
Inverse Comorbidity between Down Syndrome and Solid Tumors: Insights from In Silico Analyses of Down Syndrome Critical Region Genes

Abstract
:1. Introduction
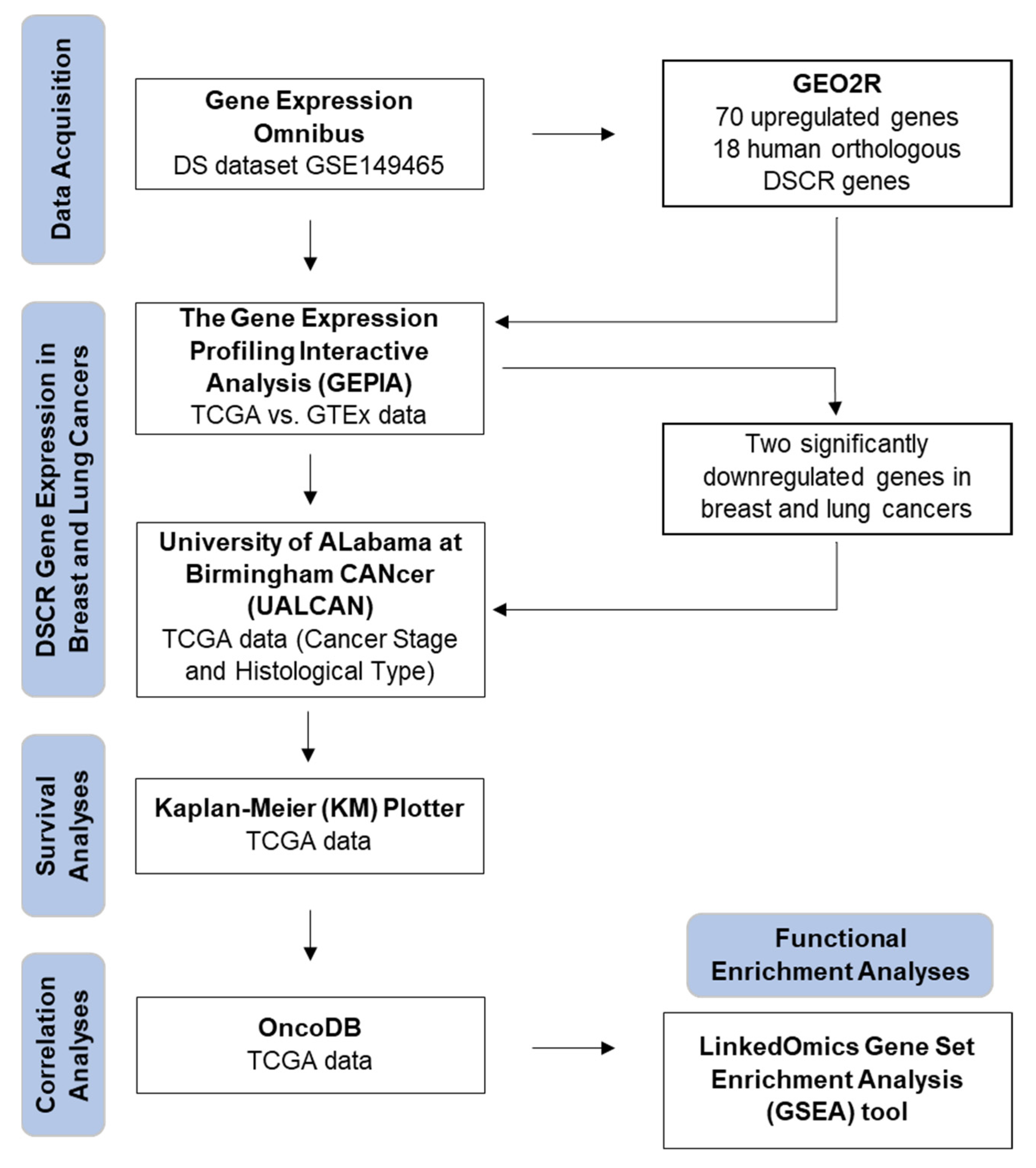
2. Materials and Methods
2.1. Gene Expression Data Acquisition
2.2. Identification of Differentially Expressed DSCR Genes
2.3. Analyses of DSCR Gene Expression in Breast and Lung Cancers
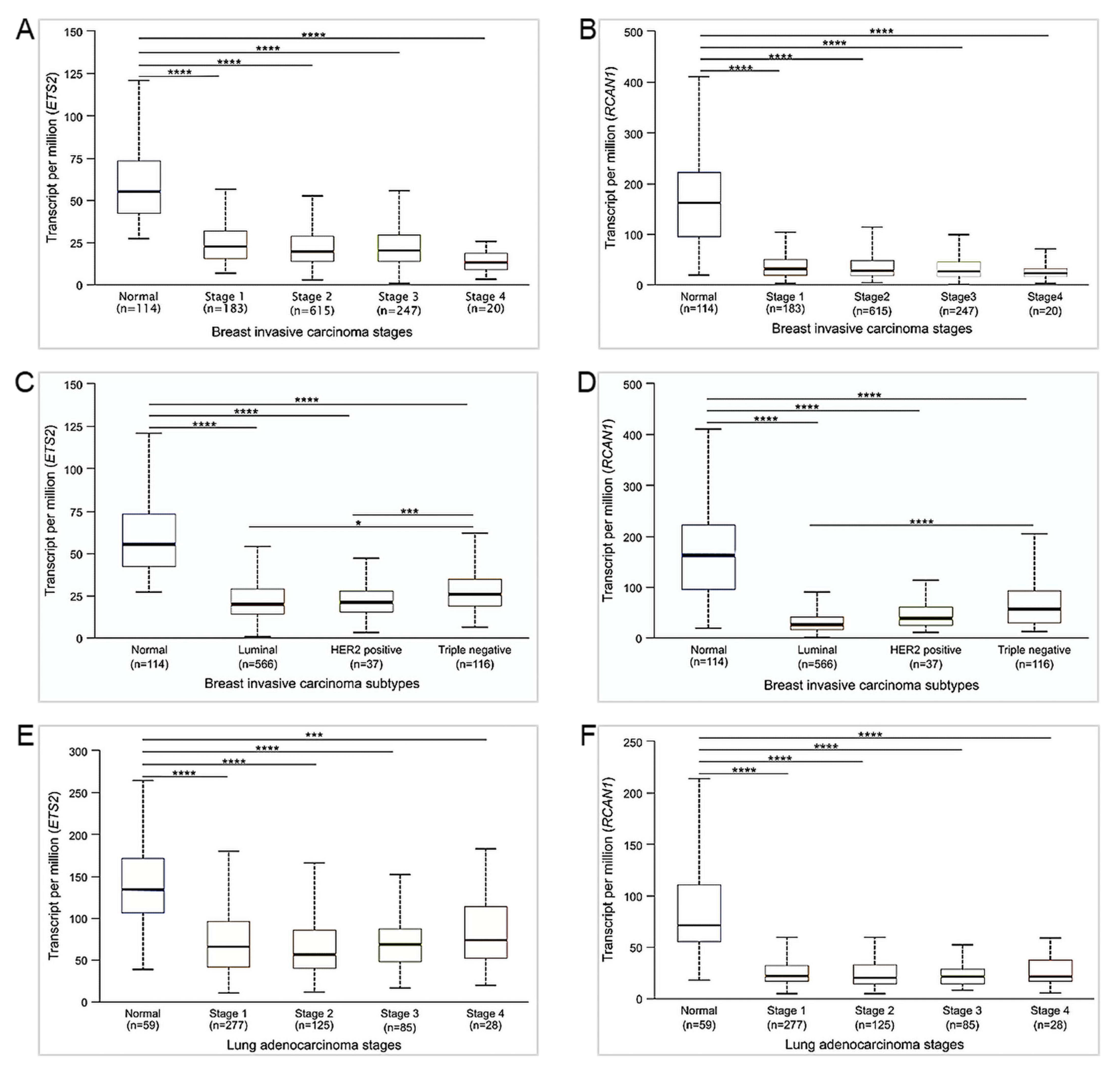
2.4. Gene Expression Analyses Based on Cancer Stage and Subtype
2.5. Survival Analyses
2.6. Correlation Analyses
2.7. Functional Enrichment Analyses
3. Results
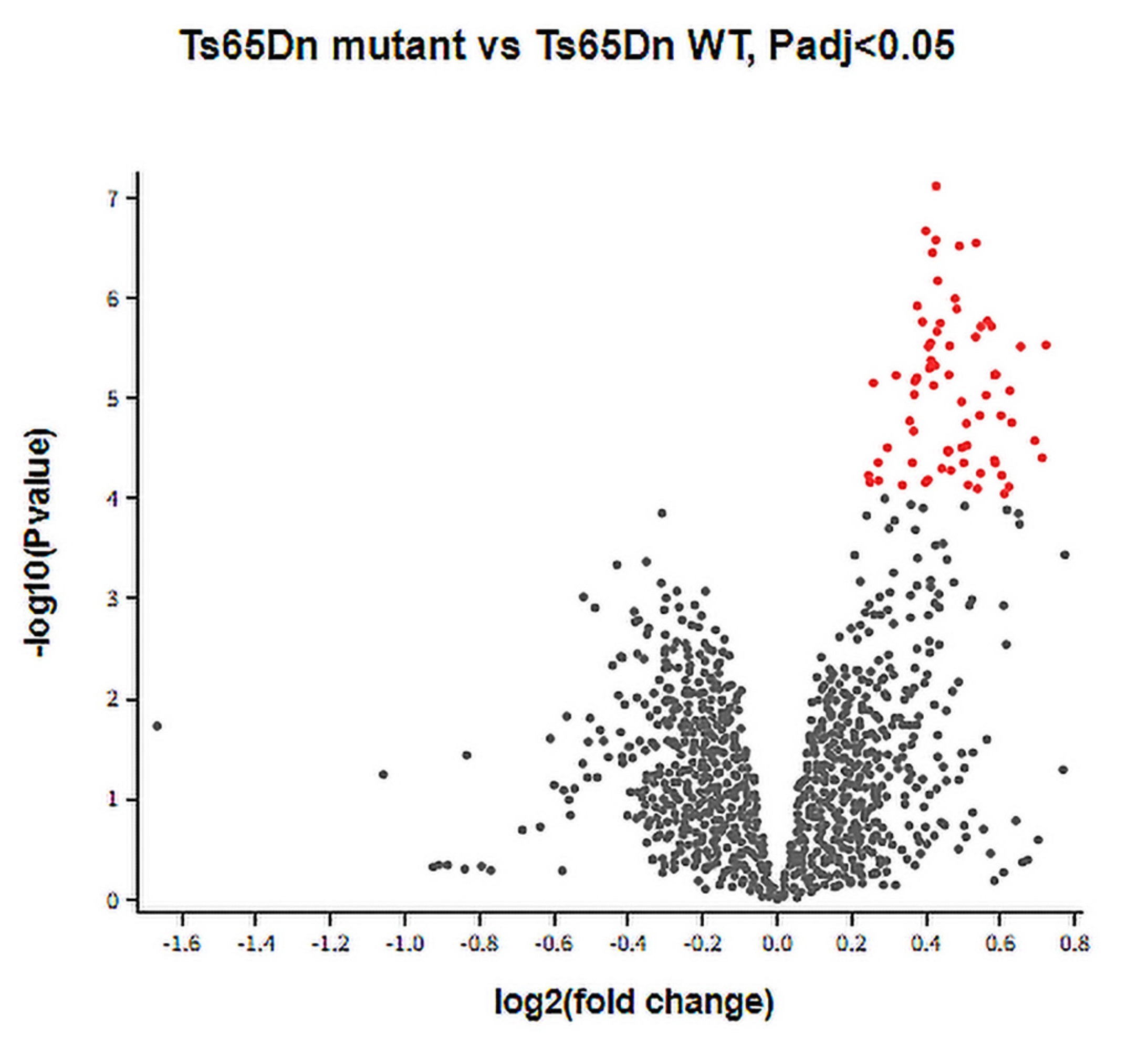
3.1. DEGs between Ts65Dn Mutant and Ts65Dn WT Groups
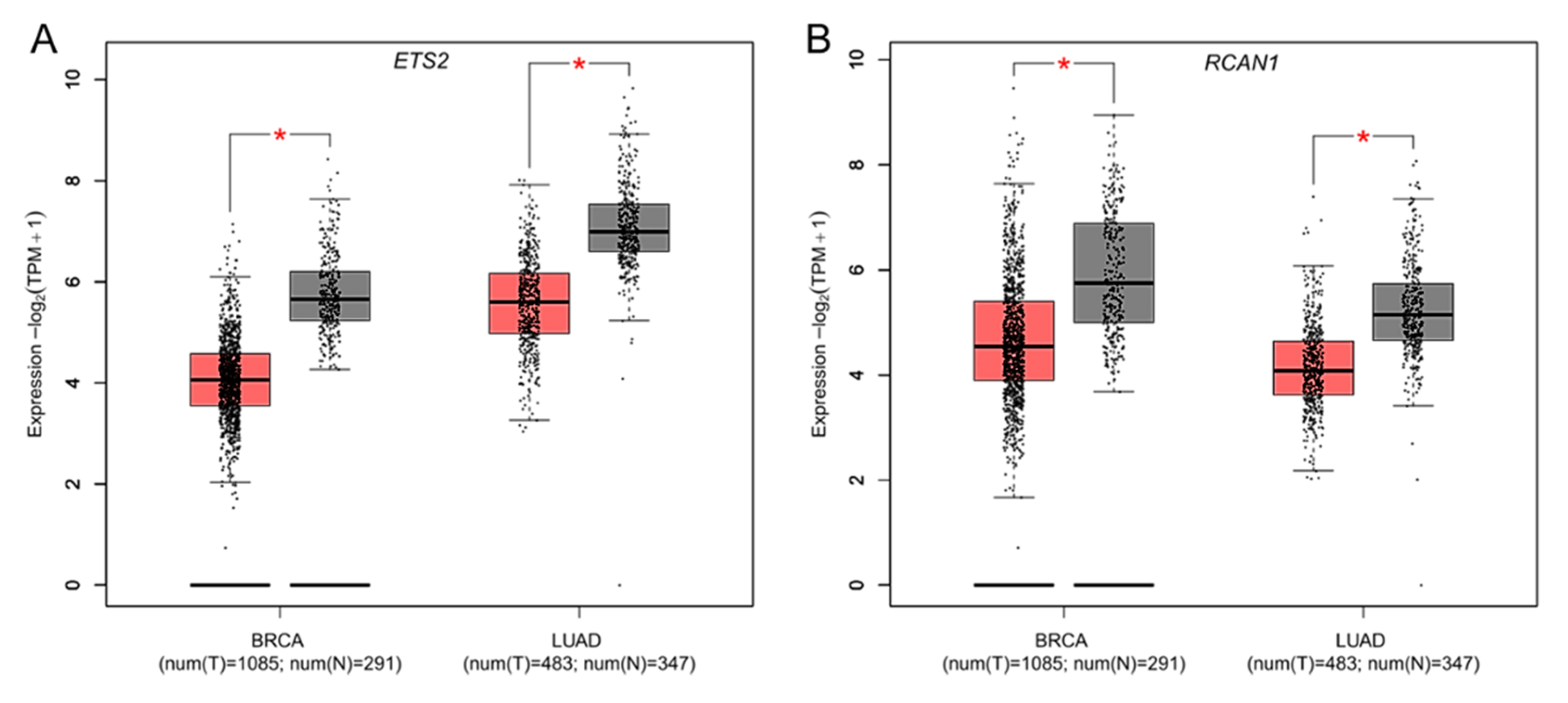
3.2. Differential Expression of DSCR Genes in Breast and Lung Cancers
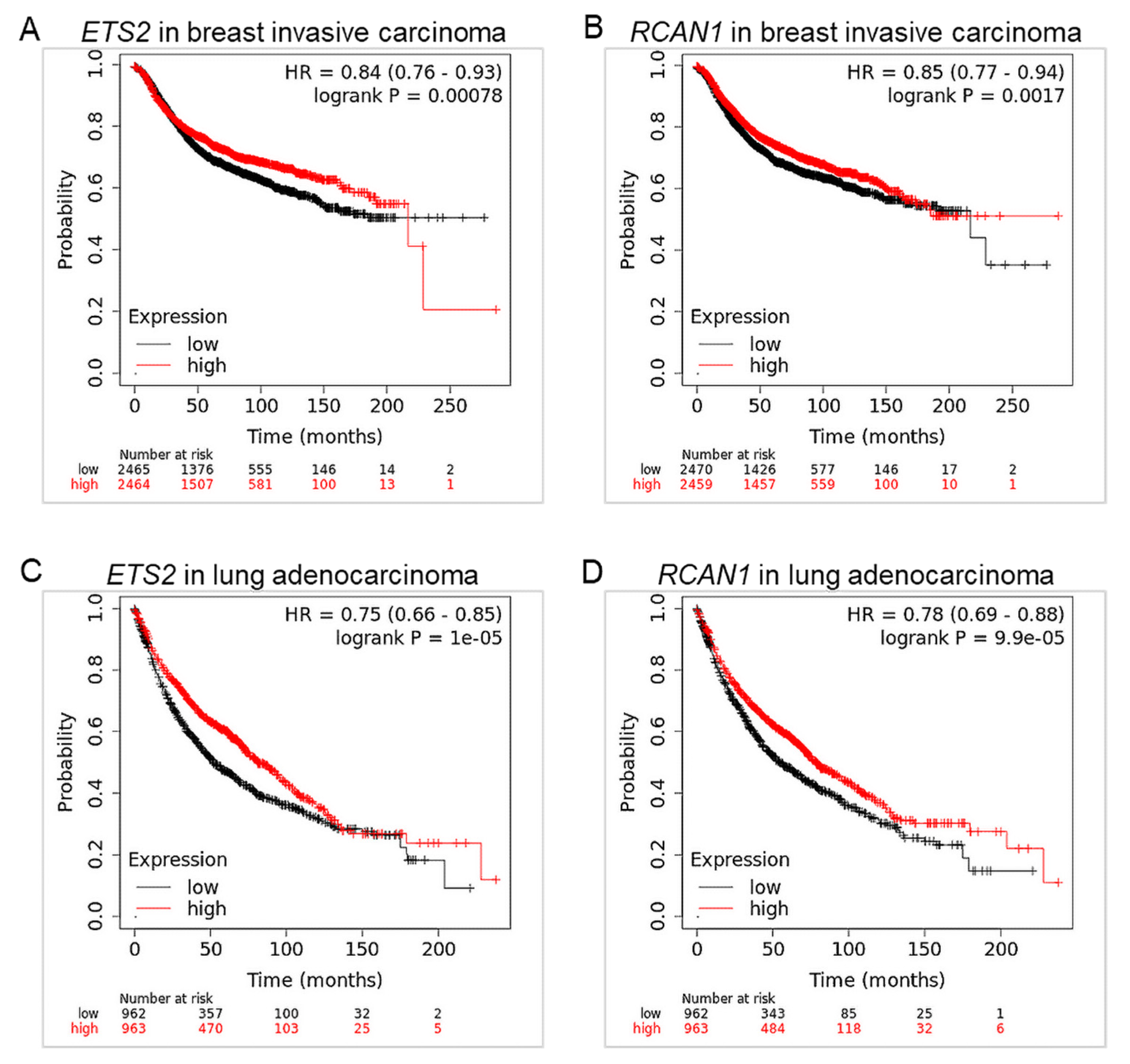
3.3. ETS2 and RCAN1 Are Associated with Low Overall Survival in Breast and Lung Cancers
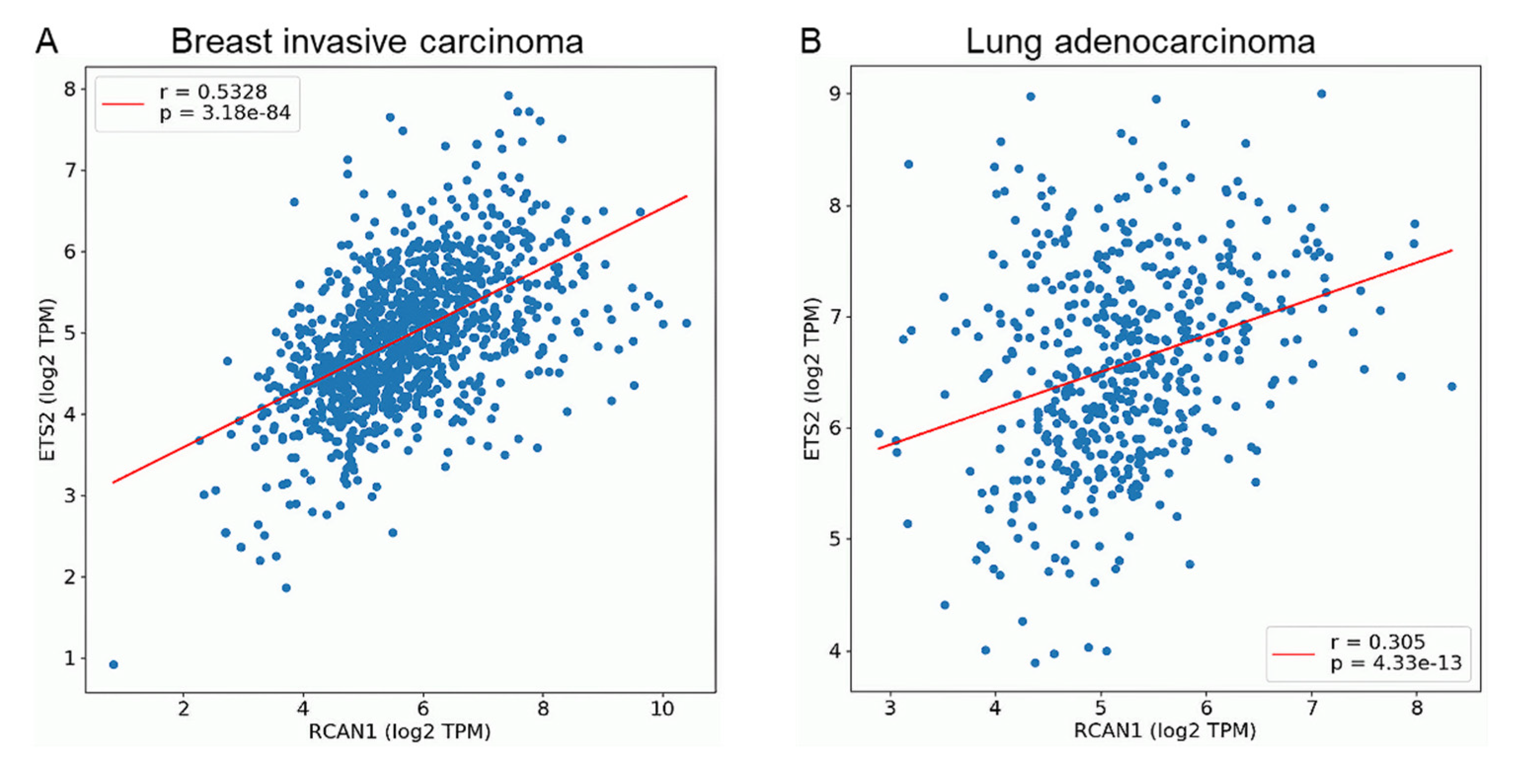
3.4. ETS2 and RCAN1 Expression Are Positively Correlated in Breast and Lung Cancers
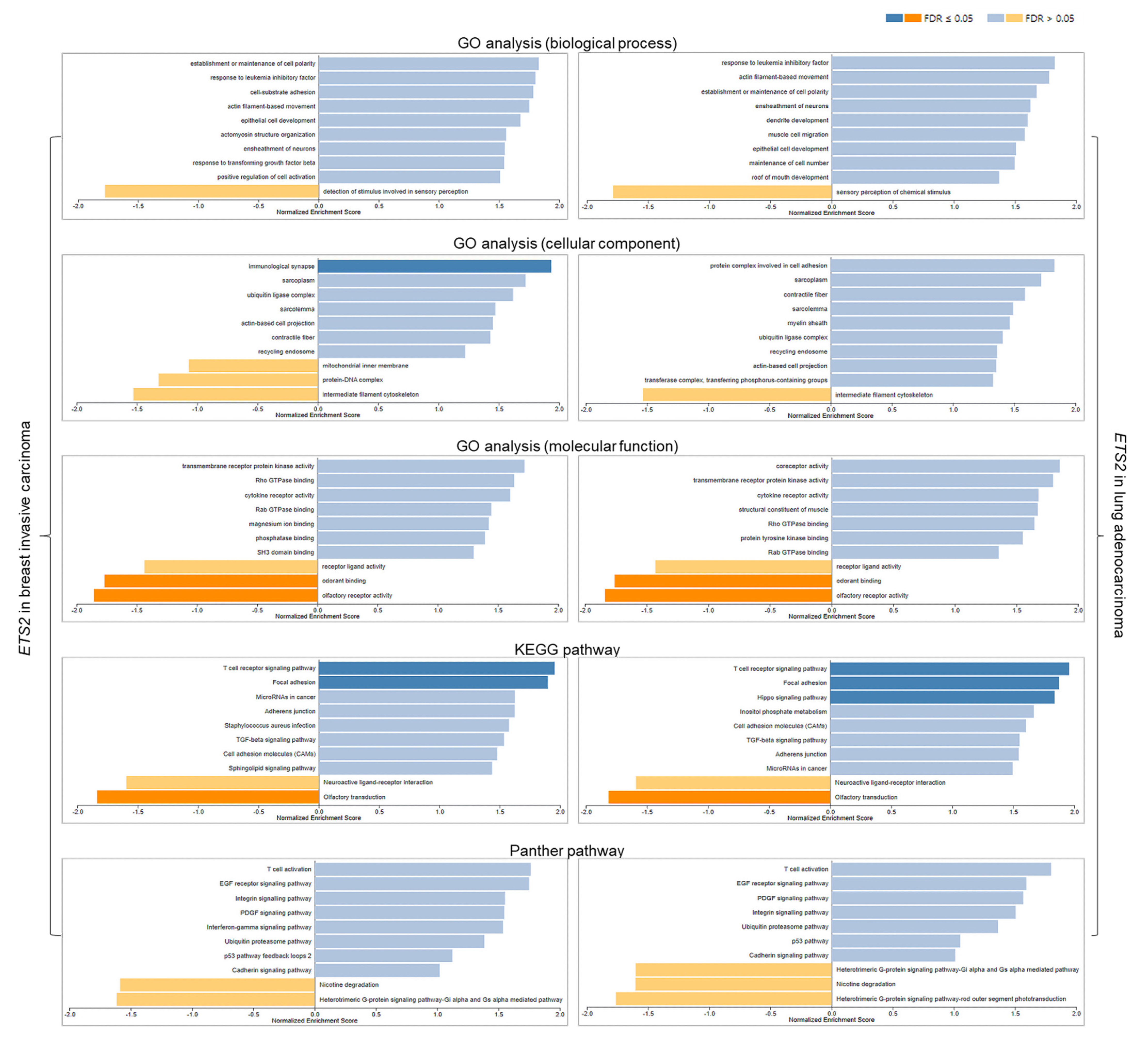
3.5. Functional Analyses of ETS2 in Breast and Lung Cancers
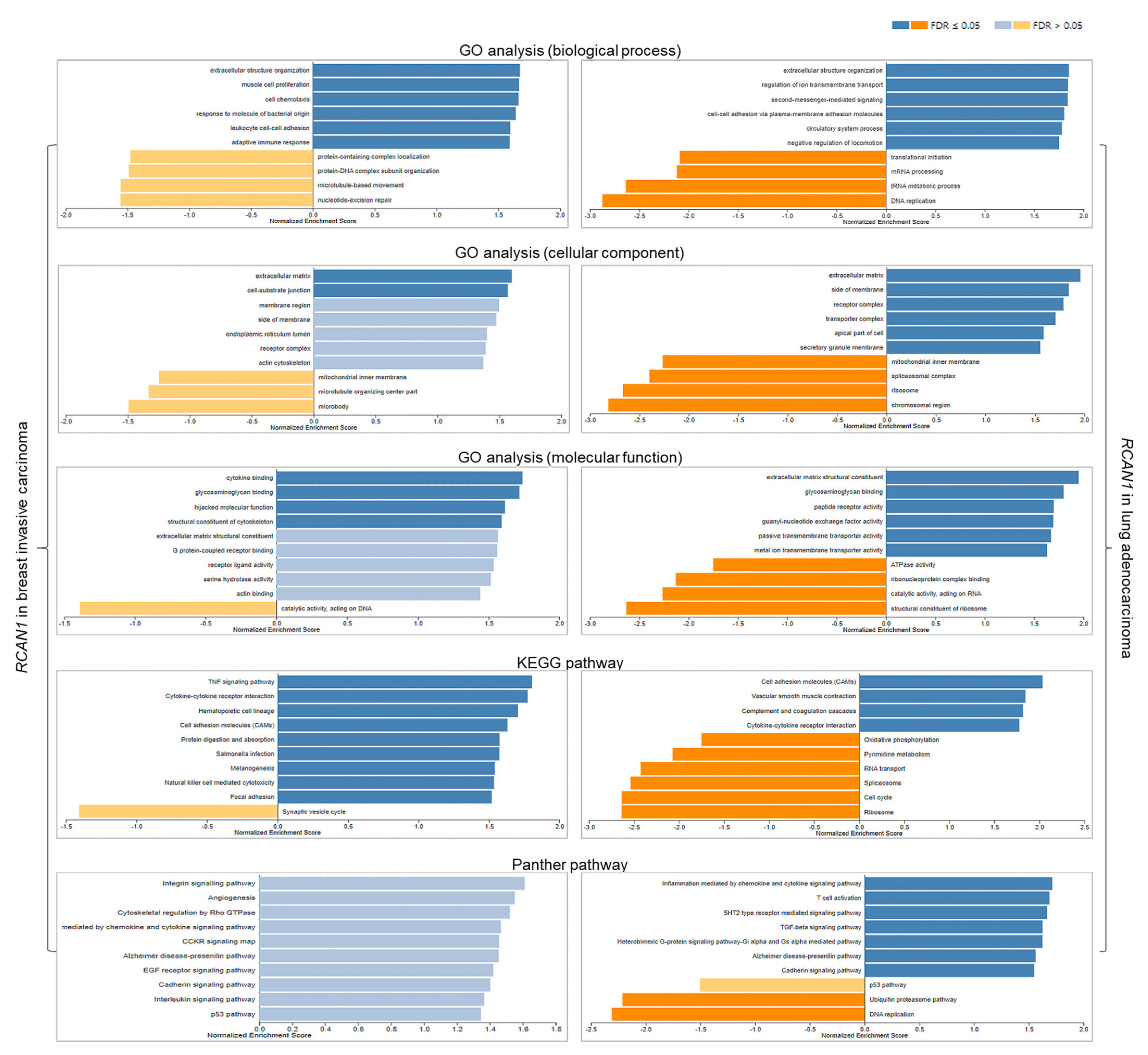
3.6. Functional Analyses of RCAN1 in Breast and Lung Cancers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- CDC. Facts about Down Syndrome|CDC; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2022.
- Bull, M.J. Down Syndrome. N. Engl. J. Med. 2020, 382, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, G.; Buckley, F.; Skotko, B.G. Estimation of the Number of People with Down Syndrome in Europe. Eur. J. Hum. Genet. 2021, 29, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down Syndrome. Nat. Rev. Dis. Prim. 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Vega. Chromosome 21: 1-1—Chromosome Summary—Homo sapiens—Vega Genome Browser 68. Available online: http://vega.archive.ensembl.org/Homo_sapiens/Location/Chromosome?r=21 (accessed on 17 March 2023).
- Osuna-Marco, M.P.; López-Barahona, M.; López-Ibor, B.; Tejera, Á.M. Ten Reasons Why People with Down Syndrome Are Protected from the Development of Most Solid Tumors-A Review. Front. Genet. 2021, 12, 749480. [Google Scholar] [CrossRef]
- Di Cunto, F.; Berto, G. Molecular Pathways of Down Syndrome Critical Region Genes. In Down Syndrome; IntechOpen: Rijeka, Croatia, 2013. [Google Scholar]
- Allen, E.G.; Freeman, S.B.; Druschel, C.; Hobbs, C.A.; O’Leary, L.A.; Romitti, P.A.; Royle, M.H.; Torfs, C.P.; Sherman, S.L. Maternal Age and Risk for Trisomy 21 Assessed by the Origin of Chromosome Nondisjunction: A Report from the Atlanta and National Down Syndrome Projects. Hum. Genet. 2009, 125, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Yoon, P.W.; Freeman, S.B.; Sherman, S.L.; Taft, L.F.; Gu, Y.; Pettay, D.; Flanders, W.D.; Khoury, M.J.; Hassold, T.J. Advanced Maternal Age and the Risk of Down Syndrome Characterized by the Meiotic Stage of Chromosomal Error: A Population-Based Study. Am. J. Hum. Genet. 1996, 58, 628–633. [Google Scholar]
- Oliver, T.R.; Feingold, E.; Yu, K.; Cheung, V.; Tinker, S.; Yadav-Shah, M.; Masse, N.; Sherman, S.L. New Insights into Human Nondisjunction of Chromosome 21 in Oocytes. PLoS Genet. 2008, 4, e1000033. [Google Scholar] [CrossRef] [Green Version]
- Coppedè, F. Segmental Trisomy of Murine Chromosome 16: A New Model System for Studying Down Syndrome. Arch. Toxicol. 2016, 90, 2917–2929. [Google Scholar] [CrossRef]
- Shimada, A. Profile of down Syndrome–Associated Malignancies: Epidemiology, Clinical Features and Therapeutic Aspects. Pediatr. Hematol. Oncol. J. 2021, 6, 63–72. [Google Scholar] [CrossRef]
- Xavier, A.C.; Ge, Y.; Taub, J.W. Down Syndrome and Malignancies: A Unique Clinical Relationship—A Paper from the 2008 William Beaumont Hospital Symposium on Molecular Pathology. J. Mol. Diagn. 2009, 11, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Hasle, H.; Friedman, J.M.; Olsen, J.H.; Rasmussen, S.A. Low Risk of Solid Tumors in Persons with Down Syndrome. Genet. Med. 2016, 18, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Patja, K.; Pukkala, E.; Sund, R.; Iivanainen, M.; Kaski, M. Cancer Incidence of Persons with Down Syndrome in Finland: A Population-Based Study. Int. J. Cancer 2006, 118, 1769–1772. [Google Scholar] [CrossRef]
- Yu, Y.E.; Xing, Z.; Do, C.; Pao, A.; Lee, E.J.; Krinsky-McHale, S.; Silverman, W.; Schupf, N.; Tycko, B. Genetic and Epigenetic Pathways in Down Syndrome: Insights to the Brain and Immune System from Humans and Mouse Models. Prog. Brain Res. 2020, 251, 1–28. [Google Scholar]
- Dekker, A.D.; De Deyn, P.P.; Rots, M.G. Epigenetics: The Neglected Key to Minimize Learning and Memory Deficits in Down Syndrome. Neurosci. Biobehav. Rev. 2014, 45, 72–84. [Google Scholar] [CrossRef]
- Sussan, T.E.; Yang, A.; Li, F.; Ostrowski, M.C.; Reeves, R.H. Trisomy Represses ApcMin-Mediated Tumours in Mouse Models of Down’s Syndrome. Nature 2008, 451, 73–75. [Google Scholar] [CrossRef]
- Yang, A.; Reeves, R.H. Increased Survival Following Tumorigenesis in Ts65Dn Mice That Model Down Syndrome. Cancer Res. 2011, 71, 3573–3581. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, L.E.; Watson, A.R.; Baker, M.; Jones, T.A.; D’Amico, G.; Robinson, S.D.; Joffre, C.; Garrido-Urbani, S.; Rodriguez-Manzaneque, J.C.; Martino-Echarri, E.; et al. Tumour Angiogenesis Is Reduced in the Tc1 Mouse Model of Down’s Syndrome. Nature 2010, 465, 813–817. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Lee, J.C.; Baek, K.-H. A Single Extra Copy of Dscr1 Improves Survival of Mice Developing Spontaneous Lung Tumors through Suppression of Tumor Angiogenesis. Cancer Lett. 2014, 342, 70–81. [Google Scholar] [CrossRef]
- Duchon, A.; Del Mar Muniz Moreno, M.; Martin Lorenzo, S.; Silva de Souza, M.P.; Chevalier, C.; Nalesso, V.; Meziane, H.; Loureiro de Sousa, P.; Noblet, V.; Armspach, J.-P.; et al. Multi-Influential Genetic Interactions Alter Behaviour and Cognition through Six Main Biological Cascades in Down Syndrome Mouse Models. Hum. Mol. Genet. 2021, 30, 771–788. [Google Scholar] [CrossRef]
- Aziz, N.M.; Guedj, F.; Pennings, J.L.A.; Olmos-Serrano, J.L.; Siegel, A.; Haydar, T.F.; Bianchi, D.W. Lifespan Analysis of Brain Development, Gene Expression and Behavioral Phenotypes in the Ts1Cje, Ts65Dn and Dp(16)1/Yey Mouse Models of Down Syndrome. Dis. Model. Mech. 2018, 11, dmm031013. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An Update to the Integrated Cancer Data Analysis Platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Cho, M.; Wang, X. OncoDB: An Interactive Online Database for Analysis of Gene Expression and Viral Infection in Cancer. Nucleic Acids Res. 2022, 50, D1334–D1339. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing Multi-Omics Data within and across 32 Cancer Types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [Green Version]
- Fry, E.A.; Inoue, K. Aberrant Expression of ETS1 and ETS2 Proteins in Cancer. Cancer Rep. Rev. 2018, 2, 1–10. [Google Scholar] [CrossRef]
- Ohtani, N.; Zebedee, Z.; Huot, T.J.; Stinson, J.A.; Sugimoto, M.; Ohashi, Y.; Sharrocks, A.D.; Peters, G.; Hara, E. Opposing Effects of Ets and Id Proteins on p16INK4a Expression during Cellular Senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef]
- Baker, K.M.; Wei, G.; Schaffner, A.E.; Ostrowski, M.C. Ets-2 and Components of Mammalian SWI/SNF Form a Repressor Complex That Negatively Regulates the BRCA1 Promoter. J. Biol. Chem. 2003, 278, 17876–17884. [Google Scholar] [CrossRef] [Green Version]
- Schedin, P.J.; Eckel-Mahan, K.L.; McDaniel, S.M.; Prescott, J.D.; Brodsky, K.S.; Tentler, J.J.; Gutierrez-Hartmann, A. ESX Induces Transformation and Functional Epithelial to Mesenchymal Transition in MCF-12A Mammary Epithelial Cells. Oncogene 2004, 23, 1766–1779. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Dwyer, J.; Li, H.; Duan, W.; Liu, J.-P. Ets2 Maintains HTERT Gene Expression and Breast Cancer Cell Proliferation by Interacting with C-Myc. J. Biol. Chem. 2008, 283, 23567–23580. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Kobayashi, S.; Borczuk, A.C.; Leidner, R.S.; Laframboise, T.; Levine, A.D.; Halmos, B. Dual Specificity Phosphatase 6 (DUSP6) Is an ETS-Regulated Negative Feedback Mediator of Oncogenic ERK Signaling in Lung Cancer Cells. Carcinogenesis 2010, 31, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Kabbout, M.; Garcia, M.M.; Fujimoto, J.; Liu, D.D.; Woods, D.; Chow, C.-W.; Mendoza, G.; Momin, A.A.; James, B.P.; Solis, L.; et al. ETS2 Mediated Tumor Suppressive Function and MET Oncogene Inhibition in Human Non-Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 3383–3395. [Google Scholar] [CrossRef] [Green Version]
- Lao, M.; Zhang, X.; Yang, H.; Bai, X.; Liang, T. RCAN1-Mediated Calcineurin Inhibition as a Target for Cancer Therapy. Mol. Med. 2022, 28, 69. [Google Scholar] [CrossRef]
- Wang, C.; Saji, M.; Justiniano, S.E.; Yusof, A.M.; Zhang, X.; Yu, L.; Fernández, S.; Wakely, P., Jr.; La Perle, K.; Nakanishi, H.; et al. RCAN1-4 Is a Thyroid Cancer Growth and Metastasis Suppressor. JCI Insight 2017, 2, e90651. [Google Scholar] [CrossRef]
- Jin, H.; Wang, C.; Jin, G.; Ruan, H.; Gu, D.; Wei, L.; Wang, H.; Wang, N.; Arunachalam, E.; Zhang, Y.; et al. Regulator of Calcineurin 1 Gene Isoform 4, Down-Regulated in Hepatocellular Carcinoma, Prevents Proliferation, Migration, and Invasive Activity of Cancer Cells and Metastasis of Orthotopic Tumors by Inhibiting Nuclear Translocation of NFAT1. Gastroenterology 2017, 153, 799–811.e33. [Google Scholar] [CrossRef] [Green Version]
- Behtaji, S.; Ghafouri-Fard, S.; Sayad, A.; Sattari, A.; Rederstorff, M.; Taheri, M. Identification of Oxytocin-Related LncRNAs and Assessment of Their Expression in Breast Cancer. Sci. Rep. 2021, 11, 6471. [Google Scholar] [CrossRef]
- Deng, R.; Huang, J.-H.; Wang, Y.; Zhou, L.-H.; Wang, Z.-F.; Hu, B.-X.; Chen, Y.-H.; Yang, D.; Mai, J.; Li, Z.-L.; et al. Disruption of Super-Enhancer-Driven Tumor Suppressor Gene RCAN1. 4 Expression Promotes the Malignancy of Breast Carcinoma. Mol. Cancer 2020, 19, 122. [Google Scholar] [CrossRef]
- Ma, N.; Shen, W.; Pang, H.; Zhang, N.; Shi, H.; Wang, J.; Zhang, H. The Effect of RCAN1 on the Biological Behaviors of Small Cell Lung Cancer. Tumor Biol. 2017, 39, 1010428317700405. [Google Scholar] [CrossRef] [Green Version]
- Minami, T.; Jiang, S.; Schadler, K.; Suehiro, J.-I.; Osawa, T.; Oike, Y.; Miura, M.; Naito, M.; Kodama, T.; Ryeom, S. The Calcineurin-NFAT-Angiopoietin-2 Signaling Axis in Lung Endothelium Is Critical for the Establishment of Lung Metastases. Cell Rep. 2013, 4, 709–723. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Park, S.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Sung, K.J.; Sung, Y.H.; Choi, C.-M.; Yun, M.; Yi, Y.-S.; et al. Tumor-Derived Exosomal MiR-619-5p Promotes Tumor Angiogenesis and Metastasis through the Inhibition of RCAN1.4. Cancer Lett. 2020, 475, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Forés-Martos, J.; Cervera-Vidal, R.; Chirivella, E.; Ramos-Jarero, A.; Climent, J. A Genomic Approach to Study Down Syndrome and Cancer Inverse Comorbidity: Untangling the Chromosome 21. Front. Physiol. 2015, 6, 10. [Google Scholar] [PubMed] [Green Version]
- Ren, Y.; Chen, B.; Zhang, M. Distinct Prognostic and Immunological Roles of ETS1 and ETS2: A Pan-Cancer Analysis. Biomed Res. Int. 2023, 2023, 4343350. [Google Scholar] [CrossRef] [PubMed]
- Bassuk, A.G.; Anandappa, R.T.; Leiden, J.M. Physical Interactions between Ets and NF-KappaB/NFAT Proteins Play an Important Role in Their Cooperative Activation of the Human Immunodeficiency Virus Enhancer in T Cells. J. Virol. 1997, 71, 3563–3573. [Google Scholar] [CrossRef] [Green Version]
- Tsao, H.-W.; Tai, T.-S.; Tseng, W.; Chang, H.-H.; Grenningloh, R.; Miaw, S.-C.; Ho, I.-C. Ets-1 Facilitates Nuclear Entry of NFAT Proteins and Their Recruitment to the IL-2 Promoter. Proc. Natl. Acad. Sci. USA 2013, 110, 15776–15781. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Jiang, N.; May, H.I.; Luo, X.; Ferdous, A.; Schiattarella, G.G.; Chen, G.; Li, Q.; Li, C.; Rothermel, B.A.; et al. Cooperative Binding of ETS2 and NFAT Links Erk1/2 and Calcineurin Signaling in the Pathogenesis of Cardiac Hypertrophy. Circulation 2021, 144, 34–51. [Google Scholar] [CrossRef]
- Yuan, M.; Zhao, Y.; Arkenau, H.-T.; Lao, T.; Chu, L.; Xu, Q. Signal Pathways and Precision Therapy of Small-Cell Lung Cancer. Signal Transduct. Target. Ther. 2022, 7, 187. [Google Scholar] [CrossRef]
- Yousefnia, S.; Seyed Forootan, F.; Seyed Forootan, S.; Nasr Esfahani, M.H.; Gure, A.O.; Ghaedi, K. Mechanistic Pathways of Malignancy in Breast Cancer Stem Cells. Front. Oncol. 2020, 10, 452. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Chromosomal Position | |
|---|---|---|---|
| Human | Mouse | ||
| Bace2 | β-site app-cleaving enzyme 2 | chr16:97157942-97244136 | chr21:39556492-39670904 |
| Brwd1 | Bromodomain and WD repeat-containing protein 1 | chr16:95793292-95883628 | chr21:37568748-37698102 |
| B3galt5 | β-1,3-galactosyltransferase 5 | chr16:96037001-96121059 | chr21:38042256-38057589 |
| Cbr1 | Carbonyl reductase 1 | chr16:93404725-93407393 | chr21:34453172-34455941 |
| Cbr3 | Carbonyl reductase 3 | chr16:93480103-93487878 | chr21:34518104-34529412 |
| Dopey2 | Dopey protein 2 | chr16:93508792-93607476 | |
| Dscam | Down syndrome cell adhesion molecule | chr16:96392040-96971952 | chr21:38398213-39235595 |
| Dscr3 | Down syndrome critical region gene 3 | chr16:94298642-94327689 | chr21:37223425-37267532 |
| Dyrk1a | Dual-specificity tyrosine-phosphorylation-regulated kinase 1a | chr16:94370869-94495926 | chr21:35802642-35897561 |
| Ets2 | Ets proto-oncogene 2 transcription factor | chr16:95502942-95522095 | chr21:37188977-37208711 |
| Hlcs | Holocarboxylase synthetase | chr16:93931271-94091145 | chr21:35130820-35348858 |
| Kcnj6 | Potassium inwardly-rectifying channel 6 | chr16:94561928-94798555 | chr21:36006636-36299912 |
| Morc3 | Microrchidia 3 | chr16:93629009-93672961 | chr21:34702785-34759216 |
| Pigp | Phosphatidylinositol N-acetylglucosaminyltransferase subunit P | chr16:94165494-94172701 | chr21:35447565-35455253 |
| Psmg1 | Proteasome assembly chaperone 1 | chr16:95781133-95792160 | chr21:37559338-37568021 |
| Rcan1 | Regulator of calcineurin 1 | chr16:92188843-92196965 | chr21:32899008-32908032 |
| Ttc3 | Tetratricopeptide protein ligase | chr16:94171477-94270079 | chr21:35455544-35585333 |
| Wrb | Tryptophan-rich basic protein | chr16:95946607-95959052 | chr21:37764912-37900121 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fosu, K.; Quarshie, J.T.; Sarpong, K.A.N.; Aikins, A.R. Inverse Comorbidity between Down Syndrome and Solid Tumors: Insights from In Silico Analyses of Down Syndrome Critical Region Genes. Genes 2023, 14, 800. https://doi.org/10.3390/genes14040800
Fosu K, Quarshie JT, Sarpong KAN, Aikins AR. Inverse Comorbidity between Down Syndrome and Solid Tumors: Insights from In Silico Analyses of Down Syndrome Critical Region Genes. Genes. 2023; 14(4):800. https://doi.org/10.3390/genes14040800
Chicago/Turabian StyleFosu, Kwadwo, Jude Tetteh Quarshie, Kwabena Amofa Nketia Sarpong, and Anastasia Rosebud Aikins. 2023. "Inverse Comorbidity between Down Syndrome and Solid Tumors: Insights from In Silico Analyses of Down Syndrome Critical Region Genes" Genes 14, no. 4: 800. https://doi.org/10.3390/genes14040800