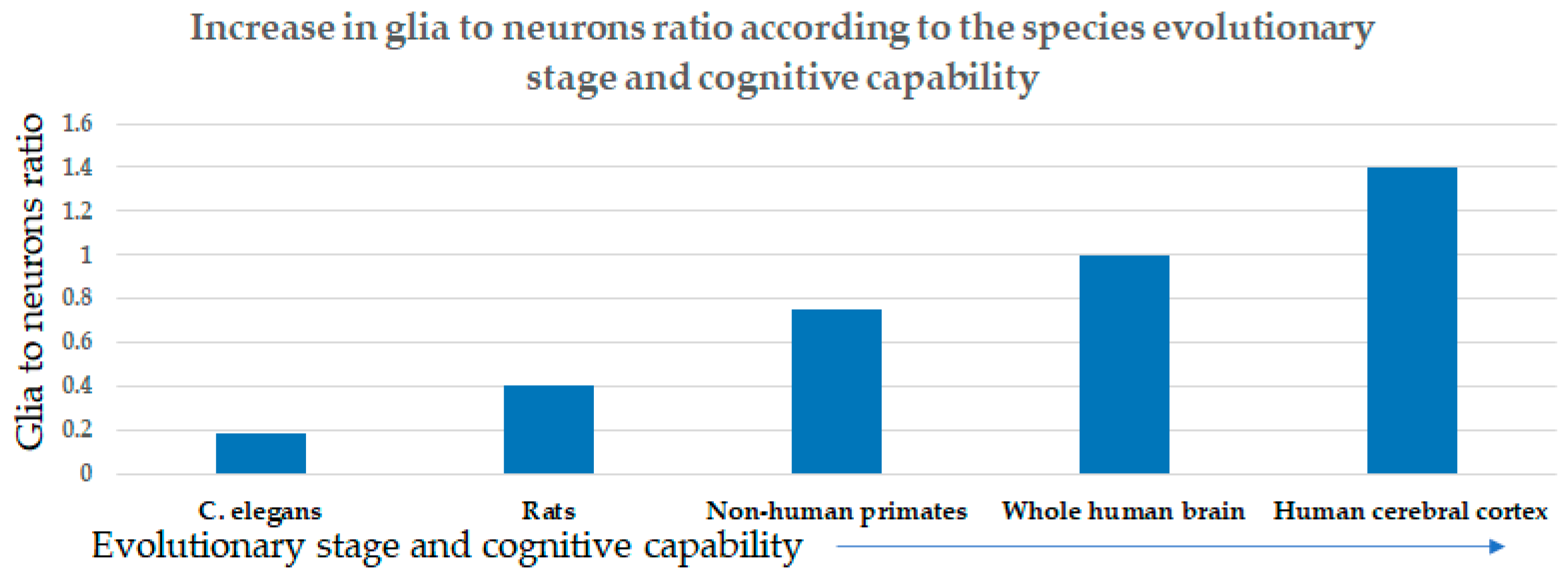
:1. Introduction
2. Non-Neuronal Cells of the Brain
3. Glia Dysfunction in Autism
4. Glia Dysfunction in Schizophrenia and Bipolar Disorder
5. Astroglia Pathology and Dysfunction in Depression
6. Microglia and Astrocytes, the Ambassadors of Microbiome Communication with the Brain
7. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jäkel, S.; Dimou, L. Glial cells and their function in the adult brain: A journey through the history of their ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar]
- Zeng, H.; Sanes, J.R. Neuronal cell-type classification: Challenges, opportunities and the path forward. Nat. Rev. Neurosci. 2017, 18, 530–546. [Google Scholar]
- Farrelly, L.A.; Zheng, S.; Schrode, N.; Topol, A.; Bhanu, N.V.; Bastle, R.M.; Ramakrishnan, A.; Chan, J.C.; Cetin, B.; Flaherty, E. Chromatin profiling in human neurons reveals aberrant roles for histone acetylation and BET family proteins in schizophrenia. Nat. Commun. 2022, 13, 2195. [Google Scholar]
- Gandal, M.J.; Haney, J.R.; Wamsley, B.; Yap, C.X.; Parhami, S.; Emani, P.S.; Chang, N.; Chen, G.T.; Hoftman, G.D.; de Alba, D. Broad transcriptomic dysregulation occurs across the cerebral cortex in ASD. Nature 2022, 611, 532–539. [Google Scholar]
- Gusev, F.E.; Reshetov, D.A.; Mitchell, A.C.; Andreeva, T.V.; Dincer, A.; Grigorenko, A.P.; Fedonin, G.; Halene, T.; Aliseychik, M.; Filippova, E. Chromatin profiling of cortical neurons identifies individual epigenetic signatures in schizophrenia. Transl. Psychiatry 2019, 9, 256. [Google Scholar]
- Jeong, H.; Mendizabal, I.; Berto, S.; Chatterjee, P.; Layman, T.; Usui, N.; Toriumi, K.; Douglas, C.; Singh, D.; Huh, I. Evolution of DNA methylation in the human brain. Nat. Commun. 2021, 12, 2021. [Google Scholar]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246. [Google Scholar]
- Progatzky, F.; Shapiro, M.; Chng, S.H.; Garcia-Cassani, B.; Classon, C.H.; Sevgi, S.; Laddach, A.; Bon-Frauches, A.C.; Lasrado, R.; Rahim, M. Regulation of intestinal immunity and tissue repair by enteric glia. Nature 2021, 599, 125–130. [Google Scholar]
- Mendizabal, I.; Berto, S.; Usui, N.; Toriumi, K.; Chatterjee, P.; Douglas, C.; Huh, I.; Jeong, H.; Layman, T.; Tamminga, C.A. Cell type-specific epigenetic links to schizophrenia risk in the brain. Genome Biol. 2019, 20, 135. [Google Scholar]
- Zhan, L.; Krabbe, G.; Du, F.; Jones, I.; Reichert, M.C.; Telpoukhovskaia, M.; Kodama, L.; Wang, C.; Cho, S.-h.; Sayed, F. Proximal recolonization by self-renewing microglia re-establishes microglial homeostasis in the adult mouse brain. PLoS Biol. 2019, 17, e3000134. [Google Scholar]
- Abdolmaleky, H.M.; Zhou, J.-R.; Thiagalingam, S. An update on the epigenetics of psychotic diseases and autism. Epigenomics 2015, 7, 427–449. [Google Scholar]
- Abdolmaleky, H.M.; Zhou, J.-R.; Thiagalingam, S. Cataloging recent advances in epigenetic alterations in major mental disorders and autism. Epigenomics 2021, 13, 1231–1245. [Google Scholar]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar]
- Mazzanti, M.; Haydon, P.G. Astrocytes selectively enhance N-type calcium current in hippocampal neurons. Glia 2003, 41, 128–136. [Google Scholar]
- Thameem Dheen, S.; Kaur, C.; Ling, E.-A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar]
- Verkhratsky, A.; Nedergaard, M. The homeostatic astroglia emerges from evolutionary specialization of neural cells. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150428. [Google Scholar]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar]
- Sherwood, C.C.; Stimpson, C.D.; Raghanti, M.A.; Wildman, D.E.; Uddin, M.; Grossman, L.I.; Goodman, M.; Redmond, J.C.; Bonar, C.J.; Erwin, J.M. Evolution of increased glia–neuron ratios in the human frontal cortex. Proc. Natl. Acad. Sci. USA 2006, 103, 13606–13611. [Google Scholar]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar]
- Theodosis, D.T.; Poulain, D.A.; Oliet, S.H. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol. Rev. 2008, 88, 983–1008. [Google Scholar]
- Ge, W.-P.; Jia, J.-M. Local production of astrocytes in the cerebral cortex. Neuroscience 2016, 323, 3–9. [Google Scholar]
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar]
- Liu, S.; Yuan, Y.-b; Guan, L.-l.; Wei, H.; Cheng, Z.; Han, X.; Yang, L. MiRNA-365 and miRNA-520c-3p respond to risperidone treatment in first-episode schizophrenia after a 1 year remission. Chin. Med. J. 2013, 126, 2676–2680. [Google Scholar]
- Mo, J.L.; Liu, Q.; Kou, Z.W.; Wu, K.W.; Yang, P.; Chen, X.H.; Sun, F.Y. MicroRNA-365 modulates astrocyte conversion into neuron in adult rat brain after stroke by targeting Pax6. Glia 2018, 66, 1346–1362. [Google Scholar]
- Verkhratsky, A. Physiology of neuronal–glial networking. Neurochem. Int. 2010, 57, 332–343. [Google Scholar]
- Li, F.; Sami, A.; Noristani, H.N.; Slattery, K.; Qiu, J.; Groves, T.; Wang, S.; Veerasammy, K.; Chen, Y.X.; Morales, J. Glial metabolic rewiring promotes axon regeneration and functional recovery in the central nervous system. Cell Metab. 2020, 32, 767–785.e767. [Google Scholar]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar]
- Füger, P.; Hefendehl, J.K.; Veeraraghavalu, K.; Wendeln, A.-C.; Schlosser, C.; Obermüller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376. [Google Scholar]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H. The lifespan and turnover of microglia in the human brain. Cell Rep. 2017, 20, 779–784. [Google Scholar]
- Uriarte Huarte, O.; Richart, L.; Mittelbronn, M.; Michelucci, A. Microglia in health and disease: The strength to be diverse and reactive. Front. Cell. Neurosci. 2021, 15, 660523. [Google Scholar]
- Alam, R.; Abdolmaleky, H.M.; Zhou, J.R. Microbiome, inflammation, epigenetic alterations, and mental diseases. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 651–660. [Google Scholar]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar]
- Chrobak, A.A.; Soltys, Z. Bergmann glia, long-term depression, and autism spectrum disorder. Mol. Neurobiol. 2017, 54, 1156–1166. [Google Scholar]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar]
- Wong, C.C.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K. The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 2018, 48, 979–991.e978. [Google Scholar]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar]
- Patnala, R.; Arumugam, T.V.; Gupta, N.; Dheen, S.T. HDAC inhibitor sodium butyrate-mediated epigenetic regulation enhances neuroprotective function of microglia during ischemic stroke. Mol. Neurobiol. 2017, 54, 6391–6411. [Google Scholar]
- Alexandrov, P.N.; Zhao, Y.; Jones, B.M.; Bhattacharjee, S.; Lukiw, W.J. Expression of the phagocytosis-essential protein TREM2 is down-regulated by an aluminum-induced miRNA-34a in a murine microglial cell line. J. Inorg. Biochem. 2013, 128, 267–269. [Google Scholar]
- Mills, J.D.; Iyer, A.M.; van Scheppingen, J.; Bongaarts, A.; Anink, J.J.; Janssen, B.; Zimmer, T.S.; Spliet, W.G.; van Rijen, P.C.; Jansen, F.E. Coding and small non-coding transcriptional landscape of tuberous sclerosis complex cortical tubers: Implications for pathophysiology and treatment. Sci. Rep. 2017, 7, 8089. [Google Scholar]
- Ozaki, Y.; Yoshino, Y.; Yamazaki, K.; Sao, T.; Mori, Y.; Ochi, S.; Yoshida, T.; Mori, T.; Iga, J.-I.; Ueno, S.-I. DNA methylation changes at TREM2 intron 1 and TREM2 mRNA expression in patients with Alzheimer’s disease. J. Psychiatr. Res. 2017, 92, 74–80. [Google Scholar]
- Yoshino, Y.; Ozaki, Y.; Yamazaki, K.; Sao, T.; Mori, Y.; Ochi, S.; Iga, J.-I.; Ueno, S.-I. DNA methylation changes in intron 1 of Triggering receptor expressed on myeloid cell 2 in Japanese schizophrenia subjects. Front. Neurosci. 2017, 11, 275. [Google Scholar]
- Smith, A.R.; Smith, R.G.; Condliffe, D.; Hannon, E.; Schalkwyk, L.; Mill, J.; Lunnon, K. Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol. Aging 2016, 47, 35–40. [Google Scholar]
- Celarain, N.; Sánchez-Ruiz de Gordoa, J.; Zelaya, M.V.; Roldán, M.; Larumbe, R.; Pulido, L.; Echavarri, C.; Mendioroz, M. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer’s disease hippocampus. Clin. Epigenetics 2016, 8, 37. [Google Scholar]
- Nagarajan, R.; Hogart, A.; Gwye, Y.; Martin, M.R.; LaSalle, J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006, 1, 172–182. [Google Scholar]
- Liyanage, V.R.; Olson, C.O.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. DNA methylation contributes to the differential expression levels of Mecp2 in male mice neurons and astrocytes. Int. J. Mol. Sci. 2019, 20, 1845. [Google Scholar]
- Lioy, D.T.; Garg, S.K.; Monaghan, C.E.; Raber, J.; Foust, K.D.; Kaspar, B.K.; Hirrlinger, P.G.; Kirchhoff, F.; Bissonnette, J.M.; Ballas, N. A role for glia in the progression of Rett’s syndrome. Nature 2011, 475, 497–500. [Google Scholar]
- Derecki, N.C.; Cronk, J.C.; Lu, Z.; Xu, E.; Abbott, S.B.; Guyenet, P.G.; Kipnis, J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 2012, 484, 105–109. [Google Scholar]
- Cronk, J.C.; Derecki, N.C.; Ji, E.; Xu, Y.; Lampano, A.E.; Smirnov, I.; Baker, W.; Norris, G.T.; Marin, I.; Coddington, N. Methyl-CpG binding protein 2 regulates microglia and macrophage gene expression in response to inflammatory stimuli. Immunity 2015, 42, 679–691. [Google Scholar]
- Presumey, J.; Bialas, A.R.; Carroll, M.C. Complement system in neural synapse elimination in development and disease. Adv. Immunol. 2017, 135, 53–79. [Google Scholar]
- Fagan, K.; Crider, A.; Ahmed, A.O.; Pillai, A. Complement C3 expression is decreased in autism spectrum disorder subjects and contributes to behavioral deficits in rodents. Complex Psychiatry 2017, 3, 19–27. [Google Scholar]
- Lin, P.; Nicholls, L.; Assareh, H.; Fang, Z.; Amos, T.G.; Edwards, R.J.; Assareh, A.A.; Voineagu, I. Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q Genes in Rett syndrome. BMC Genom. 2016, 17, 427. [Google Scholar]
- Rey, R.; Suaud-Chagny, M.-F.; Bohec, A.-L.; Dorey, J.-M.; d’Amato, T.; Tamouza, R.; Leboyer, M. Overexpression of complement component C4 in the dorsolateral prefrontal cortex, parietal cortex, superior temporal gyrus and associative striatum of patients with schizophrenia. Brain Behav. Immun. 2020, 90, 216–225. [Google Scholar]
- Woo, J.J.; Pouget, J.G.; Zai, C.C.; Kennedy, J.L. The complement system in schizophrenia: Where are we now and what’s next? Mol. Psychiatry 2020, 25, 114–130. [Google Scholar]
- Nardone, S.; Sharan Sams, D.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar]
- Jenkins, A.K.; Lewis, D.A.; Volk, D.W. Altered expression of microglial markers of phagocytosis in schizophrenia. Schizophr. Res. 2023, 251, 22–29. [Google Scholar]
- Lardenoije, R.; Roubroeks, J.A.; Pishva, E.; Leber, M.; Wagner, H.; Iatrou, A.; Smith, A.R.; Smith, R.G.; Eijssen, L.M.; Kleineidam, L. Alzheimer’s disease-associated (hydroxy) methylomic changes in the brain and blood. Clin. Epigenetics 2019, 11, 164. [Google Scholar]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.-M. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 2019, 28, 2111–2123.e2116. [Google Scholar]
- Melbourne, J.K.; Rosen, C.; Feiner, B.; Sharma, R.P. C4A mRNA expression in PBMCs predicts the presence and severity of delusions in schizophrenia and bipolar disorder with psychosis. Schizophr. Res. 2018, 197, 321–327. [Google Scholar]
- Hatzimanolis, A.; Foteli, S.; Stefanatou, P.; Ntigrintaki, A.-A.; Ralli, I.; Kollias, K.; Nikolaou, C.; Gazouli, M.; Stefanis, N.C. Deregulation of complement components C4A and CSMD1 peripheral expression in first-episode psychosis and links to cognitive ability. Eur. Arch. Psychiatry Clin. Neurosci. 2022, 272, 1219–1228. [Google Scholar]
- Abdolmaleky, H.M.; Gower, A.C.; Wong, C.K.; Cox, J.W.; Zhang, X.; Thiagalingam, A.; Shafa, R.; Sivaraman, V.; Zhou, J.R.; Thiagalingam, S. Aberrant transcriptomes and DNA methylomes define pathways that drive pathogenesis and loss of brain laterality/asymmetry in schizophrenia and bipolar disorder. Am. J. Med. Genet. Part B Neuropsychiatr. Genetics 2019, 180, 138–149. [Google Scholar]
- Kim, R.; Sepulveda-Orengo, M.T.; Healey, K.L.; Williams, E.A.; Reissner, K.J. Regulation of glutamate transporter 1 (GLT-1) gene expression by cocaine self-administration and withdrawal. Neuropharmacology 2018, 128, 1–10. [Google Scholar]
- Perisic, T.; Holsboer, F.; Rein, T.; Zschocke, J. The CpG island shore of the GLT-1 gene acts as a methylation-sensitive enhancer. Glia 2012, 60, 1345–1355. [Google Scholar]
- Hoye, M.L.; Regan, M.R.; Jensen, L.A.; Lake, A.M.; Reddy, L.V.; Vidensky, S.; Richard, J.-P.; Maragakis, N.J.; Rothstein, J.D.; Dougherty, J.D. Motor neuron-derived microRNAs cause astrocyte dysfunction in amyotrophic lateral sclerosis. Brain 2018, 141, 2561–2575. [Google Scholar]
- Cui, W.; Mizukami, H.; Yanagisawa, M.; Aida, T.; Nomura, M.; Isomura, Y.; Takayanagi, R.; Ozawa, K.; Tanaka, K.; Aizawa, H. Glial dysfunction in the mouse habenula causes depressive-like behaviors and sleep disturbance. J. Neurosci. 2014, 34, 16273–16285. [Google Scholar]
- Cui, Y.; Yang, Y.; Ni, Z.; Dong, Y.; Cai, G.; Foncelle, A.; Ma, S.; Sang, K.; Tang, S.; Li, Y. Astroglial Kir4. 1 in the lateral habenula drives neuronal bursts in depression. Nature 2018, 554, 323–327. [Google Scholar]
- Aleksovska, K.; Leoncini, E.; Bonassi, S.; Cesario, A.; Boccia, S.; Frustaci, A. Systematic review and meta-analysis of circulating S100B blood levels in schizophrenia. PLoS ONE 2014, 9, e106342. [Google Scholar]
- Gasparoni, G.; Bultmann, S.; Lutsik, P.; Kraus, T.F.; Sordon, S.; Vlcek, J.; Dietinger, V.; Steinmaurer, M.; Haider, M.; Mulholland, C.B. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 2018, 11, 41. [Google Scholar]
- Kano, S.-I.; Nwulia, E.; Niwa, M.; Chen, Y.; Sawa, A.; Cascella, N. Altered MHC class I expression in dorsolateral prefrontal cortex of nonsmoker patients with schizophrenia. Neurosci. Res. 2011, 71, 289–293. [Google Scholar]
- Garg, P.; Sharp, A.J. Screening for rare epigenetic variations in autism and schizophrenia. Hum. Mutat. 2019, 40, 952–961. [Google Scholar]
- Boni, J.L.; Kahanovitch, U.; Nwaobi, S.E.; Floyd, C.L.; Olsen, M.L. DNA methylation: A mechanism for sustained alteration of KIR4. 1 expression following central nervous system insult. Glia 2020, 68, 1495–1512. [Google Scholar]
- Ernst, C.; Deleva, V.; Deng, X.; Sequeira, A.; Pomarenski, A.; Klempan, T.; Ernst, N.; Quirion, R.; Gratton, A.; Szyf, M. Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Arch. Gen. Psychiatry 2009, 66, 22–32. [Google Scholar]
- Kaut, O.; Schmitt, I.; Hofmann, A.; Hoffmann, P.; Schlaepfer, T.E.; Wüllner, U.; Hurlemann, R. Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 331–341. [Google Scholar]
- Shan, W.; Xu, L.; Qiu, Z.; Wang, J.; Shao, J.; Feng, J.; Zhao, J. Increased high-mobility group box 1 levels are associated with depression after acute ischemic stroke. Neurol. Sci. 2022, 43, 3131–3137. [Google Scholar]
- Franklin, T.C.; Wohleb, E.S.; Zhang, Y.; Fogaça, M.; Hare, B.; Duman, R.S. Persistent Increase in Microglial RAGE Contributes to Chronic Stress-Induced Priming of Depressive-like Behavior. Biol. Psychiatry 2018, 83, 50–60. [Google Scholar]
- Rohde, K.; Rønningen, T.; la Cour Poulsen, L.; Keller, M.; Blüher, M.; Böttcher, Y. Role of the DNA repair genes H2AX and HMGB1 in human fat distribution and lipid profiles. BMJ Open Diabetes Res. Care 2020, 8, e000831. [Google Scholar]
- Su, J.; Fang, M.; Tian, B.; Luo, J.; Jin, C.; Wang, X.; Ning, Z.; Li, X. Hypoxia induces hypomethylation of the HMGB1 promoter via the MAPK/DNMT1/HMGB1 pathway in cardiac progenitor cells. Acta Biochim. Et Biophys. Sin. 2018, 50, 1121–1130. [Google Scholar]
- He, M.; Zhang, B.; Wei, X.; Wang, Z.; Fan, B.; Du, P.; Zhang, Y.; Jian, W.; Chen, L.; Wang, L. HDAC 4/5-HMGB 1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J. Cell. Mol. Med. 2013, 17, 531–542. [Google Scholar]
- Yang, Y.; Huang, J.-Q.; Zhang, X.; Shen, L.-F. MiR-129-2 functions as a tumor suppressor in glioma cells by targeting HMGB1 and is down-regulated by DNA methylation. Mol. Cell. Biochem. 2015, 404, 229–239. [Google Scholar]
- Karthikeyan, A.; Patnala, R.; PJadhav, S.; Eng-Ang, L.; Thameem Dheen, S. MicroRNAs: Key players in microglia and astrocyte mediated inflammation in CNS pathologies. Curr. Med. Chem. 2016, 23, 3528–3546. [Google Scholar]
- Han, V.X.; Patel, S.; Jones, H.F.; Dale, R.C. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat. Rev. Neurol. 2021, 17, 564–579. [Google Scholar]
- Toker, L.; Mancarci, B.O.; Tripathy, S.; Pavlidis, P. Transcriptomic evidence for alterations in astrocytes and parvalbumin interneurons in subjects with bipolar disorder and schizophrenia. Biol. Psychiatry 2018, 84, 787–796. [Google Scholar]
- Bellesi, M.; Melone, M.; Gubbini, A.; Battistacci, S.; Conti, F. GLT-1 upregulation impairs prepulse inhibition of the startle reflex in adult rats. Glia 2009, 57, 703–713. [Google Scholar]
- Torres-Berrío, A.; Lopez, J.P.; Bagot, R.C.; Nouel, D.; Dal Bo, G.; Cuesta, S.; Zhu, L.; Manitt, C.; Eng, C.; Cooper, H.M. DCC confers susceptibility to depression-like behaviors in humans and mice and is regulated by miR-218. Biol. Psychiatry 2017, 81, 306–315. [Google Scholar]
- Torres-Berrío, A.; Nouel, D.; Cuesta, S.; Parise, E.M.; Restrepo-Lozano, J.M.; Larochelle, P.; Nestler, E.J.; Flores, C. MiR-218: A molecular switch and potential biomarker of susceptibility to stress. Mol. Psychiatry 2020, 25, 951–964. [Google Scholar]
- Chong, J.R.; Chai, Y.L.; Lee, J.H.; Howlett, D.; Attems, J.; Ballard, C.G.; Aarsland, D.; Francis, P.T.; Chen, C.P.; Lai, M.K. Increased transforming growth factor β2 in the neocortex of Alzheimer’s disease and dementia with lewy bodies is correlated with disease severity and soluble Aβ 42 load. J. Alzheimer’s Dis. 2017, 56, 157–166. [Google Scholar]
- Gast, H.; Gordic, S.; Petrzilka, S.; Lopez, M.; Müller, A.; Gietl, A.; Hock, C.; Birchler, T.; Fontana, A. Transforming growth factor-β inhibits the expression of clock genes. Ann. N. Y. Acad. Sci. 2012, 1261, 79–87. [Google Scholar]
- Noguchi, A.; Nawa, M.; Aiso, S.; Okamoto, K.; Matsuoka, M. Transforming growth factor β2 level is elevated in neurons of Alzheimer’s disease brains. Int. J. Neurosci. 2010, 120, 168–175. [Google Scholar]
- Hashimoto, Y.; Chiba, T.; Yamada, M.; Nawa, M.; Kanekura, K.; Suzuki, H.; Terashita, K.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Transforming growth factor β2 is a neuronal death-inducing ligand for amyloid-β precursor protein. Mol. Cell. Biol. 2005, 25, 9304–9317. [Google Scholar]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar]
- Da Rosa, M.I.; Simon, C.; Grande, A.J.; Barichello, T.; Oses, J.P.; Quevedo, J. Serum S100B in manic bipolar disorder patients: Systematic review and meta-analysis. J. Affect. Disord. 2016, 206, 210–215. [Google Scholar]
- Baecker, J.; Wartchow, K.; Sehm, T.; Ghoochani, A.; Buchfelder, M.; Kleindienst, A. Treatment with the neurotrophic protein S100B increases synaptogenesis after traumatic brain injury. J. Neurotrauma 2020, 37, 1097–1107. [Google Scholar]
- Montoya, A.; Elgueta, D.; Campos, J.; Chovar, O.; Falcón, P.; Matus, S.; Alfaro, I.; Bono, M.R.; Pacheco, R. Dopamine receptor D3 signalling in astrocytes promotes neuroinflammation. J. Neuroinflammation 2019, 16, 258. [Google Scholar]
- Melka, M.G.; Castellani, C.A.; Laufer, B.I.; Rajakumar, N.; O’Reilly, R.; Singh, S.M. Olanzapine induced DNA methylation changes support the dopamine hypothesis of psychosis. J. Mol. Psychiatry 2013, 1, 19. [Google Scholar]
- Bergen, S.; O’dushlaine, C.; Ripke, S.; Lee, P.; Ruderfer, D.; Akterin, S.; Moran, J.; Chambert, K.; Handsaker, R.; Backlund, L. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry 2012, 17, 880–886. [Google Scholar]
- Yue, W.-H.; Wang, H.-F.; Sun, L.-D.; Tang, F.-L.; Liu, Z.-H.; Zhang, H.-X.; Li, W.-Q.; Zhang, Y.-L.; Zhang, Y.; Ma, C.-C. Genome-wide association study identifies a susceptibility locus for schizophrenia in Han Chinese at 11p11. 2. Nat. Genet. 2011, 43, 1228–1231. [Google Scholar]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; van Doren, V. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar]
- Windrem, M.S.; Osipovitch, M.; Liu, Z.; Bates, J.; Chandler-Militello, D.; Zou, L.; Munir, J.; Schanz, S.; McCoy, K.; Miller, R.H. Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 2017, 21, 195–208.e196. [Google Scholar]
- Mallya, A.P.; Wang, H.-D.; Lee HN, R.; Deutch, A.Y. Microglial pruning of synapses in the prefrontal cortex during adolescence. Cereb. Cortex 2019, 29, 1634–1643. [Google Scholar]
- Inta, D.; Lang, U.E.; Borgwardt, S.; Meyer-Lindenberg, A.; Gass, P. Microglia activation and schizophrenia: Lessons from the effects of minocycline on postnatal neurogenesis, neuronal survival and synaptic pruning. Schizophr. Bull. 2017, 43, 493–496. [Google Scholar]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar]
- Chaudhry, I.B.; Hallak, J.; Husain, N.; Minhas, F.; Stirling, J.; Richardson, P.; Dursun, S.; Dunn, G.; Deakin, B. Minocycline benefits negative symptoms in early schizophrenia: A randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J. Psychopharmacol. 2012, 26, 1185–1193. [Google Scholar]
- Levkovitz, Y.; Mendlovich, S.; Riwkes, S.; Braw, Y.; Levkovitch-Verbin, H.; Gal, G.; Fennig, S.; Treves, I.; Kron, S. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J. Clin. Psychiatry 2009, 70, 4863. [Google Scholar]
- Laskaris, L.; Zalesky, A.; Weickert, C.S.; di Biase, M.A.; Chana, G.; Baune, B.T.; Bousman, C.; Nelson, B.; McGorry, P.; Everall, I. Investigation of peripheral complement factors across stages of psychosis. Schizophr. Res. 2019, 204, 30–37. [Google Scholar]
- Reginia, A.; Kucharska-Mazur, J.; Jabłoński, M.; Budkowska, M.; Dołȩgowska, B.; Sagan, L.; Misiak, B.; Ratajczak, M.Z.; Rybakowski, J.K.; Samochowiec, J. Assessment of complement cascade components in patients with bipolar disorder. Front. Psychiatry 2018, 9, 614. [Google Scholar]
- Akcan, U.; Karabulut, S.; Küçükali, C.İ.; Çakır, S.; Tüzün, E. Bipolar disorder patients display reduced serum complement levels and elevated peripheral blood complement expression levels. Acta Neuropsychiatr. 2018, 30, 70–78. [Google Scholar]
- Van Dongen, J.; Zilhão, N.R.; Sugden, K.; Heijmans, B.T.; AC’t Hoen, P.; van Meurs, J.; Isaacs, A.; Jansen, R.; Franke, L.; Boomsma, D.I. Epigenome-wide association study of attention-deficit/hyperactivity disorder symptoms in adults. Biol. Psychiatry 2019, 86, 599–607. [Google Scholar]
- Schirwani, S.; McConnell, V.; Willoughby, J.; Study, D.; Balasubramanian, M. Exploring the association between SRPX2 variants and neurodevelopment: How causal is it? Gene 2019, 685, 50–54. [Google Scholar]
- Cong, Q.; Soteros, B.M.; Wollet, M.; Kim, J.H.; Sia, G.-M. The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat. Neurosci. 2020, 23, 1067–1078. [Google Scholar]
- Øster, B.; Linnet, L.; Christensen, L.L.; Thorsen, K.; Ongen, H.; Dermitzakis, E.T.; Sandoval, J.; Moran, S.; Esteller, M.; Hansen, T.F. Non-CpG island promoter hypomethylation and miR-149 regulate the expression of SRPX2 in colorectal cancer. Int. J. Cancer 2013, 132, 2303–2315. [Google Scholar]
- Orlic-Milacic, M.; Kaufman, L.; Mikhailov, A.; Cheung, A.Y.; Mahmood, H.; Ellis, J.; Gianakopoulos, P.J.; Minassian, B.A.; Vincent, J.B. Over-expression of either MECP2_e1 or MECP2_e2 in neuronally differentiated cells results in different patterns of gene expression. PLoS ONE 2014, 9, e91742. [Google Scholar]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar]
- Pantazatos, S.P.; Huang, Y.; Rosoklija, G.B.; Dwork, A.J.; Arango, V.; Mann, J.J. Whole-transcriptome brain expression and exon-usage profiling in major depression and suicide: Evidence for altered glial, endothelial and ATPase activity. Mol. Psychiatry 2017, 22, 760–773. [Google Scholar]
- Nagy, C.; Suderman, M.; Yang, J.; Szyf, M.; Mechawar, N.; Ernst, C.; Turecki, G. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol. Psychiatry 2015, 20, 320–328. [Google Scholar]
- Fachim, H.A.; Loureiro, C.M.; Corsi-Zuelli, F.; Shuhama, R.; Louzada-Junior, P.; Menezes, P.R.; Dalton, C.F.; Del-Ben, C.M.; Reynolds, G.P. GRIN2B promoter methylation deficits in early-onset schizophrenia and its association with cognitive function. Epigenomics 2019, 11, 401–410. [Google Scholar]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317–322. [Google Scholar]
- Liu, W.-X.; Wang, J.; Xie, Z.-M.; Xu, N.; Zhang, G.-F.; Jia, M.; Zhou, Z.-Q.; Hashimoto, K.; Yang, J.-J. Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a role in the anti-apoptotic and antidepressant effects of ketamine in chronic unpredictable stress model of depression. Psychopharmacology 2016, 233, 405–415. [Google Scholar]
- Ju, L.-S.; Yang, J.-J.; Lei, L.; Xia, J.-Y.; Luo, D.; Ji, M.-H.; Martynyuk, A.E.; Yang, J.-J. The combination of long-term ketamine and extinction training contributes to fear erasure by Bdnf methylation. Front. Cell. Neurosci. 2017, 11, 100. [Google Scholar]
- Holt, L.M.; Hernandez, R.D.; Pacheco, N.L.; Torres Ceja, B.; Hossain, M.; Olsen, M.L. Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB. T1. Elife 2019, 8, e44667. [Google Scholar]
- Zhang, H.; Ding, L.; Shen, T.; Peng, D. HMGB1 involved in stress-induced depression and its neuroinflammatory priming role: A systematic review. Gen. Psychiatry 2019, 32, e100084. [Google Scholar]
- Kreisel, T.; Frank, M.; Licht, T.; Reshef, R.; Ben-Menachem-Zidon, O.; Baratta, M.; Maier, S.; Yirmiya, R. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 2014, 19, 699–709. [Google Scholar]
- Wu, C.; Zhang, X.; Chen, P.; Ruan, X.; Liu, W.; Li, Y.; Sun, C.; Hou, L.; Yin, B.; Qiang, B. MicroRNA-129 modulates neuronal migration by targeting Fmr1 in the developing mouse cortex. Cell Death Dis. 2019, 10, 287. [Google Scholar]
- Hicks, S.D.; Carpenter, R.L.; Wagner, K.E.; Pauley, R.; Barros, M.; Tierney-Aves, C.; Barns, S.; Greene, C.D.; Middleton, F.A. Saliva microRNA differentiates children with autism from peers with typical and atypical development. J. Am. Acad. Child Adolesc. Psychiatry 2020, 59, 296–308. [Google Scholar]
- Hicks, S.D.; Uhlig, R.; Afshari, P.; Williams, J.; Chroneos, M.; Tierney-Aves, C.; Wagner, K.; Middleton, F.A. Oral microbiome activity in children with autism spectrum disorder. Autism Research 2018, 11, 1286–1299. [Google Scholar]
- Tomova, A.; Husarova, V.; Lakatosova, S.; Bakos, J.; Vlkova, B.; Babinska, K.; Ostatnikova, D. Gastrointestinal microbiota in children with autism in Slovakia. Physiol. Behav. 2015, 138, 179–187. [Google Scholar]
- Nguyen, T.T.; Kosciolek, T.; Maldonado, Y.; Daly, R.E.; Martin, A.S.; McDonald, D.; Knight, R.; Jeste, D.V. Differences in gut microbiome composition between persons with chronic schizophrenia and healthy comparison subjects. Schizophr. Res. 2019, 204, 23–29. [Google Scholar]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 2016, 21, 786–796. [Google Scholar]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A. Alzheimer’s disease microbiome is associated with dysregulation of the anti-inflammatory P-glycoprotein pathway. MBio 2019, 10, e00632-19. [Google Scholar]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar]
- Qing, Y.; Xu, L.; Cui, G.; Sun, L.; Hu, X.; Yang, X.; Jiang, J.; Zhang, J.; Zhang, T.; Wang, T. Salivary microbiome profiling reveals a dysbiotic schizophrenia-associated microbiota. NPJ Schizophr. 2021, 7, 51. [Google Scholar]
- Sharon, G.; Cruz, N.J.; Kang, D.-W.; Gandal, M.J.; Wang, B.; Kim, Y.-M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 2019, 177, 1600–1618.e1617. [Google Scholar]
- Zheng, P.; Zeng, B.; Liu, M.; Chen, J.; Pan, J.; Han, Y.; Liu, Y.; Cheng, K.; Zhou, C.; Wang, H. The gut microbiome from patients with schizophrenia modulates the glutamate-glutamine-GABA cycle and schizophrenia-relevant behaviors in mice. Sci. Adv. 2019, 5, eaau8317. [Google Scholar]
- D’Amato, A.; di Cesare Mannelli, L.; Lucarini, E.; Man, A.L.; Le Gall, G.; Branca, J.J.; Ghelardini, C.; Amedei, A.; Bertelli, E.; Regoli, M. Faecal microbiota transplant from aged donor mice affects spatial learning and memory via modulating hippocampal synaptic plasticity-and neurotransmission-related proteins in young recipients. Microbiome 2020, 8, 140. [Google Scholar]
- Chevalier, G.; Siopi, E.; Guenin-Macé, L.; Pascal, M.; Laval, T.; Rifflet, A.; Boneca, I.G.; Demangel, C.; Colsch, B.; Pruvost, A. Effect of gut microbiota on depressive-like behaviors in mice is mediated by the endocannabinoid system. Nat. Commun. 2020, 11, 6363. [Google Scholar]
- Chu, C.; Murdock, M.H.; Jing, D.; Won, T.H.; Chung, H.; Kressel, A.M.; Tsaava, T.; Addorisio, M.E.; Putzel, G.G.; Zhou, L. The microbiota regulate neuronal function and fear extinction learning. Nature 2019, 574, 543–548. [Google Scholar]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.; Shanahan, F.; Dinan, T.; Cryan, J. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar]
- Razazan, A.; Karunakar, P.; Mishra, S.P.; Sharma, S.; Miller, B.; Jain, S.; Yadav, H. Activation of microbiota sensing–free fatty acid receptor 2 signaling ameliorates amyloid-β induced neurotoxicity by modulating proteolysis-senescence axis. Front. Aging Neurosci. 2021, 13, 735933. [Google Scholar]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar]
- Asbury, M.R.; Butcher, J.; Copeland, J.K.; Unger, S.; Bando, N.; Comelli, E.M.; Forte, V.; Kiss, A.; LeMay-Nedjelski, L.; Sherman, P.M. Mothers of preterm infants have individualized breast milk microbiota that changes temporally based on maternal characteristics. Cell Host Microbe 2020, 28, 669–682.e664. [Google Scholar]
- LeMay-Nedjelski, L.; Asbury, M.R.; Butcher, J.; Ley, S.H.; Hanley, A.J.; Kiss, A.; Unger, S.; Copeland, J.K.; Wang, P.W.; Stintzi, A. Maternal diet and infant feeding practices are associated with variation in the human milk microbiota at 3 months postpartum in a cohort of women with high rates of gestational glucose intolerance. J. Nutr. 2021, 151, 320–329. [Google Scholar]
- Hamshere, M.L.; Walters, J.T.R.; Smith, R.; Richards, A.L.; Green, E.; Grozeva, D.; Jones, I.; Forty, L.; Jones, L.; Gordon-Smith, K. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Mol. Psychiatry 2013, 18, 708–712. [Google Scholar]
- Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F. The International Schizophrenia Consortium: Common polygenic variation contributes to risk of schizophrenia that overlaps with bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar]
- Pegg, C.E.; Zaichick, S.V.; Bomba-Warczak, E.; Jovasevic, V.; Kim, D.; Kharkwal, H.; Wilson, D.W.; Walsh, D.; Sollars, P.J.; Pickard, G.E. Herpesviruses assimilate kinesin to produce motorized viral particles. Nature 2021, 599, 662–666. [Google Scholar]
- Qian, Y.; Yang, X.; Xu, S.; Wu, C.; Qin, N.; Chen, S.-D.; Xiao, Q. Detection of microbial 16S rRNA gene in the blood of patients with Parkinson’s disease. Front. Aging Neurosci. 2018, 10, 156. [Google Scholar]
- Sabri, F.; Titanji, K.; de Milito, A.; Chiodi, F. Astrocyte activation and apoptosis: Their roles in the neuropathology of HIV infection. Brain Pathol. 2003, 13, 84–94. [Google Scholar]
- Barichello, T.; Simoes, L.R.; Quevedo, J.; Zhang, X.Y. Microglial activation and psychotic disorders: Evidence from pre-clinical and clinical studies. Neuroinflammation Schizophr. 2020, 44, 161–205. [Google Scholar]
- Prata, J.; Santos, S.G.; Almeida, M.I.; Coelho, R.; Barbosa, M.A. Bridging Autism Spectrum Disorders and Schizophrenia through inflammation and biomarkers-pre-clinical and clinical investigations. J. Neuroinflammation 2017, 14, 179. [Google Scholar]
- Giovanoli, S.; Engler, H.; Engler, A.; Richetto, J.; Feldon, J.; Riva, M.A.; Schedlowski, M.; Meyer, U. Preventive effects of minocycline in a neurodevelopmental two-hit model with relevance to schizophrenia. Transl. Psychiatry 2016, 6, e772. [Google Scholar]
- Miyauchi, E.; Kim, S.W.; Suda, W.; Kawasumi, M.; Onawa, S.; Taguchi-Atarashi, N.; Morita, H.; Taylor, T.D.; Hattori, M.; Ohno, H. Gut microorganisms act together to exacerbate inflammation in spinal cords. Nature 2020, 585, 102–106. [Google Scholar]
- Leonardi, I.; Gao, I.H.; Lin, W.-Y.; Allen, M.; Li, X.V.; Fiers, W.D.; de Celie, M.B.; Putzel, G.G.; Yantiss, R.K.; Johncilla, M. Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 2022, 185, 831–846.e814. [Google Scholar]
- Dodiya, H.B.; Lutz, H.L.; Weigle, I.Q.; Patel, P.; Michalkiewicz, J.; Roman-Santiago, C.J.; Zhang, C.M.; Liang, Y.; Srinath, A.; Zhang, X. Gut microbiota–driven brain Aβ amyloidosis in mice requires microglia. J. Exp. Med. 2021, 219, e20200895. [Google Scholar]
- Sun, J.; Xu, J.; Yang, B.; Chen, K.; Kong, Y.; Fang, N.; Gong, T.; Wang, F.; Ling, Z.; Liu, J. Effect of Clostridium butyricum against microglia-mediated neuroinflammation in Alzheimer’s disease via regulating gut microbiota and metabolites butyrate. Mol. Nutr. Food Res. 2020, 64, 1900636. [Google Scholar]
- Lebovitz, Y.; Kowalski, E.A.; Wang, X.; Kelly, C.; Lee, M.; McDonald, V.; Ward, R.; Creasey, M.; Mills, W.; Basso, E.K.G. Lactobacillus rescues postnatal neurobehavioral and microglial dysfunction in a model of maternal microbiome dysbiosis. Brain Behav. Immun. 2019, 81, 617–629. [Google Scholar]
- Li, S.-B.; Borniger, J.C.; Yamaguchi, H.; Hédou, J.; Gaudilliere, B.; de Lecea, L. Hypothalamic circuitry underlying stress-induced insomnia and peripheral immunosuppression. Sci. Adv. 2020, 6, eabc2590. [Google Scholar]
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z. Microglia require CD4 T cells to complete the fetal-to-adult transition. Cell 2020, 182, 625–640.e624. [Google Scholar]
- Kular, L.; Ewing, E.; Needhamsen, M.; Pahlevan Kakhki, M.; Covacu, R.; Gomez-Cabrero, D.; Brundin, L.; Jagodic, M. DNA methylation changes in glial cells of the normal-appearing white matter in Multiple Sclerosis patients. Epigenetics 2022, 17, 1311–1330. [Google Scholar]
- Bagga, D.; Reichert, J.L.; Koschutnig, K.; Aigner, C.S.; Holzer, P.; Koskinen, K.; Moissl-Eichinger, C.; Schöpf, V. Probiotics drive gut microbiome triggering emotional brain signatures. Gut Microbes 2018, 9, 486–496. [Google Scholar]
- Fond, G.B.; Lagier, J.-C.; Honore, S.; Lancon, C.; Korchia, T.; Verville, P.-L.S.D.; Llorca, P.-M.; Auquier, P.; Guedj, E.; Boyer, L. Microbiota-orientated treatments for major depression and schizophrenia. Nutrients 2020, 12, 1024. [Google Scholar]
- Kang, D.-W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci. Rep. 2019, 9, 5821. [Google Scholar]
- Liu, R.T.; Walsh, R.F.; Sheehan, A.E. Prebiotics and probiotics for depression and anxiety: A systematic review and meta-analysis of controlled clinical trials. Neurosci. Biobehav. Rev. 2019, 102, 13–23. [Google Scholar]
- Yamamura, R.; Okubo, R.; Katsumata, N.; Odamaki, T.; Hashimoto, N.; Kusumi, I.; Xiao, J.; Matsuoka, Y.J. Lipid and energy metabolism of the gut microbiota is associated with the response to probiotic Bifidobacterium breve strain for anxiety and depressive symptoms in schizophrenia. J. Pers. Med. 2021, 11, 987. [Google Scholar]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Active in | Functions | Phenotypes | Expression Status | Epigenetic Alteration(s) | References |
|---|---|---|---|---|---|---|
| TREM2 | Microglia | synapse pruning | Autism | Decreased in brain | Increased miRNA-34a | [39,43] |
| Alzheimer’s Disease | Increased in blood cells | DNA hypo-methylation | [44] | |||
| SCZ | Increased in blood cells | DNA hypo-methylation | [45] | |||
| Superior temporal gyrus (No expression study) | DNA hyper-methylation | [46] | ||||
| Increased in hippocampus | DNA hypermethylation (higher 5-hydroxymethylcytosine) | [47] | ||||
| MECP2 | Astrocytes | Neurodevelopment and regulation of microglia genes | Autism | Reduced in the frontal cortex | DNA hyper-methylation | [48,49] |
| C1q, C3 and C4 | Microglia and astrocytes | Synapse pruning | Autism and SCZ | Increased in several brain areas (e.g., DLPFC) | Different DNA methylation alterations | [54,55,56,57,58,59] |
| C3 | Microglia | synapse pruning | Alzheimer’s Disease | Increased in brain and middle temporal gyrus | DNA hypo-methylation | [60,61] |
| C4a | Microglia | synapse pruning | SCZ | Increased in blood cells | Not defined in SCZ (regulated by DNA methylation in ADHD) | [62,63] |
| SLC1A2/GLT1 | Astrocytes | Glutamate transporter and extracellular synapse glutamate removal | SCZ, BD | Increased in brain | Regulated by miR-218, DNA methylation and histone acetylation | [64,65,66,67] |
| Depression | Reduced in lateral habenula | ? | [68,69] | |||
| S100B | Mainly astrocytes | Hippocampal synaptogenesis | SCZ | Increased in blood cells and serum | DNA methylation alterations | [70,71] |
| MHC class I | Microglia | Synaptic pruning | SCZ | Reduced in brain (DLPFC) and blood | DNA methylation alterations | [58,72] |
| NDN | Astrocytes | Neurodevelopment, spine formation | SCZ and autism | ? | DNA Hypo-methylation (Imprinted gene) | [73] |
| KCNJ10 | Astrocytes | A potassium channel | Depressive symptoms | Increased in lateral habenula | Regulated by DNA methylation | [69,74] |
| NTRK2 | Astrocytes | Astrocyte maturation | Suicide | Decreased in brain | DNA hyper-methylation | [75] |
| SCZ | Increased in DLPFC | ? | [64] | |||
| GRIN2A | Astrocytes | Aβ cleanup | Depression | ? | DNA hypermethylation | [76] |
| HMGB1 | Microglia | Inflammation. stimulates microglia | Depression | Increased in hippocampal microglia and serum | Regulated by DNA methylation, HDAC4&5, and miR-129 | [77,78,79,80,81,82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdolmaleky, H.M.; Martin, M.; Zhou, J.-R.; Thiagalingam, S. Epigenetic Alterations of Brain Non-Neuronal Cells in Major Mental Diseases. Genes 2023, 14, 896. https://doi.org/10.3390/genes14040896
Abdolmaleky HM, Martin M, Zhou J-R, Thiagalingam S. Epigenetic Alterations of Brain Non-Neuronal Cells in Major Mental Diseases. Genes. 2023; 14(4):896. https://doi.org/10.3390/genes14040896
Chicago/Turabian StyleAbdolmaleky, Hamid Mostafavi, Marian Martin, Jin-Rong Zhou, and Sam Thiagalingam. 2023. "Epigenetic Alterations of Brain Non-Neuronal Cells in Major Mental Diseases" Genes 14, no. 4: 896. https://doi.org/10.3390/genes14040896