
Evaluation of Genetic Testing in a Cohort of Diverse Pediatric Patients in the United States with Congenital Cataracts
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Assessment
2.2. Molecular Investigations
2.3. Analysis
3. Results
3.1. Microarray Results
3.2. Targeted Genetic Testing for Ocular Phenotypes
3.3. Whole Exome Sequencing/Expanded Testing
3.4. Genetic Testing for Other Systemic Phenotypes
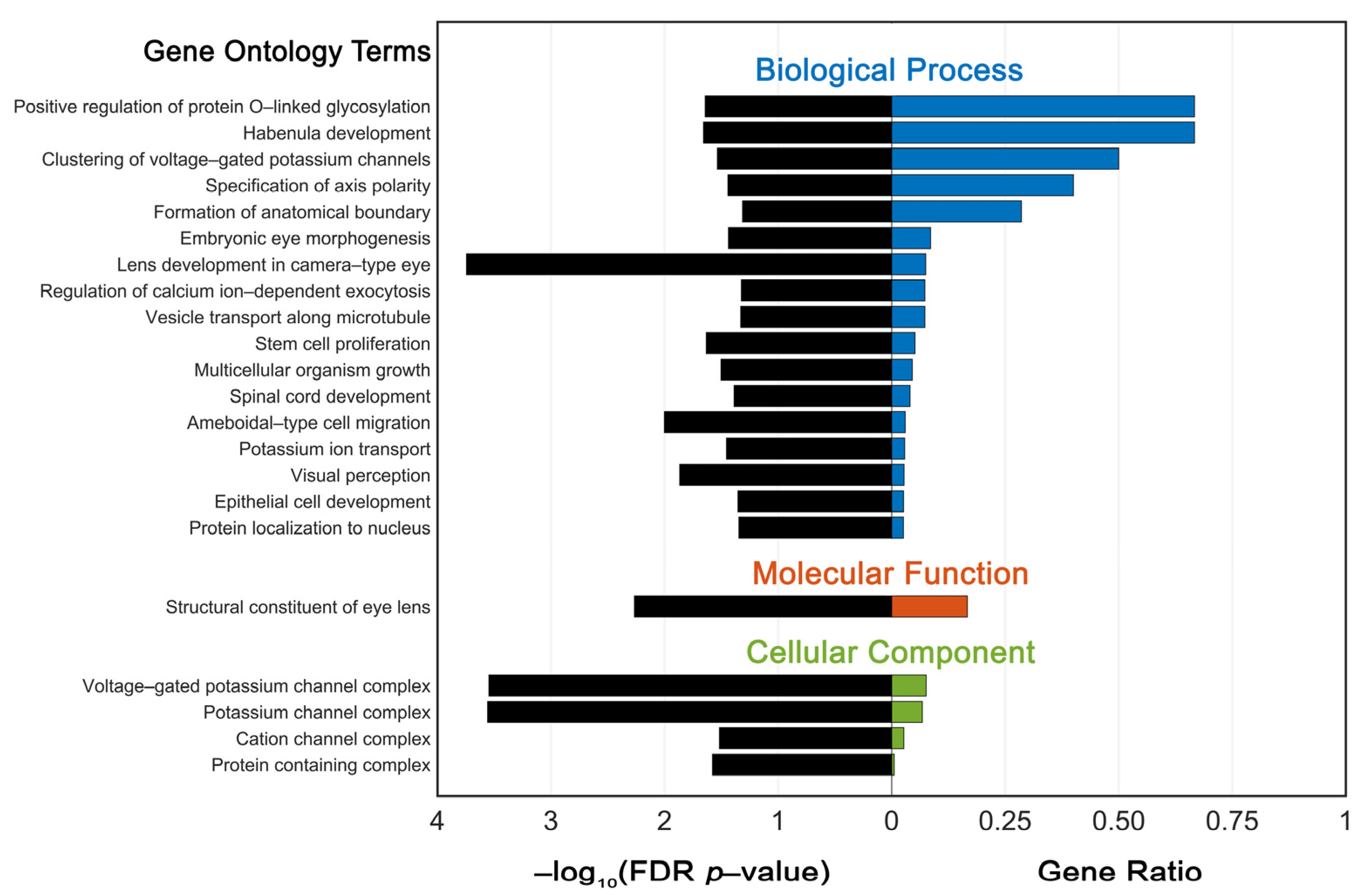
3.5. Functional Enrichment Analysis of Variant Genes
3.6. Variant Reclassification
4. Discussion
4.1. Diagnostic Yield
4.2. Inconclusive/Negative Results
4.3. GO Enrichment Analysis
4.4. Variant Reclassification
4.5. Future Research Aims
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Fry, M.; Al-Samarraie, M.; Gilbert, C.; Steinkuller, P.G. An update on progress and the changing epidemiology of causes of childhood blindness worldwide. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2012, 16, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, W.W.; Aleman, T.S.; Kaushal, S.; Cideciyan, A.V.; Schwartz, S.B.; Wang, L.; Conlon, T.J.; Boye, S.L.; Flotte, T.R.; Byrne, B.J.; et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: Short-term results of a phase I trial. Hum Gene Ther. 2008, 19, 979–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idriss, L.T.; Hussain, M.; Khan, M.; Ahmad, T.; Muhammad, K.; Baig, M.; Khan, M.M.; Inamullah. Mapping of global research output in congenital cataracts from 1903 to 2021. Medicine 2021, 100, e27756. [Google Scholar] [CrossRef]
- Sheeladevi, S.; Lawrenson, J.G.; Fielder, A.R.; Suttle, C.M. Global prevalence of childhood cataract: A systematic review. Eye 2016, 30, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Iancu, I.F.; Avila-Fernandez, A.; Arteche, A.; Trujillo-Tiebas, M.J.; Riveiro-Alvarez, R.; Almoguera, B.; Martin-Merida, I.; Del Pozo-Valero, M.; Perea-Romero, I.; Corton, M.; et al. Prioritizing variants of uncertain significance for reclassification using a rule-based algorithm in inherited retinal dystrophies. NPJ Genom Med. 2021, 6, 18. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Yan, Y.; Yao, K. Molecular genetics of congenital cataracts. Exp Eye Res. 2020, 191, 107872. [Google Scholar] [CrossRef]
- Nie, S.; Chen, G.; Cao, X.; Zhang, Y. Cerebrotendinous xanthomatosis: A comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet. J. Rare Dis. 2014, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Castañeda, Y.S.; Cheng-Patel, C.S.; Leske, D.A.; Wernimont, S.M.; Hatt, S.R.; Liebermann, L.; Birch, E.E.; Holmes, J.M. Quality of life and functional vision concerns of children with cataracts and their parents. Eye 2016, 30, 1251–1259. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Shiels, A.; Bennett, T.M.; Hejtmancik, J.F. Cat-Map Complete List. Available online: https://cat-map.wustl.edu/ (accessed on 28 April 2022).
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Fokkema, I.F.A.C.; Kroon, M.; López Hernández, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Correction: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2021, 590, E53. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Pejaver, V.; Byrne, A.B.; Feng, B.J.; Pagel, K.A.; Mooney, S.D.; Karchin, R.; O’Donnell-Luria, A.; Harrison, S.M.; Tavtigian, S.V.; Greenblatt, M.S.; et al. ClinGen Sequence Variant Interpretation Working Group. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 2022, 109, 2163–2177. [Google Scholar] [CrossRef]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méjécase, C.; Way, C.M.; Owen, N.; Moosajee, M. Ocular Phenotype Associated with DYRK1A Variants. Genes 2021, 12, 234. [Google Scholar] [CrossRef]
- Peng, Y.; Ye, W.; Chen, Z.; Peng, H.; Wang, P.; Hou, X.; Wang, C.; Zhou, X.; Hou, X.; Li, T.; et al. Identifying SYNE1 Ataxia With Novel Mutations in a Chinese Population. Front. Neurol. 2018, 9, 1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, L.M.; Tyler, R.C.; Muheisen, S.; Raggio, V.; Salviati, L.; Han, D.P.; Costakos, D.; Yonath, H.; Hall, S.; Power, P.; et al. Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum. Genet. 2013, 132, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, A.; Grigg, J.R.; Flaherty, M.; Smith, J.; Minoche, A.E.; Cowley, M.J.; Nash, B.M.; Ho, G.; Gayagay, T.; Lai, T.; et al. Genome sequencing in congenital cataracts improves diagnostic yield. Hum. Mutat. 2021, 42, 1173–1183. [Google Scholar] [CrossRef]
- Gillespie, R.L.; O’Sullivan, J.; Ashworth, J.; Bhaskar, S.; Williams, S.; Biswas, S.; Kehdi, E.; Ramsden, S.C.; Clayton-Smith, J.; Black, G.C.; et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 2014, 121, e1–e2. [Google Scholar] [CrossRef]
- Ma, A.S.; Grigg, J.R.; Ho, G.; Prokudin, I.; Farnsworth, E.; Holman, K.; Cheng, A.; Billson, F.A.; Martin, F.; Fraser, C.; et al. Sporadic and Familial Congenital Cataracts: Mutational Spectrum and New Diagnoses Using Next-Generation Sequencing. Hum. Mutat. 2016, 37, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Xiao, X.; Li, S.; Guo, X.; Zhang, Q. Mutation analysis of 12 genes in Chinese families with congenital cataracts. Mol. Vis. 2011, 17, 2197–2206. [Google Scholar]
- Weihbrecht, K.; Goar, W.A.; Pak, T.; Garrison, J.E.; DeLuca, A.P.; Stone, E.M.; Scheetz, T.E.; Sheffield, V.C. Keeping an Eye on Bardet-Biedl Syndrome: A Comprehensive Review of the Role of Bardet-Biedl Syndrome Genes in the Eye. Med. Res. Arch. 2017, 5. [Google Scholar] [CrossRef]
- Patel, N.; Anand, D.; Monies, D.; Maddirevula, S.; Khan, A.O.; Algoufi, T.; Alowain, M.; Faqeih, E.; Alshammari, M.; Qudair, A.; et al. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum. Genet. 2017, 136, 205–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, R.; Pascoal, C.; Marques-da-Silva, D.; Morava, E.; Gole, G.A.; Coman, D.; Jaeken, J.; Dos Reis Ferreira, V.J. Keeping an eye on congenital disorders of O-glycosylation: A systematic literature review. Inherit. Metab. Dis. 2019, 42, 29–48. [Google Scholar] [CrossRef] [PubMed]
- van Reeuwijk, J.; Janssen, M.; van den Elzen, C.; Beltran-Valero de Bernabe, D.; Sabatelli, P.; Merlini, L.; Boon, M.; Scheffer, H.; Brockington, M.; Muntoni, F.; et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 2005, 42, 907–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| N (%) | |
|---|---|
| Race/Ethnicity | |
| White | 22 (42%) |
| Hispanic | 19 (37%) |
| Black | 4 (8%) |
| Asian | 3 (6%) |
| Other | 4 (8%) |
| Systemic Illness | |
| Yes | 40 (77%) |
| No | 12 (23%) |
| Family History | |
| Yes | 4 (8%) |
| No | 48 (92%) |
| Lensectomy | |
| No | 22 (42%) |
| Yes | 30 (58%) |
| Unilateral | 5 (17%) |
| Bilateral | 25 (83%) |
| <1 year-old | 16 (53%) |
| 1–10 years-old | 8 (27%) |
| >10 years-old | 6 (20%) |
| Patient # | Date of Test | Microarray Findings | Diagnostic Yield Positive |
|---|---|---|---|
| 1 | 2011 | 11p14.3p11.2(25,958,860–43,778,471)×1 | Yes |
| 2 | 2013 | 7q31.1(110,950,859–111,234,120)×1 | No |
| 3 | 2020 | 21q22.12q22.13(35,903,896–39,545,357)×1 | Yes |
| 4 | 2021 | 2p25.3(1,741,827–1,842,843)×3 | No |
| 5 | 2016 | 7q35(146,051,998–147,171,974)×3 7q35q36.3(147,171,974–159,138,663)×1 | No |
| 6 | 2010 | 22q11.22(20,640,000–20,905,000)×1 | No |
| 7 | 2019 | Normal | No |
| 8 | 2009 | Normal | No |
| 9 | 2011 | Normal | No |
| 10 | 2016 | Normal | No |
| 11 | 2018 | Normal | No |
| 12 | 2014 | Normal | No |
| 13 | 2010 | Normal | No |
| 14 | 2008 | Normal | No |
| 15 | 2020 | Normal | No |
| 16 | 2012 | Normal | No |
| 17 | 2005 | Normal | No |
| 18 | 2011 | Normal | No |
| Patient # | Test (Laboratory) | Date of Test | Gene (Variant Classification) | Diagnostic Yield Positive |
|---|---|---|---|---|
| 19 | Comprehensive Cataracts Panel (Prevention Genetics) | 2020 | CAPN15 (VUS), CYP27A1 (VUS), FYCO1 (VUS) | No |
| 20 | Early-Onset Bilateral Cataracts Panel (Prevention Genetics) | 2021 | CRYBB2 (VUS) | No |
| 21 | Comprehensive Cataracts Panel (Prevention Genetics) | 2021 | INPP5B (VUS), LTBP2 (VUS), POMT2 (VUS) | No |
| 22 | Microphthalmia, Anophthalmia, and Anterior Segment Dysgenesis Gene Panel (Blueprint Genetics) | 2019 | PAX6 (LP) | Yes |
| 23 | Comprehensive Cataracts Panel (Prevention Genetics) | 2019 | NHS (P) | Yes |
| 24 | PAX6 Gene/Aniridia/Developmental Eye Disorders (GeneDx) | 2019 | 11p13 deletion (P) | Yes |
| 25 | Comprehensive Cataract Panel (Prevention Genetics) | 2021 | RAB3GAP1 (LP), RAB3GAP1 (VUS), BFSP1 (VUS), BFSP1 (VUS), ADAMTS10 (VUS), AGPS (VUS), ERCC1 (VUS) | Yes |
| 26 | Early-Onset Bilateral Cataracts Panel (Prevention Genetics) | 2021 | COL18A1 (VUS), GALK1 (VUS) | No |
| 27 | PAX6 Sequencing (Emory Genetics Laboratory) | 2015 | PAX6 (P) | Yes |
| 28 | Microphthalmia, Anophthalmia, and Anterior Segment Dysgenesis Gene Panel (Blueprint Genetics) | 2020 | No P, LP or VUS | No |
| 29 | :Custom sequencing panel with CNV detection for Coloboma Genes (Prevention Genetics) | 2018 | No P, LP or VUS | No |
| 30 | Cataract Panel (Invitae) | 2022 | CRYAA (P), CRYBB1 ((VUS) | Yes |
| 31 | Early-Onset Bilateral Cataracts Panel (Prevention Genetics) | 2022 | ERCC2 (VUS), ERCC2 (VUS) | No |
| Patient # | Test (Laboratory) | Date of Test | Gene (Variant Classification) | Diagnostic Yield Positive |
|---|---|---|---|---|
| 32 | XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2019 | PHACTR4 (VUS) | No |
| 33 | XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2016 | ALDH18A1 (LP) | Yes |
| 34 | XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2017 | DYNC1H1 (LP), LRP5 (VUS) | Yes |
| 35 | XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2020 | KIF1A (P) | Yes |
| 36 | XomeDx [WES] (GeneDx) | 2014 | BCOR (LP/P), ATP2A3 (VUS) | Yes |
| 37 | XomeDx [WES] (GeneDx) | 2014 | BCOR (LP.P) | Yes |
| 38 | XomeDx Reanalysis (GeneDx) | 2021 | No P, LP or VUS | No |
| 39 | XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2019 | No P, LP or VUS | No |
| 40 | XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2017 | SYNE1 (P), AAAS (P) | Inconclusive |
| 41 | XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2018 | PIK3R1 (VUS) | No |
| 42 | Cerebral Palsy Spectrum Disorders Panel (Invitae) & XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2021 | RARB (LP) | Yes |
| 43 | XomeDx Plus [WES + mitochondrial genome analyses] (GeneDx) | 2021 | WDPCP (VUS), WDPCP (VUS) | Inconclusive |
| 44 | XomeDx [WES] (GeneDx) | 2022 | 13q12.11 duplication which includes GJA3 (VUS) | Inconclusive |
| Patient # | Test (Laboratory) | Date of Test | Gene (Variant Classification) | Diagnostic Yield Positive |
|---|---|---|---|---|
| 46 | Dystroglycan-Related Congenital Muscular Dystrophy Panel (Prevention Genetics) | 2019 | POMT1 (P) | Yes |
| 47 | Cholestasis Panel (EGL Genetics) | 2017 | NOTCH2 (VUS) | Inconclusive |
| 48 | Autism/ID Xpanded Panel (GeneDx) | 2019 | CSNK2B (LP), KCNB1 (LP) | No |
| 49 | Comprehensive Epilepsy Panel (Lurie Molecular Diagnostic Laboratory) & ExomeNext [WES] (Ambry Genetics) | 2018 | NR4A2 (P), DYNC1H1 (VUS), PNKP (VUS), SCN2A (VUS) | Inconclusive |
| 50 | GRB10 gene to confirm variant identified in lab (GeneDx | 2014 | GRB10 (LP/P) | No |
| 51 | NGLY1 evaluation for research found variant (GeneDx) | 2015 | NGLY1 (LP/P) | No |
| 52 | PGL/PCC panel, evaluating for familial variant (GeneDx) | 2013 | SDHD (LP/P) | Inconclusive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossen, J.L.; Bohnsack, B.L.; Zhang, K.X.; Ing, A.; Drackley, A.; Castelluccio, V.; Ralay-Ranaivo, H. Evaluation of Genetic Testing in a Cohort of Diverse Pediatric Patients in the United States with Congenital Cataracts. Genes 2023, 14, 608. https://doi.org/10.3390/genes14030608
Rossen JL, Bohnsack BL, Zhang KX, Ing A, Drackley A, Castelluccio V, Ralay-Ranaivo H. Evaluation of Genetic Testing in a Cohort of Diverse Pediatric Patients in the United States with Congenital Cataracts. Genes. 2023; 14(3):608. https://doi.org/10.3390/genes14030608
Chicago/Turabian StyleRossen, Jennifer L., Brenda L. Bohnsack, Kevin X. Zhang, Alexander Ing, Andy Drackley, Valerie Castelluccio, and Hanta Ralay-Ranaivo. 2023. "Evaluation of Genetic Testing in a Cohort of Diverse Pediatric Patients in the United States with Congenital Cataracts" Genes 14, no. 3: 608. https://doi.org/10.3390/genes14030608