
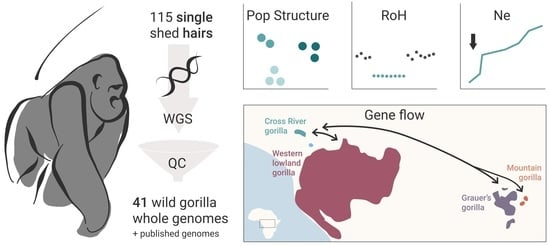
Past Connectivity but Recent Inbreeding in Cross River Gorillas Determined Using Whole Genomes from Single Hairs
 , , , , , and
, , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Samples, DNA Extraction, and Library Preparation
2.2. Sequencing Data Processing (FASTQ to Genotypes/Likelihoods)
2.3. Quality Control (Depth of Coverage and Contamination)
2.4. Genotype Likelihoods and SNP Calling
2.5. Genetic Sexing from Coverage
2.6. Relatedness Test
2.7. Population Structure Assessment
2.8. Heterozygosity Estimates
2.9. Estimation of Runs of Homozygosity and Inbreeding Coefficients
2.10. Past and Present Demographic History
2.11. Assessment of Gene Flow and Connectivity
3. Results
3.1. Single Hairs as a Source for Genomic DNA
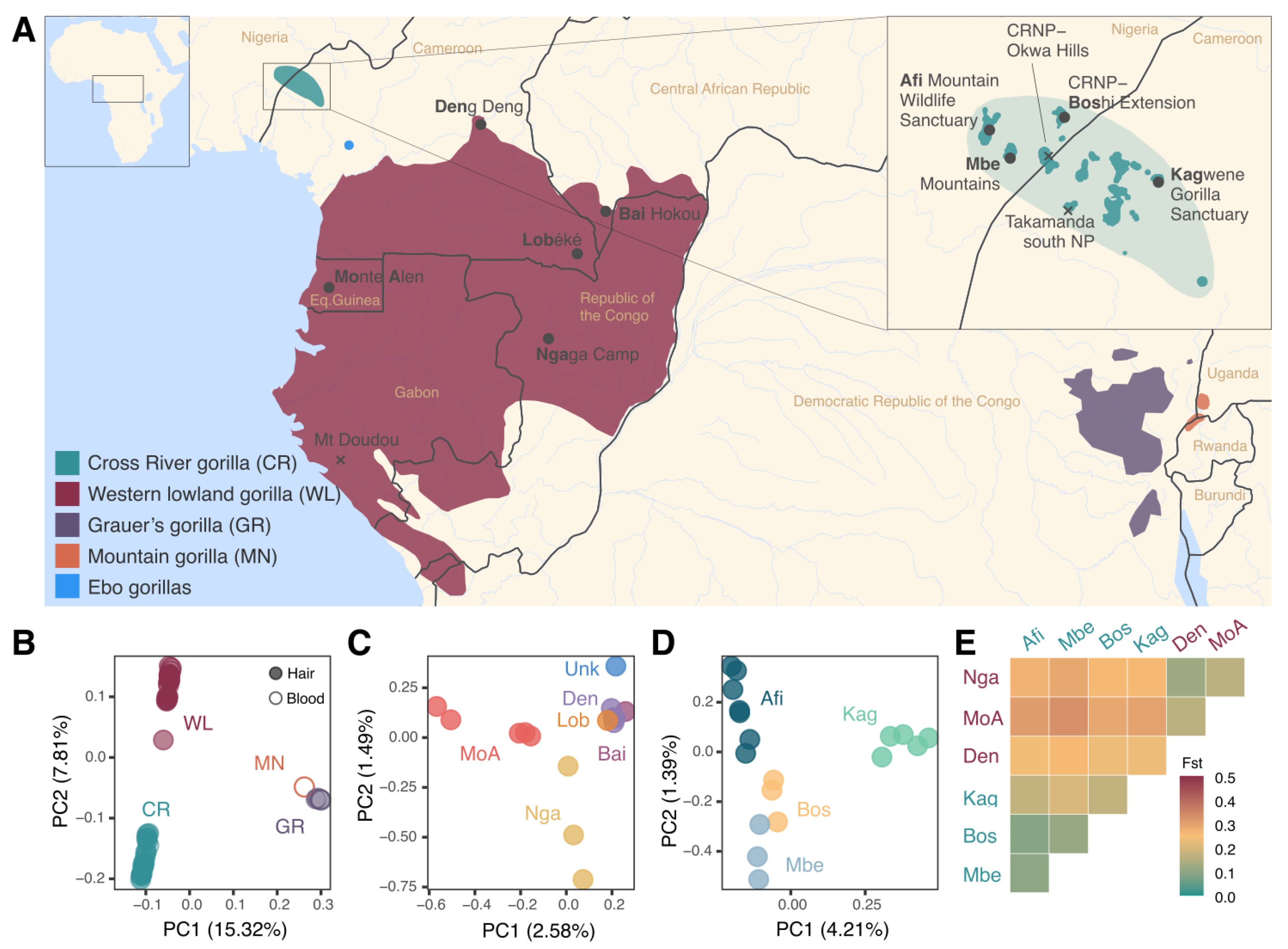
3.2. Increased Resolution of Western Gorilla Population Structure
3.3. Genetic Diversity among Gorilla Species
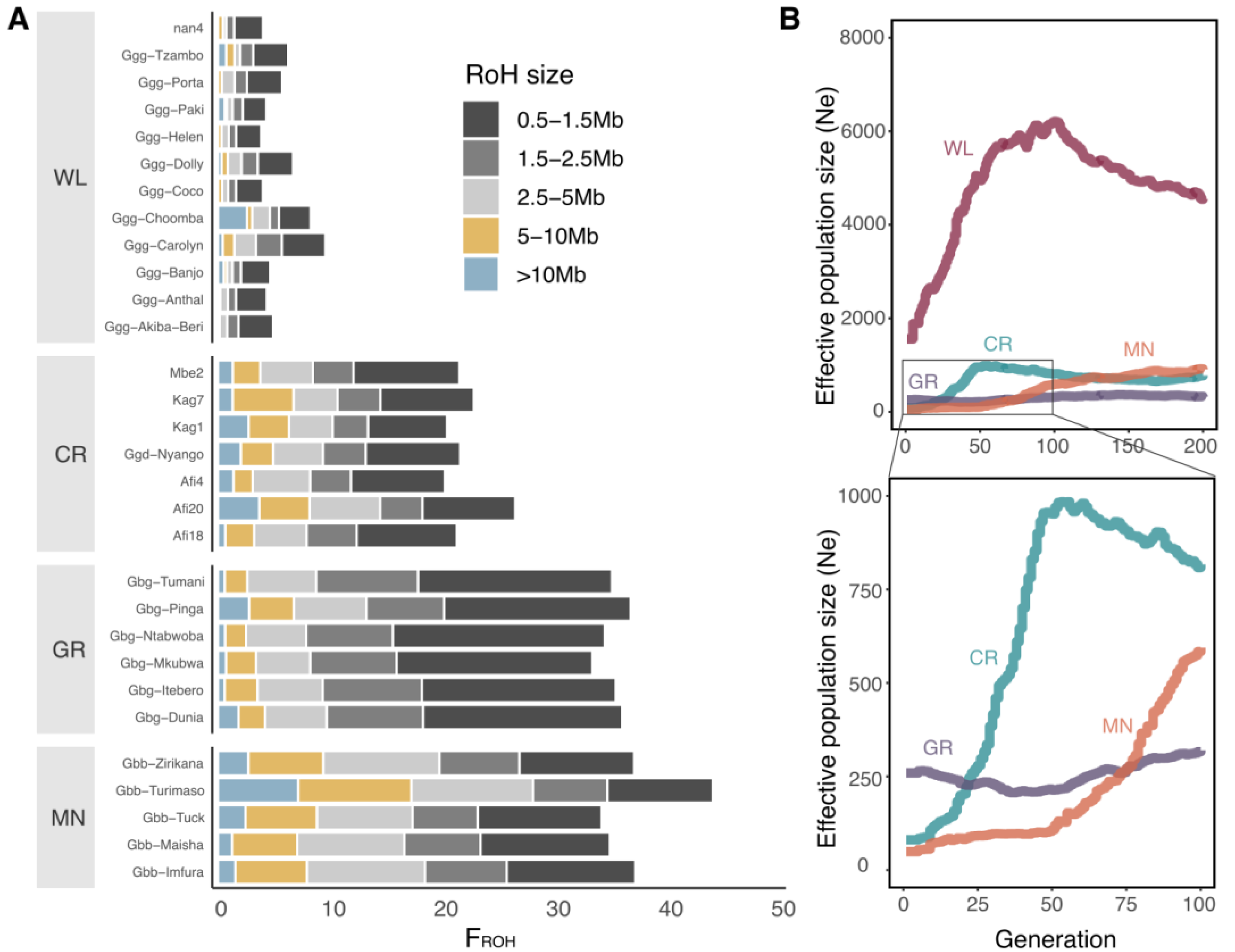
3.4. Strong Bottleneck in Cross River Gorillas
3.5. Past Gene Flow among Gorilla Subspecies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strindberg, S.; Maisels, F.; Williamson, E.A.; Blake, S.; Stokes, E.J.; Aba’a, R.; Abitsi, G.; Agbor, A.; Ambahe, R.D.; Bakabana, P.C.; et al. Guns, Germs, and Trees Determine Density and Distribution of Gorillas and Chimpanzees in Western Equatorial Africa. Sci. Adv. 2018, 4, eaar2964. [Google Scholar] [CrossRef] [Green Version]
- Dunn, A.; Bergl, R.; Byler, D.; Eben-Ebai, S.; Ndeloh Etiendem, D.; Fotso, R.; Ikfuingei, R.; Imong, I.; Jameson, C.; Macfie, L.; et al. Revised Regional Action Plan for the Conservation of the Cross River Gorilla (Gorilla gorilla diehli) 2014–2019; IUCN/SSC Primate Specialist Group and Wildlife Conservation Society: New York, NY, USA, 2014; ISBN 9780963206428. [Google Scholar]
- Imong, I.; Robbins, M.M.; Mundry, R.; Bergl, R.; Kühl, H.S. Informing Conservation Management about Structural versus Functional Connectivity: A Case-Study of Cross River Gorillas. Am. J. Primatol. 2014, 76, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Bergl, R.A.; Warren, Y.; Nicholas, A.; Dunn, A.; Imong, I.; Sunderland-Groves, J.L.; Oates, J.F. Remote Sensing Analysis Reveals Habitat, Dispersal Corridors and Expanded Distribution for the Critically Endangered Cross River Gorilla Gorilla gorilla diehli. Oryx 2012, 46, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Oates, J.; Sunderland-Groves, J.; Bergl, R.; Dunn, A.; Nicholas, A.; Takang, E.; Omeni, F.; Imong, I.; Fotso, R.; Nkembi, L.; et al. Regional Action Plan for the Conservation of the Cross River Gorilla (Gorilla gorilla diehli); IUCN/SSC Primate Specialist Group and Wildlife Conservation Society: Arlington, VA, USA, 2007; ISBN 9781934151020. [Google Scholar]
- Plumptre, A.J.; Kirkby, A.; Spira, C.; Kivono, J.; Mitamba, G.; Ngoy, E.; Nishuli, R.; Strindberg, S.; Maisels, F.; Buckland, S.; et al. Changes in Grauer’s Gorilla (Gorilla beringei graueri) and Other Primate Populations in the Kahuzi-Biega National Park and Oku Community Reserve, the Heart of Grauer’s Gorilla Global Range. Am. J. Primatol. 2021, 83, e23288. [Google Scholar] [CrossRef] [PubMed]
- Granjon, A.-C.; Robbins, M.M.; Arinaitwe, J.; Cranfield, M.R.; Eckardt, W.; Mburanumwe, I.; Musana, A.; Robbins, A.M.; Roy, J.; Sollmann, R.; et al. Estimating Abundance and Growth Rates in a Wild Mountain Gorilla Population. Anim. Conserv. 2020, 23, 455–465. [Google Scholar] [CrossRef]
- Hickey, J.R.; Basabose, A.; Gilardi, K.V.; Greer, D.; Nampindo, S.; Robbins, M.M.; Stoinski, T.S. IUCN Gorilla beringei ssp. beringei. IUCN Red List Threat. Species 2018. [Google Scholar] [CrossRef]
- Maisels, F.; Strindberg, S.; Breuer, T.; Greer, D.; Jeffery, K.; Stokes, E. IUCN Gorilla gorilla ssp. gorilla. IUCN Red List Threat. Species 2016. [Google Scholar] [CrossRef]
- Bergl, R.A.; Dunn, A.; Fowler, A.; Imong, I.; Ndeloh, D.; Nicholas, A.; Oates, J.F. IUCN Gorilla gorilla ssp. diehli. IUCN Red List Threat. Species 2016. [Google Scholar]
- Plumptre, A.; Nixon, S.; Caillaud, D.; Hall, J.S.; Hart, J.A.; Nishuli, R.; Williamson, E.A. IUCN Gorilla beringei ssp. graueri. IUCN Red List Threat. Species 2016. [Google Scholar]
- Imong, I.; Robbins, M.M.; Mundry, R.; Bergl, R.; Kühl, H.S. Distinguishing Ecological Constraints from Human Activity in Species Range Fragmentation: The Case of Cross River Gorillas. Anim. Conserv. 2014, 17, 323–331. [Google Scholar] [CrossRef]
- Etiendem, D.N.; Funwi-Gabga, N.; Tagg, N.; Hens, L.; Indah, E.K. The Cross River Gorillas (Gorilla gorilla diehli) at Mawambi Hills, South-West Cameroon: Habitat Suitability and Vulnerability to Anthropogenic Disturbance. Folia Primatol. 2013, 84, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Bergl, R.A.; Bradley, B.J.; Nsubuga, A.; Vigilant, L. Effects of Habitat Fragmentation, Population Size and Demographic History on Genetic Diversity: The Cross River Gorilla in a Comparative Context. Am. J. Primatol. 2008, 70, 848–859. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.L. Ecology of Cross River Gorillas (Gorilla gorilla diehli) on Afi Mountain, Cross River State, Nigeria; City University of New York: Ann Arbor, MI, USA, 2007. [Google Scholar]
- Clifford, S.L.; Anthony, N.M.; Bawe-Johnson, M.; Abernethy, K.A.; Tutin, C.E.G.; White, L.J.T.; Bermejo, M.; Goldsmith, M.L.; McFarland, K.; Jeffery, K.J.; et al. Mitochondrial DNA Phylogeography of Western Lowland Gorillas (Gorilla gorilla gorilla). Mol. Ecol. 2004, 13, 1551–1565, 1567. [Google Scholar] [CrossRef]
- Anthony, N.M.; Johnson-Bawe, M.; Jeffery, K.; Clifford, S.L.; Abernethy, K.A.; Tutin, C.E.; Lahm, S.A.; White, L.J.T.; Utley, J.F.; Wickings, E.J.; et al. The Role of Pleistocene Refugia and Rivers in Shaping Gorilla Genetic Diversity in Central Africa. Proc. Natl. Acad. Sci. USA 2007, 104, 20432–20436. [Google Scholar] [CrossRef] [Green Version]
- Städele, V.; Arandjelovic, M.; Nixon, S.; Bergl, R.A.; Bradley, B.J.; Breuer, T.; Cameron, K.N.; Guschanski, K.; Head, J.; Kyungu, J.C.; et al. The Complex Y-Chromosomal History of Gorillas. Am. J. Primatol. 2022, 84, e23363. [Google Scholar] [CrossRef]
- Bergl, R.A.; Vigilant, L. Genetic Analysis Reveals Population Structure and Recent Migration within the Highly Fragmented Range of the Cross River Gorilla (Gorilla gorilla diehli). Mol. Ecol. 2007, 16, 501–516. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, O.; Fischer, A.; Lankester, F.; Pääbo, S.; Vigilant, L. The Complex Evolutionary History of Gorillas: Insights from Genomic Data. Mol. Biol. Evol. 2007, 24, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Prado-Martinez, J.; Sudmant, P.H.; Kidd, J.M.; Li, H.; Kelley, J.L.; Lorente-Galdos, B.; Veeramah, K.R.; Woerner, A.E.; O’Connor, T.D.; Santpere, G.; et al. Great Ape Genetic Diversity and Population History. Nature 2013, 499, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Thalmann, O.; Wegmann, D.; Spitzner, M.; Arandjelovic, M.; Guschanski, K.; Leuenberger, C.; Bergl, R.A.; Vigilant, L. Historical Sampling Reveals Dramatic Demographic Changes in Western Gorilla Populations. BMC Evol. Biol. 2011, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- McManus, K.F.; Kelley, J.L.; Song, S.; Veeramah, K.R.; Woerner, A.E.; Stevison, L.S.; Ryder, O.A.; Ape Genome Project, G.; Kidd, J.M.; Wall, J.D.; et al. Inference of Gorilla Demographic and Selective History from Whole-Genome Sequence Data. Mol. Biol. Evol. 2015, 32, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Kapp, J.D.; Green, R.E.; Shapiro, B. A Fast and Efficient Single-Stranded Genomic Library Preparation Method Optimized for Ancient DNA. J. Hered. 2021, 112, 241–249. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard Toolkit Picard Toolkit. Broad Inst. 2019.
- Meyer, M.; Arsuaga, J.-L.; de Filippo, C.; Nagel, S.; Aximu-Petri, A.; Nickel, B.; Martínez, I.; Gracia, A.; Bermúdez de Castro, J.M.; Carbonell, E.; et al. Nuclear DNA Sequences from the Middle Pleistocene Sima de Los Huesos Hominins. Nature 2016, 531, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Kuhlwilm, M.; Fontsere, C.; Han, S.; Alvarez-Estape, M.; Marques-Bonet, T. HuConTest: Testing Human Contamination in Great Ape Samples. Genome Biol. Evol. 2021, 13, evab117. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Prado-Martinez, J.; Sudmant, P.H.; Narasimhan, V.; Ayub, Q.; Szpak, M.; Frandsen, P.; Chen, Y.; Yngvadottir, B.; Cooper, D.N.; et al. Mountain Gorilla Genomes Reveal the Impact of Long-Term Population Decline and Inbreeding. Science 2015, 348, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick Coverage Calculation for Genomes and Exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap. 2015. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 15 June 2022).
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, A.A.; Rey-Iglesia, A.; Louis, M.; Skovrind, M.; Westbury, M.V.; Lorenzen, E.D. How Low Can You Go? Introducing SeXY: Sex Identification from Low-Quantity Sequencing Data despite Lacking Assembled Sex Chromosomes. Ecol. Evol. 2022, 12, e9185. [Google Scholar] [CrossRef]
- Hanghøj, K.; Moltke, I.; Andersen, P.A.; Manica, A.; Korneliussen, T.S. Fast and Accurate Relatedness Estimation from High-Throughput Sequencing Data in the Presence of Inbreeding. Gigascience 2019, 8, giz034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisner, J.; Albrechtsen, A. Inferring Population Structure and Admixture Proportions in Low-Depth NGS Data. Genetics 2018, 210, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skotte, L.; Korneliussen, T.S.; Albrechtsen, A. Estimating Individual Admixture Proportions from next Generation Sequencing Data. Genetics 2013, 195, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Erill, G.; Albrechtsen, A. Evaluation of Model Fit of Inferred Admixture Proportions. Mol. Ecol. Resour. 2020, 20, 936–949. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 1 February 2023).
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. BCFtools/RoH: A Hidden Markov Model Approach for Detecting Autozygosity from next-Generation Sequencing Data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.A. Identity by Descent: Variation in Meiosis, across Genomes, and in Populations. Genetics 2013, 194, 301–326. [Google Scholar] [CrossRef] [Green Version]
- Fontsere, C.; Kuhlwilm, M.; Morcillo-Suarez, C.; Alvarez-Estape, M.; Lester, J.D.; Gratton, P.; Schmidt, J.M.; Dieguez, P.; Aebischer, T.; Álvarez-Varona, P.; et al. Population Dynamics and Genetic Connectivity in Recent Chimpanzee History. Cell Genom. 2022, 2, 100133. [Google Scholar] [CrossRef]
- Van der Valk, T.; Díez-Del-Molino, D.; Marques-Bonet, T.; Guschanski, K.; Dalén, L. Historical Genomes Reveal the Genomic Consequences of Recent Population Decline in Eastern Gorillas. Curr. Biol. 2019, 29, 165–170.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Barreiro, F.; Gopalakrishnan, S.; Ramos-Madrigal, J.; Westbury, M.V.; de Manuel, M.; Margaryan, A.; Ciucani, M.M.; Vieira, F.G.; Patramanis, Y.; Kalthoff, D.C.; et al. Historical Population Declines Prompted Significant Genomic Erosion in the Northern and Southern White Rhinoceros (Ceratotherium simum). Mol. Ecol. 2021, 30, 6355–6369. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Inference of Human Population History from Individual Whole-Genome Sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Besenbacher, S.; Hvilsom, C.; Marques-Bonet, T.; Mailund, T.; Schierup, M.H. Direct Estimation of Mutations in Great Apes Reconciles Phylogenetic Dating. Nat. Ecol. Evol. 2019, 3, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Santiago, E.; Novo, I.; Pardiñas, A.F.; Saura, M.; Wang, J.; Caballero, A. Recent Demographic History Inferred by High-Resolution Analysis of Linkage Disequilibrium. Mol. Biol. Evol. 2020, 37, 3642–3653. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Chang, C. PLINK 1.9. Available online: www.cog-genomics.org/plink/1.9/ (accessed on 26 October 2022).
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, s13742-015-0047–0048. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Stevison, L.S.; Woerner, A.E.; Kidd, J.M.; Kelley, J.L.; Veeramah, K.R.; McManus, K.F.; Great Ape Genome Project; Bustamante, C.D.; Hammer, M.F.; Wall, J.D. The Time Scale of Recombination Rate Evolution in Great Apes. Mol. Biol. Evol. 2016, 33, 928–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinsky, M.; Matschiner, M.; Svardal, H. Dsuite—Fast D-Statistics and Related Admixture Evidence from VCF Files. Mol. Ecol. Resour. 2021, 21, 584–595. [Google Scholar] [CrossRef]
- Soraggi, S.; Wiuf, C.; Albrechtsen, A. Powerful Inference with the D-Statistic on Low-Coverage Whole-Genome Data. G3 2018, 8, 551–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, R.; Flegontov, P.; Flegontova, O.; Changmai, P.; Reich, D. On the Limits of Fitting Complex Models of Population History to Genetic Data. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gerloff, U.; Hartung, B.; Fruth, B.; Hohmann, G.; Tautz, D. Intracommunity Relationships, Dispersal Pattern and Paternity Success in a Wild Living Community of Bonobos (Pan Paniscus) Determined from DNA Analysis of Faecal Samples. Proc. Biol. Sci. 1999, 266, 1189–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nater, A.; Arora, N.; Greminger, M.P.; van Schaik, C.P.; Singleton, I.; Wich, S.A.; Fredriksson, G.; Perwitasari-Farajallah, D.; Pamungkas, J.; Krützen, M. Marked Population Structure and Recent Migration in the Critically Endangered Sumatran Orangutan (Pongo abelii). J. Hered. 2013, 104, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fünfstück, T.; Vigilant, L. The Geographic Distribution of Genetic Diversity within Gorillas. Am. J. Primatol. 2015, 77, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lester, J.D.; Vigilant, L.; Gratton, P.; McCarthy, M.S.; Barratt, C.D.; Dieguez, P.; Agbor, A.; Álvarez-Varona, P.; Angedakin, S.; Ayimisin, E.A.; et al. Recent Genetic Connectivity and Clinal Variation in Chimpanzees. Commun. Biol. 2021, 4, 283. [Google Scholar] [CrossRef]
- Khan, A.; Patel, K.; Bhattacharjee, S.; Sharma, S.; Chugani, A.N.; Sivaraman, K.; Hosawad, V.; Sahu, Y.K.; Reddy, G.V.; Ramakrishnan, U. Are Shed Hair Genomes the Most Effective Noninvasive Resource for Estimating Relationships in the Wild? Ecol. Evol. 2020, 10, 4583–4594. [Google Scholar] [CrossRef]
- Baas, P.; van der Valk, T.; Vigilant, L.; Ngobobo, U.; Binyinyi, E.; Nishuli, R.; Caillaud, D.; Guschanski, K. Population-Level Assessment of Genetic Diversity and Habitat Fragmentation in Critically Endangered Grauer’s Gorillas. Am. J. Phys. Anthropol. 2018, 165, 565–575. [Google Scholar] [CrossRef]
- Kardos, M.; Armstrong, E.E.; Fitzpatrick, S.W.; Hauser, S.; Hedrick, P.W.; Miller, J.M.; Tallmon, D.A.; Funk, W.C. The Crucial Role of Genome-Wide Genetic Variation in Conservation. Proc. Natl. Acad. Sci. USA 2021, 118, e2104642118. [Google Scholar] [CrossRef]
- Bertorelle, G.; Raffini, F.; Bosse, M.; Bortoluzzi, C.; Iannucci, A.; Trucchi, E.; Morales, H.E.; van Oosterhout, C. Genetic Load: Genomic Estimates and Applications in Non-Model Animals. Nat. Rev. Genet. 2022, 23, 492–503. [Google Scholar] [CrossRef]
- Robinson, J.; Kyriazis, C.C.; Yuan, S.C.; Lohmueller, K.E. Deleterious Variation in Natural Populations and Implications for Conservation Genetics. Annu. Rev. Anim. Biosci. 2022, 11, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, R.R.; Bishop, J.M. Morphological and Molecular Evidence Reveals Recent Hybridization between Gorilla Taxa. Evolution 2010, 64, 271–290. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez-Estape, M.; Pawar, H.; Fontsere, C.; Trujillo, A.E.; Gunson, J.L.; Bergl, R.A.; Bermejo, M.; Linder, J.M.; McFarland, K.; Oates, J.F.; et al. Past Connectivity but Recent Inbreeding in Cross River Gorillas Determined Using Whole Genomes from Single Hairs. Genes 2023, 14, 743. https://doi.org/10.3390/genes14030743
Alvarez-Estape M, Pawar H, Fontsere C, Trujillo AE, Gunson JL, Bergl RA, Bermejo M, Linder JM, McFarland K, Oates JF, et al. Past Connectivity but Recent Inbreeding in Cross River Gorillas Determined Using Whole Genomes from Single Hairs. Genes. 2023; 14(3):743. https://doi.org/10.3390/genes14030743
Chicago/Turabian StyleAlvarez-Estape, Marina, Harvinder Pawar, Claudia Fontsere, Amber E. Trujillo, Jessica L. Gunson, Richard A. Bergl, Magdalena Bermejo, Joshua M. Linder, Kelley McFarland, John F. Oates, and et al. 2023. "Past Connectivity but Recent Inbreeding in Cross River Gorillas Determined Using Whole Genomes from Single Hairs" Genes 14, no. 3: 743. https://doi.org/10.3390/genes14030743