
Design and Outcomes of a Novel Multidisciplinary Ophthalmic Genetics Clinic
 , , , and
, , , and
Abstract
:1. Introduction
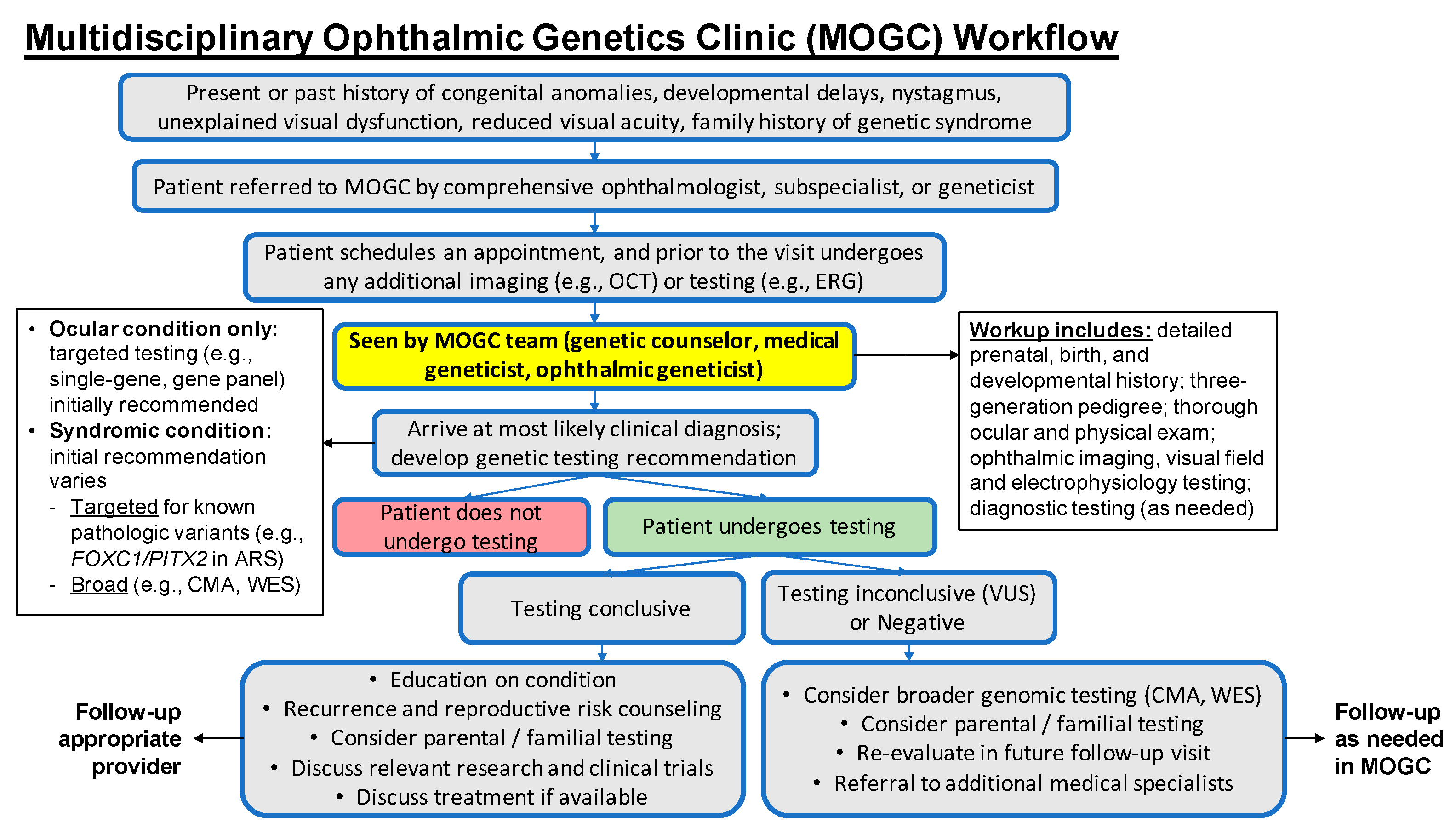
2. Materials and Methods
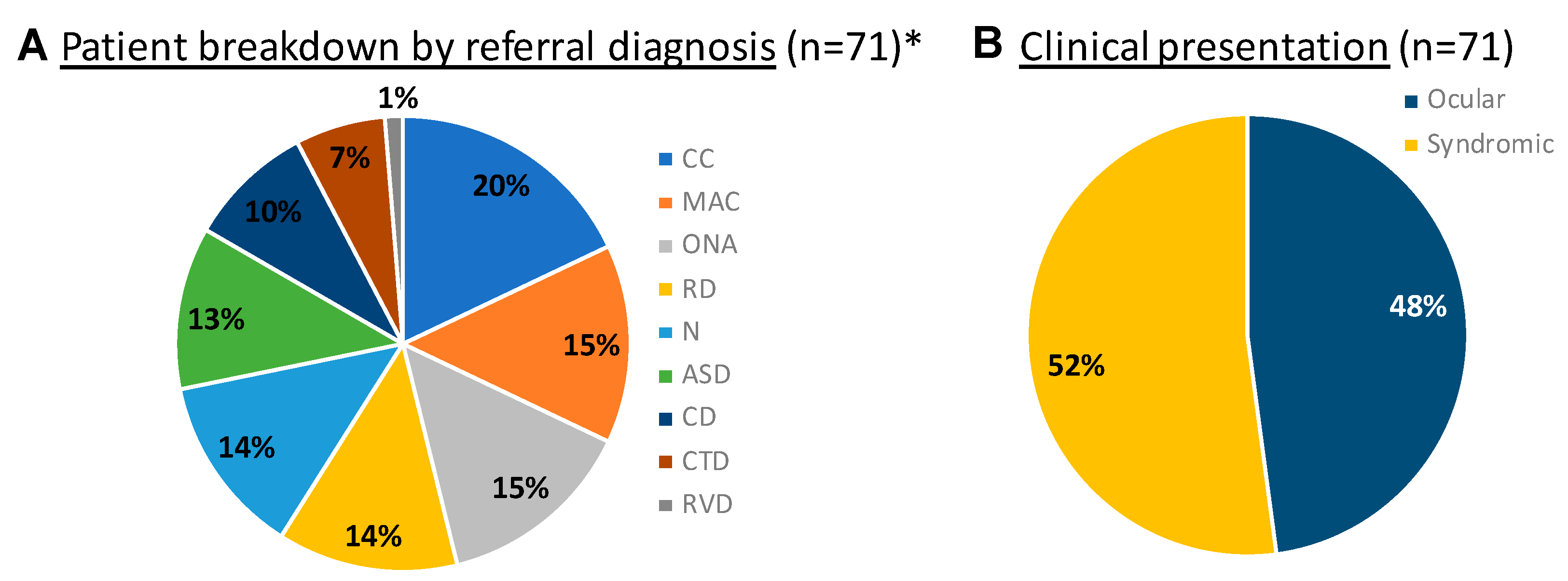
3. Results
3.1. The Multidisciplinary Ophthalmic Genetics Clinic
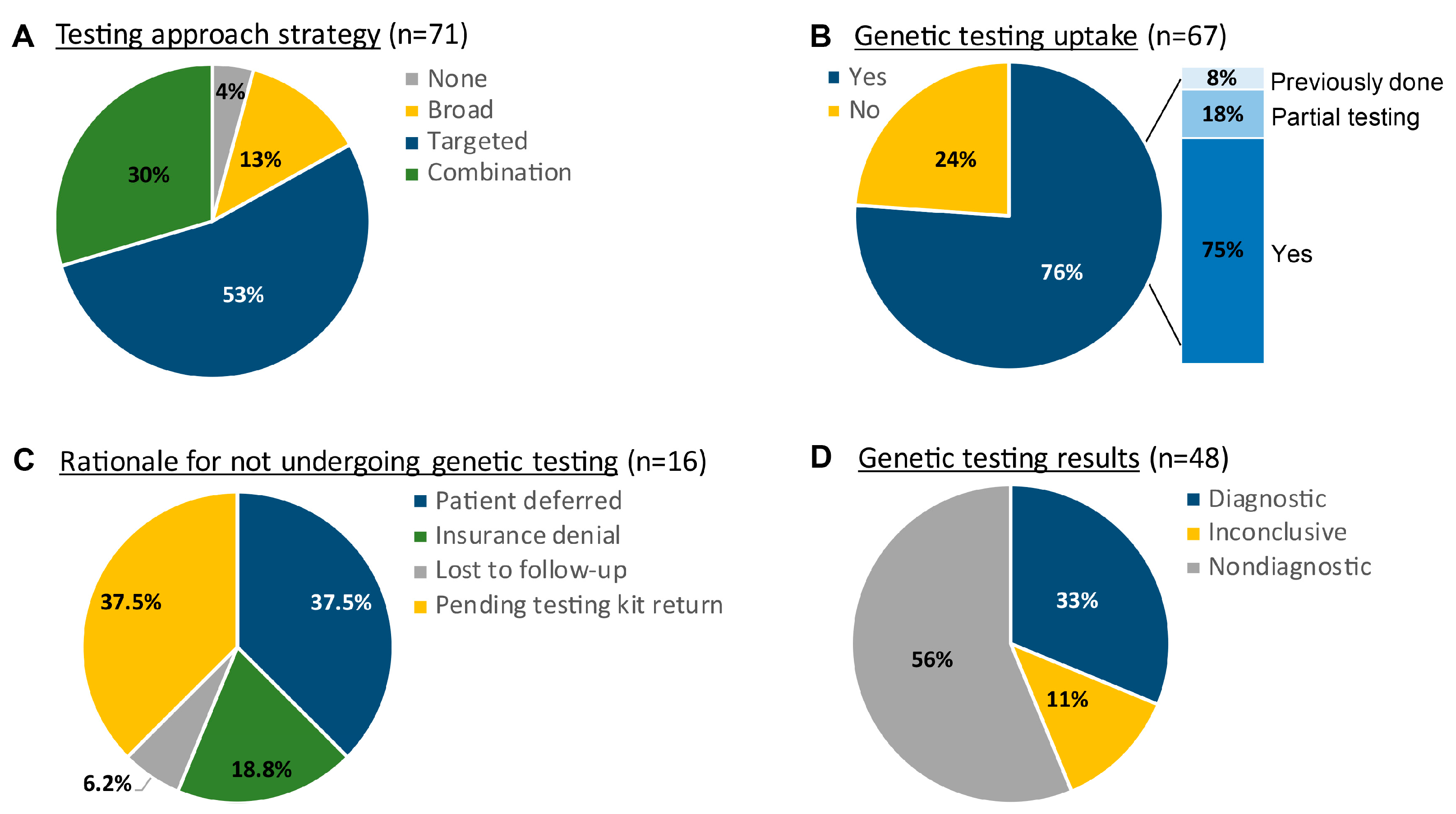
3.2. Approaches to Genetic Testing and Diagnostic Yield
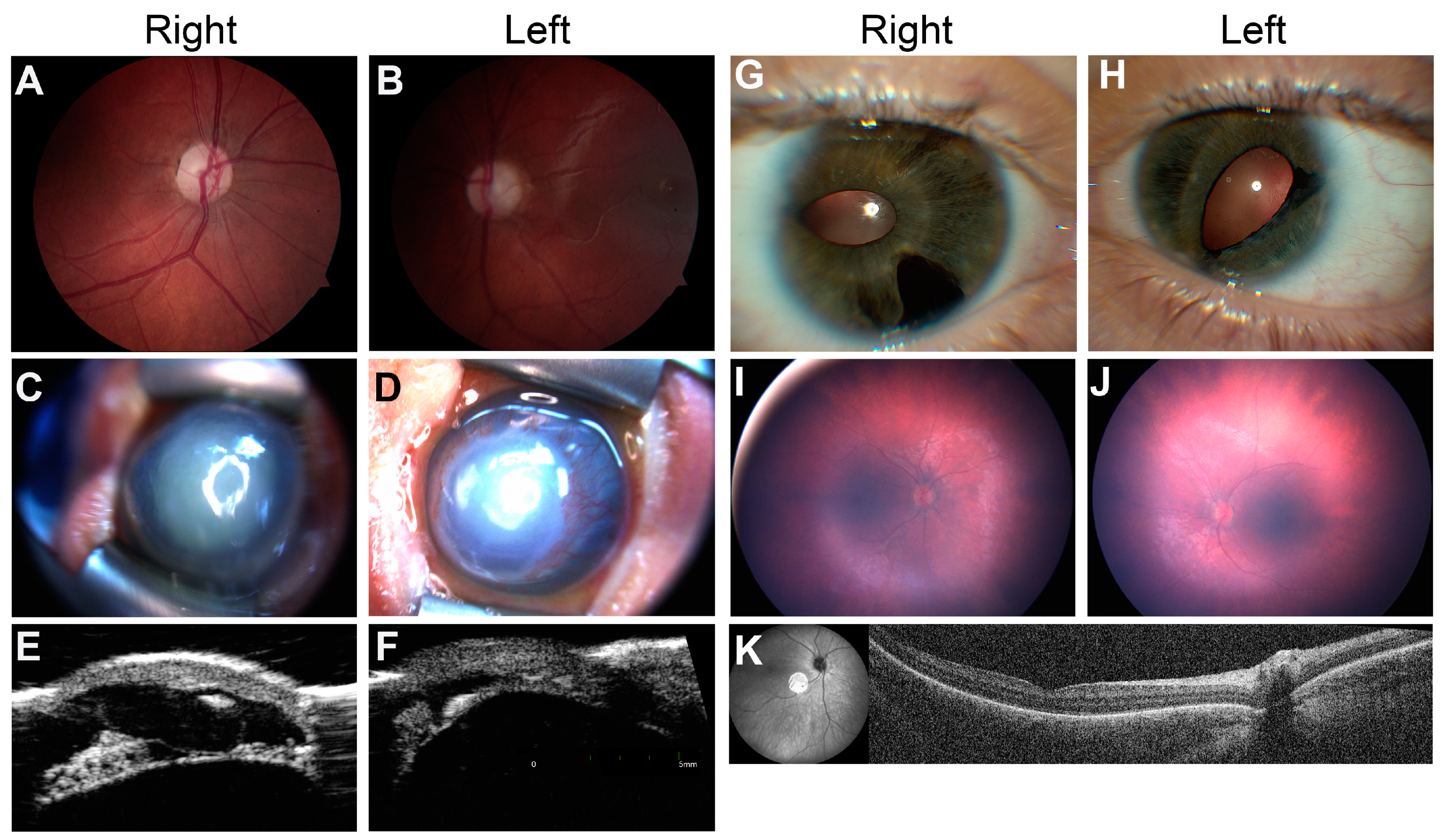
3.3. Case Reports
3.3.1. Syndromic Optic Neuropathy
3.3.2. Familial Anterior Segment Dysgenesis
3.3.3. Infantile Nystagmus Secondary to Retinal Disorder
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morad, Y.; Sutherland, J.; DaSilva, L.; Ulster, A.; Shik, J.; Gallie, B.; Héon, E.; Levin, A.V. Ocular Genetics Program: Multidisciplinary care of patients with ocular genetic eye disease. Can. J. Ophthalmol. 2007, 42, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Couser, N.L.; Brooks, B.P.; Drack, A.V.; Shankar, S.P. The evolving role of genetics in ophthalmology. Ophthalmic Genet. 2021, 42, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Stiff, H.A.; Sloan-Heggen, C.M.; Ko, A.; Pfeifer, W.L.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Booth, K.T.; Wang, D.; Weaver, A.E.; et al. Is it Usher syndrome? Collaborative diagnosis and molecular genetics of patients with visual impairment and hearing loss. Ophthalmic Genet. 2020, 41, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, L.Z.; Glinton, K.; Yuan, B.; Liu, P.; Pillai, N.; Mizerik, E.; Magoulas, P.; Rosenfeld, J.A.; Karaviti, L.; Sutton, V.R.; et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am. J. Med. Genet. Part A 2019, 179, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Uster, T.; Nezarati, M.; Schnur, R.; Bhoj, E. An Additional Individual with a De Novo Variant in Myelin Regulatory Factor (MYRF) with Cardiac and Urogenital Anomalies: Further Proof of Causality: Comments on the article by Pinz et al. ( ). Am. J. Med. Genet. Part A 2018, 176, 2041–2043. [Google Scholar] [CrossRef]
- Pinz, H.; Pyle, L.C.; Li, D.; Izumi, K.; Skraban, C.; Tarpinian, J.; Braddock, S.R.; Telegrafi, A.; Monaghan, K.G.; Zackai, E.; et al. De novo variants in Myelin regulatory factor (MYRF) as candidates of a new syndrome of cardiac and urogenital anomalies. Am. J. Med. Genet. Part A 2018, 176, 969–972. [Google Scholar] [CrossRef]
- Qi, H.; Yu, L.; Zhou, X.; Wynn, J.; Zhao, H.; Guo, Y.; Zhu, N.; Kitaygorodsky, A.; Hernan, R.; Aspelund, G.; et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018, 14, e1007822. [Google Scholar] [CrossRef] [Green Version]
- Garnai, S.J.; Brinkmeier, M.L.; Emery, B.; Aleman, T.S.; Pyle, L.C.; Veleva-Rotse, B.; Sisk, R.A.; Rozsa, F.W.; Ozel, A.B.; Li, J.Z.; et al. Variants in myelin regulatory factor (MYRF) cause autosomal dominant and syndromic nanophthalmos in humans and retinal degeneration in mice. PLoS Genet. 2019, 15, e1008130. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.D.; Stewart, B.; Prasov, L.; Pyle, L.C. MYRF-Related Cardiac Urogenital Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK586169/ (accessed on 6 February 2023).
- Guo, C.; Zhao, Z.; Chen, D.; He, S.; Sun, N.; Li, Z.; Liu, J.; Zhang, D.; Zhang, J.; Li, J.; et al. Detection of Clinically Relevant Genetic Variants in Chinese Patients With Nanophthalmos by Trio-Based Whole-Genome Sequencing Study. Invest Ophthalmol Vis. Sci. 2019, 60, 2904–2913. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Sun, W.; Ouyang, J.; Li, S.; Jia, X.; Tan, Z.; Hejtmancik, J.F.; Zhang, Q. Novel truncation mutations in MYRF cause autosomal dominant high hyperopia mapped to 11p12-q13.3. Hum Genet. 2019, 138, 1077–1090. [Google Scholar] [CrossRef] [Green Version]
- Prasov, L.; Bohnsack, B.L.; El Husny, A.S.; Tsoi, L.C.; Guan, B.; Kahlenberg, J.M.; Almeida, E.; Wang, H.; Cowen, E.W.; A De Jesus, A.; et al. DDX58(RIG-I)-related disease is associated with tissue-specific interferon pathway activation. J. Med. Genet. 2022, 59, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Leroy, B.P.; Black, G.; Ong, T.; Yoon, D.; Trzupek, K. Genetic testing and diagnosis of inherited retinal diseases. Orphanet J. Rare Dis. 2021, 16, 514. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, L.M.; Maheshwari, M.; Capasso, J.; Atilla, H.; Dudakova, L.; Thompson, S.; Zitano, L.; Lay-Son, G.; Lowry, R.B.; Black, J.; et al. Axenfeld-Rieger syndrome: More than meets the eye. J. Med. Genet. 2022, epub ahead of print. [CrossRef] [PubMed]
- Shiels, A.; Bennett, T.M.; Hejtmancik, J.F. Cat-Map: Putting cataract on the map. Mol. Vis. 2010, 16, 2007–2015. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Bosch, D.G.M.; Boonstra, F.N.; Gonzaga-Jauregui, C.; Xu, M.; De Ligt, J.; Jhangiani, S.; Wiszniewski, W.; Muzny, D.M.; Yntema, H.G.; Pfundt, R.; et al. NR2F1 mutations cause optic atrophy with intellectual disability. Am. J. Hum. Genet. 2014, 94, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Bertacchi, M.; Tocco, C.; Schaaf, C.P.; Studer, M. Pathophysiological Heterogeneity of the BBSOA Neurodevelopmental Syndrome. Cells 2022, 11, 1260. [Google Scholar] [CrossRef]
- McAnany, J.J.; Genead, M.A.; Walia, S.; Drack, A.; Stone, E.M.; Koenekoop, R.K.; Traboulsi, E.I.; Smith, A.; Weleber, R.G.; Jacobson, S.; et al. Visual Acuity Changes in Patients With Leber Congenital Amaurosis and Mutations in CEP290. JAMA Ophthalmol. 2013, 131, 178. [Google Scholar] [CrossRef]
- Li, T. Leber Congenital Amaurosis Caused by Mutations in RPGRIP1. Cold Spring Harb. Perspect. Med. 2015, 5, a017384. [Google Scholar] [CrossRef] [Green Version]
- Reuter, C.M.; Kohler, J.N.; Bonner, D.; Zastrow, D.; Fernandez, L.; Dries, A.; Marwaha, S.; Davidson, J.; Brokamp, E.; Herzog, M.; et al. Yield of whole exome sequencing in undiagnosed patients facing insurance coverage barriers to genetic testing. J. Genet. Couns. 2019, 28, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Atwal, P.S.; Brennan, M.-L.; Cox, R.; Niaki, M.; Platt, J.; Homeyer, M.; Kwan, A.; Parkin, S.; Schelley, S.; Slattery, L.; et al. Clinical whole-exome sequencing: Are we there yet? Genet Med. 2014, 16, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ankala, A.; Wilcox, W.R.; Hegde, M.R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet Med. 2015, 17, 444–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Referral Condition | Diagnostic Yield, % (n) |
|---|---|
| Anterior-Segment Dysgenesis (ASD) | 57% (n = 4/7) |
| Nystagmus (N) | 33% (n = 2/6) |
| Optic Nerve Anomaly/Atrophy (ONA) | 30% (n = 3/10) |
| Congenital or Juvenile Cataract (CC) | 36% (n = 4/11) |
| Retinal Dystrophy (RD) | 20% (n = 1/5) |
| Microphthalmia/Anophthalmia/Coloboma (MAC) | 13% (n = 1/8) |
| Corneal Dystrophy (CD) | 0% (n = 0/2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parekh, B.; Beil, A.; Blevins, B.; Jacobson, A.; Williams, P.; Innis, J.W.; Barone Pritchard, A.; Prasov, L. Design and Outcomes of a Novel Multidisciplinary Ophthalmic Genetics Clinic. Genes 2023, 14, 726. https://doi.org/10.3390/genes14030726
Parekh B, Beil A, Blevins B, Jacobson A, Williams P, Innis JW, Barone Pritchard A, Prasov L. Design and Outcomes of a Novel Multidisciplinary Ophthalmic Genetics Clinic. Genes. 2023; 14(3):726. https://doi.org/10.3390/genes14030726
Chicago/Turabian StyleParekh, Bela, Adelyn Beil, Bridget Blevins, Adam Jacobson, Pamela Williams, Jeffrey W. Innis, Amanda Barone Pritchard, and Lev Prasov. 2023. "Design and Outcomes of a Novel Multidisciplinary Ophthalmic Genetics Clinic" Genes 14, no. 3: 726. https://doi.org/10.3390/genes14030726