

A Missense Mutation c.1132G > A in Fumarate Hydratase (FH) Leads to Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC) Syndrome and Insights into Clinical Management in Uterine Leiomyomata
 ,
,
Abstract
:1. Introduction
2. Case Presentation
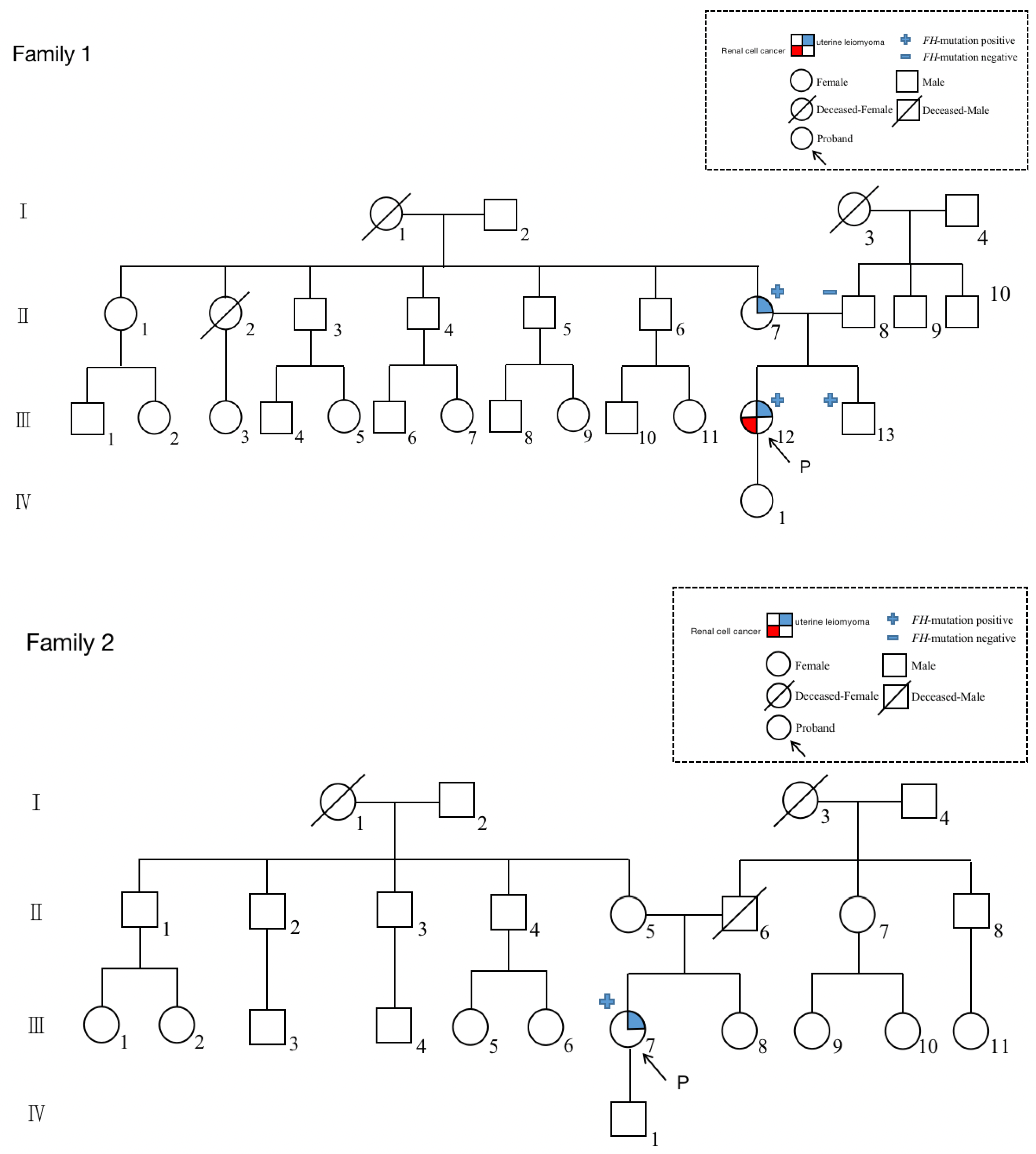
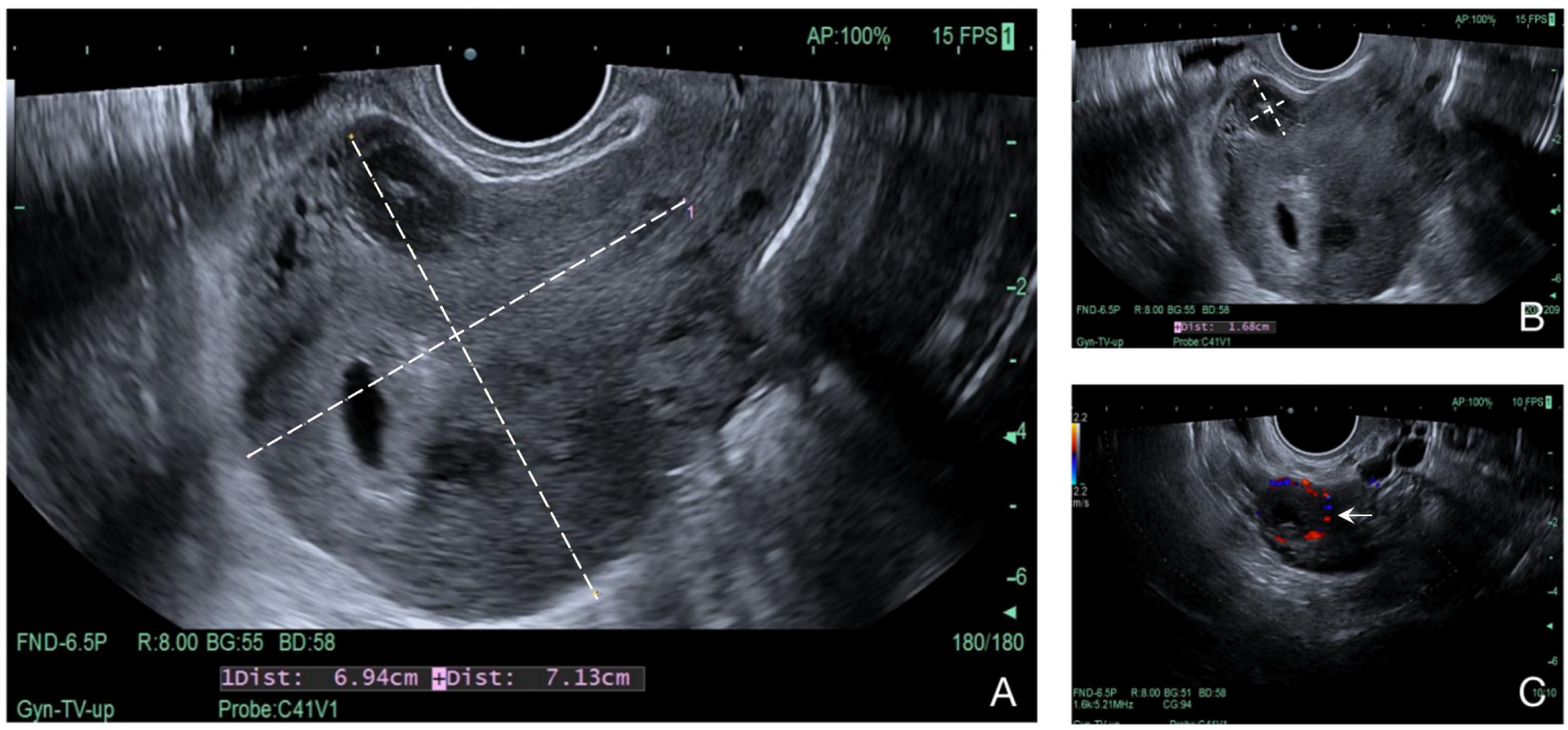
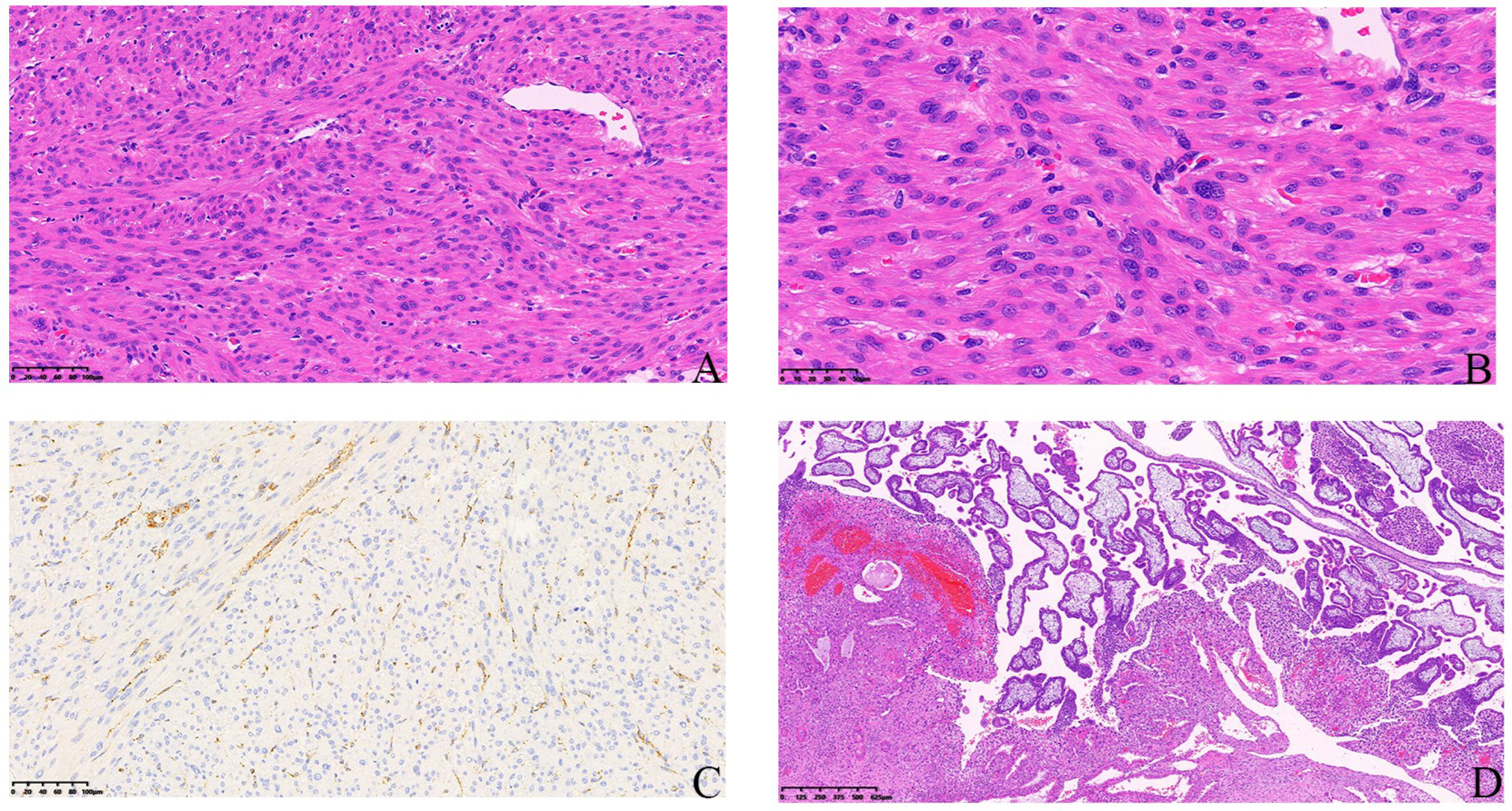
2.1. Case 1
2.2. Case 2
3. Materials and Methods
3.1. Case Selection
- (1)
- Severely symptomatic uterine leiomyomas treated with surgery before the age of 40, especially those confirmed to be FH-absent.
- (2)
- Type 2 papillary or collecting duct renal cell carcinoma before the age of 40.
- (3)
- Positive family history: A first-degree family member who meets one of the criteria. (Occurrence of severely symptomatic uterine leiomyomas < 40 years old in second-degree paternal family members may also be relevant.)
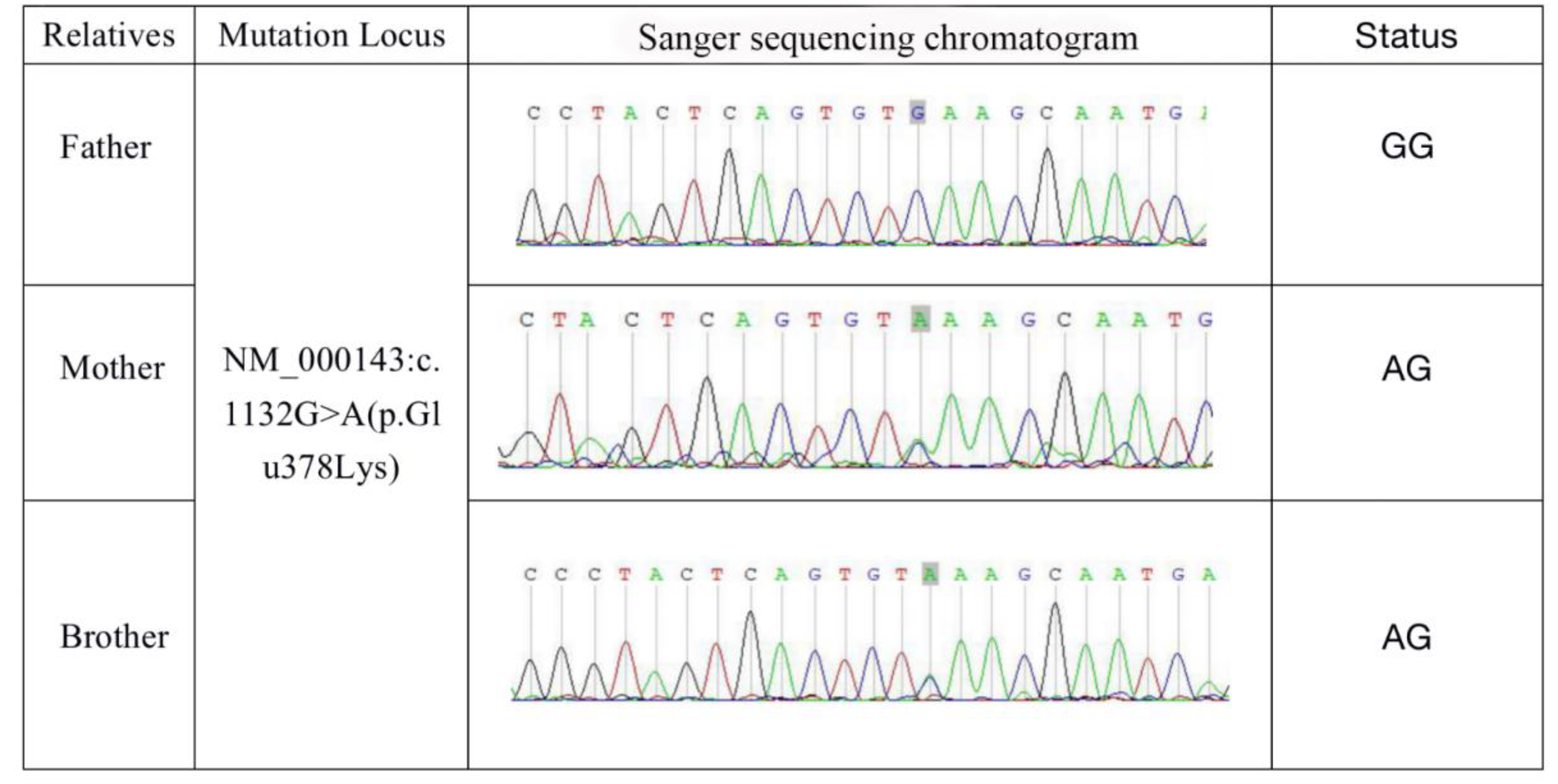
3.2. Detection of FH Mutations
3.3. In Silico Characterization of the Variant
3.4. In Vitro Functional Studies
3.4.1. Construction of FH-Wild Type (FH-WT) and FH-Mutant (FH-MUT) (c.1132G > A) Plasmid
3.4.2. Cell Cultures and Transfection
3.4.3. Proliferation Assessment
3.4.4. RNA Extraction, cDNA Synthesis, and Real-Time Quantitative Polymerase Chain Reaction (qPCR) Analysis
3.4.5. Western Blot Analysis
3.4.6. Measurement of FH Enzyme Activity
3.4.7. Statistical Analysis
4. Results
4.1. Identification of FH Mutation in Patients
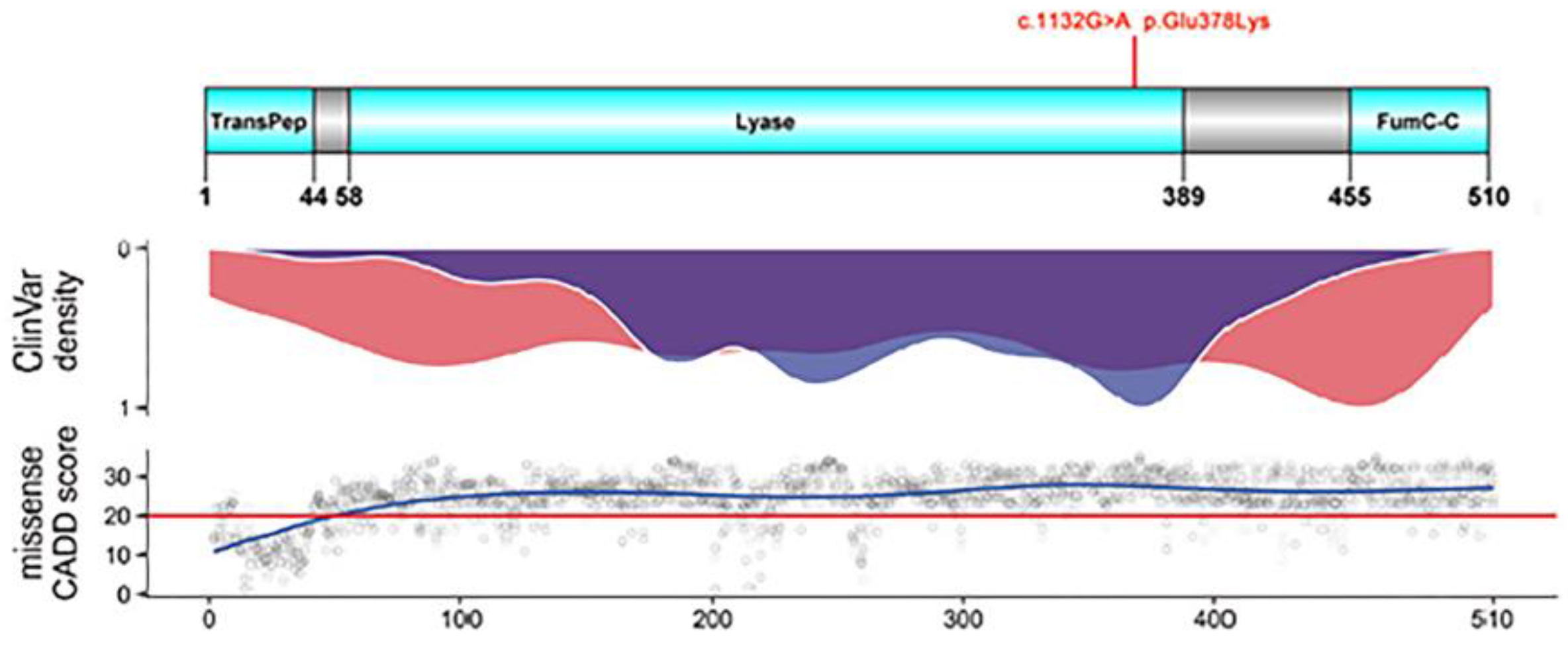
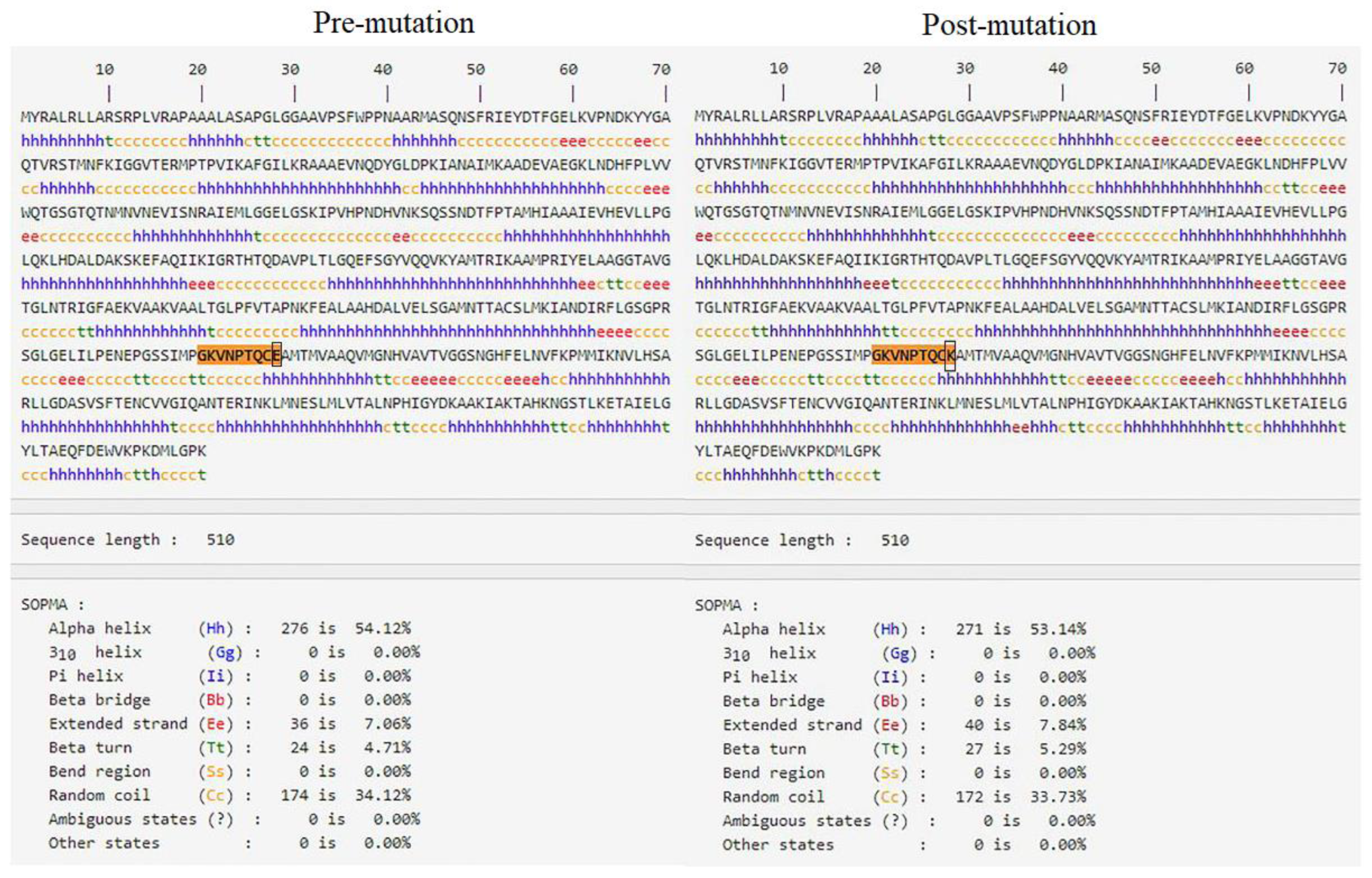
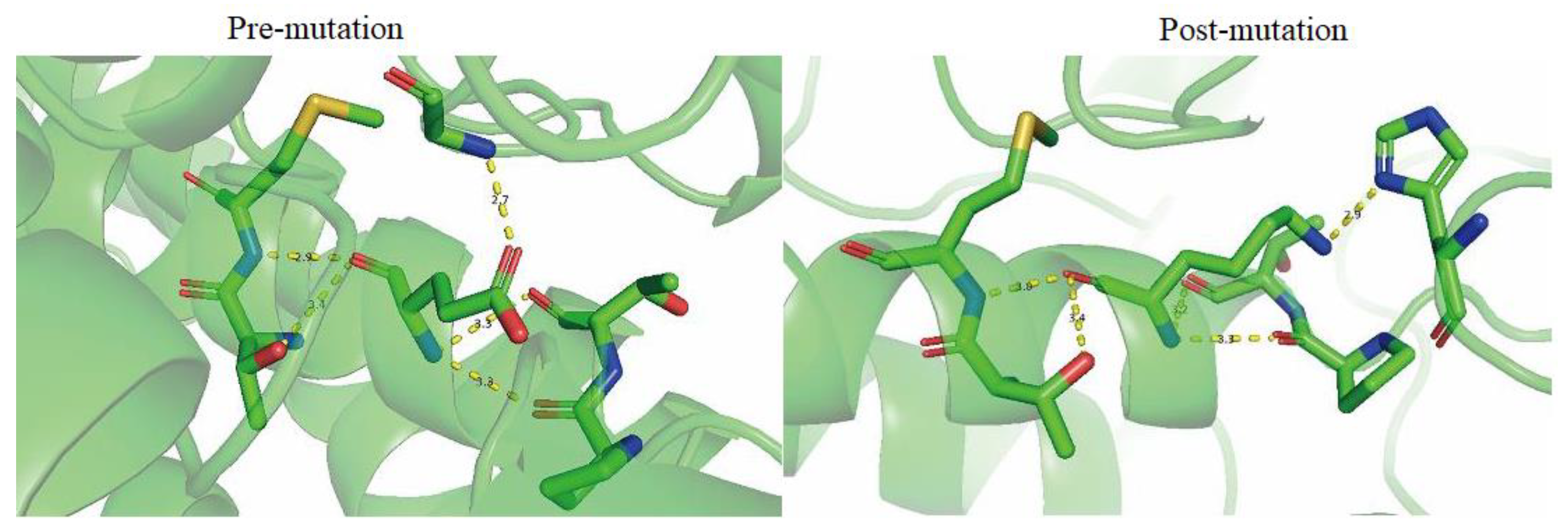
4.2. The Missense Mutation c.1132G > A of FH Is Predicted to Be Highly Damaging
4.3. In Vitro Functional Studies
4.3.1. The c.1132G > A Mutation Increases the Cell Proliferation in Uterine Leiomyoma Cell
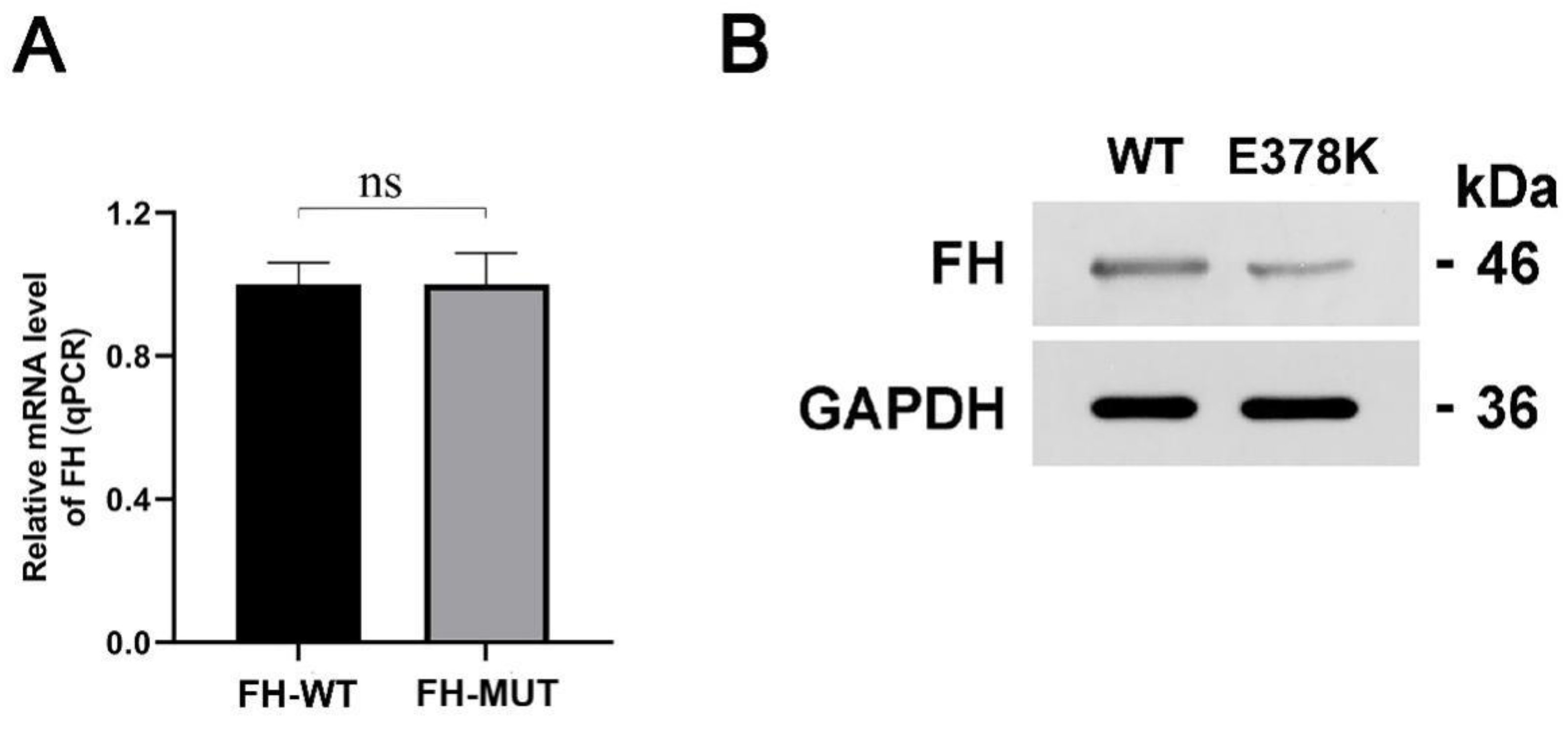
4.3.2. The E378K Mutation Does Not Influence the mRNA Transcription of FH
4.3.3. The Mutation Decreases the FH Enzyme Expression
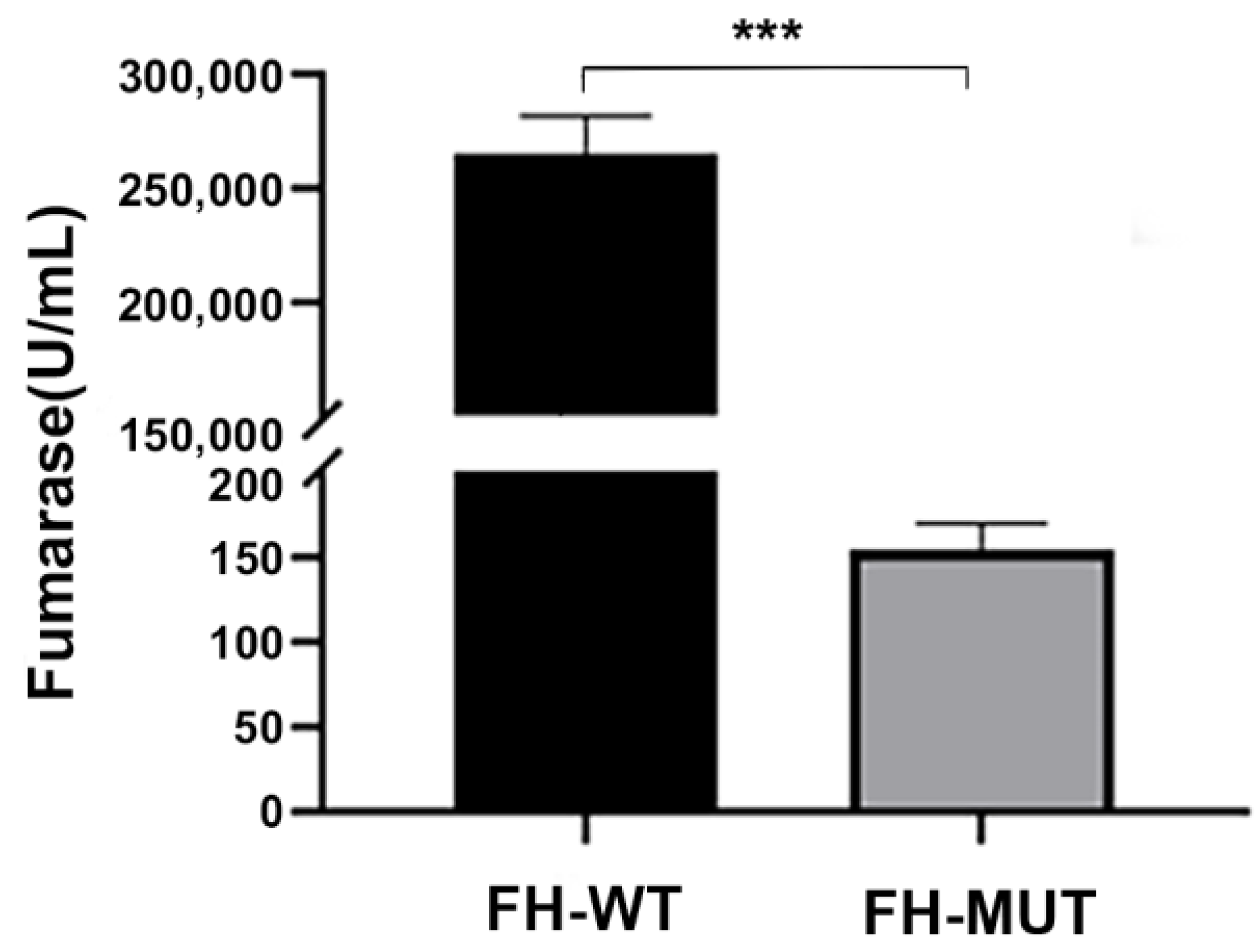
4.3.4. The E378K Mutation Is Associated with Altered FH Enzyme Activity
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Launonen, V.; Vierimaa, O.; Kiuru, M.; Isola, J.; Roth, S.; Pukkala, E.; Sistonen, P.; Herva, R.; Aaltonen, L.A. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 3387–3392. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.M.; Handler, M.Z.; Schwartz, R.A.; Lambert, W.C. Hereditary leiomyomatosis and renal cell cancer syndrome: An update and review. J. Am. Acad. Dermatol. 2017, 77, 149–158. [Google Scholar] [CrossRef]
- Muller, M.; Ferlicot, S.; Guillaud-Bataille, M.; Le Teuff, G.; Genestie, C.; Deveaux, S.; Slama, A.; Poulalhon, N.; Escudier, B.; Albiges, L.; et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin. Genet. 2017, 92, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Ricketts, C.J.; Vocke, C.D.; Valera, V.A.; Chen, C.C.; Gautam, R.; Gupta, G.N.; Macias, G.S.G.; Merino, M.J.; Bratslavsky, G.; et al. Adrenal Nodular Hyperplasia in Hereditary Leiomyomatosis and Renal Cell Cancer. J. Urol. 2013, 189, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Clark, G.R.; Sciacovelli, M.; Gaude, E.; Walsh, D.M.; Kirby, G.; Simpson, M.; Trembath, R.; Berg, J.N.; Woodward, E.R.; Kinning, E.; et al. Germline FH Mutations Presenting With Pheochromocytoma. J. Clin. Endocrinol. Metab. 2014, 99, E2046–E2050. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, W.; You, Y.; Zhu, L.; Feng, F. Novel FH mutation associated with multiple uterine leiomyomas in Chinese siblings. Mol. Genet. Genom. Med. 2019, 8, e1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.Y.; Ahn, J.-Y.; Keam, B.; Kim, M.; Yoon, S.; Lee, J.L.; Park, K.; Park, I. Genotypic and Phenotypic Characteristics of Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome in Korean Patients. Ann. Lab. Med. 2021, 41, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.; Glenn, G.M.; Stratton, P.; Goldstein, A.M.; Merino, M.J.; Tucker, M.A.; Linehan, W.M.; Toro, J.R. Association of Germline Mutations in the Fumarate Hydratase Gene and Uterine Fibroids in Women With Hereditary Leiomyomatosis and Renal Cell Cancer. Arch. Dermatol. 2008, 144, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Ortega, J.; Vocke, C.; Stratton, P.; Linehan, W.M.; Merino, M.J. Morphologic and Molecular Characteristics of Uterine Leiomyomas in Hereditary Leiomyomatosis and Renal Cancer (HLRCC) Syndrome. Am. J. Surg. Pathol. 2013, 37, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.A.; Weigelt, B.; Chiang, S.; Selenica, P.; Chen, Y.-B.; Bialik, A.; Bi, R.; Schultheis, A.M.; Lim, R.S.; Ng, C.K.Y.; et al. Leiomyoma with bizarre nuclei: A morphological, immunohistochemical and molecular analysis of 31 cases. Mod. Pathol. 2017, 30, 1476–1488. [Google Scholar] [CrossRef] [Green Version]
- Joseph, N.M.; Solomon, D.A.; Frizzell, N.; Rabban, J.T.; Zaloudek, C.; Garg, K. Morphology and Immunohistochemistry for 2SC and FH Aid in Detection of Fumarate Hydratase Gene Aberrations in Uterine Leiomyomas From Young Patients. Am. J. Surg. Pathol. 2015, 39, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Rabban, J.T.; Mak, J.; Zaloudek, C.; Garg, K. Detailed Morphologic and Immunohistochemical Characterization of Myomectomy and Hysterectomy Specimens From Women With Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome (HLRCC). Am. J. Surg. Pathol. 2019, 43, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.; Karamurzin, Y.; Frizzell, N.; Garg, K.; Nonaka, D.; Chen, Y.-B.; Soslow, R.A. Uterine smooth muscle tumors with features suggesting fumarate hydratase aberration: Detailed morphologic analysis and correlation with S-(2-succino)-cysteine immunohistochemistry. Mod. Pathol. 2013, 27, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Garg, K.; Tickoo, S.K.; Soslow, R.; Reuter, V.E. Morphologic Features of Uterine Leiomyomas Associated With Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome: A case report. Am. J. Surg. Pathol. 2011, 35, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Siegler, L.; Erber, R.; Burghaus, S.; Brodkorb, T.; Wachter, D.; Wilkinson, N.; Bolton, J.; Stringfellow, H.; Haller, F.; Beckmann, M.W.; et al. Fumarate hydratase (FH) deficiency in uterine leiomyomas: Recognition by histological features versus blind immunoscreening. Virchows Arch. 2018, 472, 789–796. [Google Scholar] [CrossRef]
- Miettinen, M.; Felisiak-Golabek, A.; Wasag, B.; Chmara, M.; Wang, Z.; Butzow, R.; Lasota, J. Fumarase-deficient Uterine Leiomyomas: An immunohistochemical, molecular genetics, and clinicopathologic study of 86 cases. Am. J. Surg. Pathol. 2016, 40, 1661–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabban, J.T.; Chan, E.; Mak, J.; Zaloudek, C.; Garg, K. Prospective Detection of Germline Mutation of Fumarate Hydratase in Women With Uterine Smooth Muscle Tumors Using Pathology-based Screening to Trigger Genetic Counseling for Hereditary Leiomyomatosis Renal Cell Carcinoma Syndrome: A 5-year single institutional experience. Am. J. Surg. Pathol. 2019, 43, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef]
- Ooi, A. Advances in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) research. Semin. Cancer Biol. 2020, 61, 158–166. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, D.; Mensenkamp, A.; Badeloe, S.; Breuning, M.; Simon, M.; Van Spaendonck, K.; Aalfs, C.; Post, J.; Shanley, S.; Krapels, I.; et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin. Genet. 2010, 79, 49–59. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayley, J.-P.; Launonen, V.; Tomlinson, I.P.M. The FH mutation database: An online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med. Genet. 2008, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Coman, D.; Kranc, K.R.; Christodoulou, J. Fumarate Hydratase Deficiency; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2006. [Google Scholar]
- Remes, A.M.; Filppula, S.A.; Rantala, H.; Leisti, J.; Ruokonen, A.; Sharma, S.; Hiltunen, J.K. A novel mutation of the fumarase gene in a family with autosomal recessive fumarase deficiency. J. Mol. Med. 2004, 82, 550–554. [Google Scholar] [CrossRef]
- Ge, X.; Li, M.; Yin, J.; Shi, Z.; Fu, Y.; Zhao, N.; Chen, H.; Meng, L.; Li, X.; Hu, Z.; et al. Fumarate inhibits PTEN to promote tumorigenesis and therapeutic resistance of type2 papillary renal cell carcinoma. Mol. Cell 2022, 82, 1249–1260.e7. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, Y.; He, H.; Shen, J. Clinical Interpretation of Sequence Variants. Curr. Protoc. Hum. Genet. 2020, 106, e98. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregová, M.; Hojný, J.; Němejcová, K.; Bártů, M.; Mára, M.; Boudová, B.; Laco, J.; Krbal, L.; Tichá, I.; Dundr, P. Leiomyoma with Bizarre Nuclei: A Study of 108 Cases Focusing on Clinicopathological Features, Morphology, and Fumarate Hydratase Alterations. Pathol. Oncol. Res. 2019, 26, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Menko, F.H.; Maher, E.R.; Schmidt, L.S.; Middelton, L.A.; Aittomäki, K.; Tomlinson, I.; Richard, S.; Linehan, W.M. Hereditary leiomyomatosis and renal cell cancer (HLRCC): Renal cancer risk, surveillance and treatment. Fam. Cancer 2014, 13, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, H.J.; Kiuru, M.; Ylisaukko-Oja, S.K.; Salovaara, R.; Herva, R.; Koivisto, P.A.; Vierimaa, O.; Aittomäki, K.; Pukkala, E.; Launonen, V.; et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J. Med. Genet. 2006, 43, 523–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, A.K.; Scott, R.J. Genetic and Environmental Modifiers of Cancer Risk in Lynch Syndrome; Springer: Cham, Switzerland, 2018; pp. 67–89. ISBN 978-3-319-74258-8. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PolyPhen2 | Provean | ||||
|---|---|---|---|---|---|
| Mutation gene | Variant | AA change | Score a | Score b | |
| Pre-Mutation | Post-Mutation | |||
|---|---|---|---|---|
| Hydrogen Bond | Distance | Hydrogen Bond | Distance | |
| Main Chain | T381 and E378 | 3.4 | T381 and K378 | 3.4 |
| M382 and E378 | 2.9 | M382 and K378 | 3.0 | |
| P374 and E378 | 3.3 | P374 and K378 | 3.3 | |
| T375 and E378 | 3.3 | T375 and K378 | 3.2 | |
| Side Chain | G144 and E378 | 2.7 | H235 and K378 | 2.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Xu, Y.; Wang, C.; Chen, Y.; Ren, X.; Kang, Y.; Wang, C. A Missense Mutation c.1132G > A in Fumarate Hydratase (FH) Leads to Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC) Syndrome and Insights into Clinical Management in Uterine Leiomyomata. Genes 2023, 14, 744. https://doi.org/10.3390/genes14030744
Shi Y, Xu Y, Wang C, Chen Y, Ren X, Kang Y, Wang C. A Missense Mutation c.1132G > A in Fumarate Hydratase (FH) Leads to Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC) Syndrome and Insights into Clinical Management in Uterine Leiomyomata. Genes. 2023; 14(3):744. https://doi.org/10.3390/genes14030744
Chicago/Turabian StyleShi, Yue, Yan Xu, Chao Wang, Yiqing Chen, Xiaojun Ren, Yu Kang, and Chao Wang. 2023. "A Missense Mutation c.1132G > A in Fumarate Hydratase (FH) Leads to Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC) Syndrome and Insights into Clinical Management in Uterine Leiomyomata" Genes 14, no. 3: 744. https://doi.org/10.3390/genes14030744