
Diagnostic Yield of Genetic Testing for Ocular and Oculocutaneous Albinism in a Diverse United States Pediatric Population
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gronskov, K.; Ek, J.; Brondum-Nielsen, K. Oculocutaneous albinism. Orphanet J. Rare Dis. 2007, 2, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.Y.; Bodurtha, J.N. The Wills Eye Handbook of Ocular Genetics, 1st ed.; Thieme Medical Publishers: New York, NY, USA, 2018; Volume 4, pp. 191–192. [Google Scholar]
- Tomita, Y.; Takeda, A.; Okinaga, S.; Tagami, H.; Shibahara, S. Human oculocutaneous albinism caused by single base insertion in the tyrosinase gene. Biochem. Biophys. Res. Commun. 1989, 164, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Rinchik, E.M.; Bultman, S.J.; Horsthemke, B.; Lee, S.T.; Strunk, K.M.; Spritz, R.A.; Avidano, K.M.; Jong, M.T.; Nicholls, R.D. A gene for the mouse pink-eyed dilution locus and for human type II oculocutaneous albinism. Nature 1993, 361, 72–76. [Google Scholar] [CrossRef]
- Rosenberg, T.; Schwartz, M. X-linked ocular albinism: Prevalence and mutations—A national study. Eur. J. Hum. Genet. 1998, 6, 570–577. [Google Scholar] [CrossRef]
- Introne, W.; Boissy, R.E.; Gahl, W.A. Clinical, Molecular, and Cell Biological Aspects of Chediak–Higashi Syndrome. Mol. Genet. Metab. 1999, 68, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Li, H.; Yang, L.; Sun, Z.; Yuan, Z.; Li, H.; Sui, R. Molecular genetic and clinical evaluation of three Chinese families with X-linked ocular albinism. Sci. Rep. 2017, 7, 33713. [Google Scholar] [CrossRef]
- Lasseaux, E.; Plaisant, C.; Michaud, V.; Pennamen, P.; Trimouille, A.; Gaston, L.; Monferme, S.; Lacombe, D.; Rooryck, C.; Morice-Picard, F.; et al. Molecular characterization of a series of 990 index patients with albinism. Pigment Cell Melanoma Res. 2018, 31, 466–474. [Google Scholar] [CrossRef]
- Campbell, P.; Ellingford, J.M.; Parry, N.R.A.; Fletcher, T.; Ramsden, S.C.; Gale, T.; Hall, G.; Smith, K.; Kasperaviciute, D.; Thomas, E.; et al. Clinical and genetic variability in children with partial albinism. Sci. Rep. 2019, 9, 16576. [Google Scholar] [CrossRef] [Green Version]
- Lenassi, E.; Clayton-Smith, J.; Douzgou, S.; Ramsden, S.C.; Ingram, S.; Hall, G.; Hardcastle, C.L.; Fletcher, T.A.; Taylor, R.L.; Ellingford, J.M.; et al. Clinical utility of genetic testing in 201 preschool children with inherited eye disorders. Genet. Med. 2020, 22, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.; Malka, S.; Harding, P.; Palma, J.; Dunbar, H.; Moosajee, M. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 578–589. [Google Scholar] [CrossRef]
- Chan, H.W.; Schiff, E.R.; Tailor, V.K.; Malka, S.; Neveu, M.M.; Theodorou, M.; Moosajee, M. Prospective Study of the Phenotypic and Mutational Spectrum of Ocular Albinism and Oculocutaneous Albinism. Genes 2021, 12, 508. [Google Scholar] [CrossRef] [PubMed]
- Hovnik, T.; Debeljak, M.; Tekavčič Pompe, M.; Bertok, S.; Battelino, T.; Stirn Kranjc, B.; Trebušak Podkrajšek, K. Genetic Variability in Slovenian Cohort of Patients with Oculocutaneous Albinism. Acta Chim. Slov. 2021, 68, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennamen, P.; Tingaud-Sequeira, A.; Gazova, I.; Keighren, M.; McKie, L.; Marlin, S.; Halem, S.G.; Kaplan, J.; Delevoye, C.; Lacombe, D.; et al. Dopachrome tautomerase variants in patients with oculocutaneous albinism. Genet. Med. 2021, 23, 479–487. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Fokkema, I.; Kroon, M.; Lopez Hernandez, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Pejaver, V.; Byrne, A.B.; Feng, B.J.; Pagel, K.A.; Mooney, S.D.; Karchin, R.; O’Donnell-Luria, A.; Harrison, S.M.; Tavtigian, S.V.; Greenblatt, M.S.; et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 2022, 109, 2163–2177. [Google Scholar] [CrossRef]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Poulter, J.A.; Al-Araimi, M.; Conte, I.; Van Genderen, M.M.; Sheridan, E.; Carr, I.M.; Parry, D.A.; Shires, M.; Carrella, S.; Bradbury, J.; et al. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am. J. Hum. Genet. 2013, 93, 1143–1150. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Characteristic | N (%) |
|---|---|
| Age (years ± standard deviation) | 1.3 ± 2.0 |
| Sex | |
| Male | 35 (66) |
| Female | 18 (34) |
| Family history of suspected albinism | 17/48 (35) |
| Clinical diagnosis | |
| Oculocutaneous albinism | 41 (77) |
| Ocular albinism | 12 (23) |
| Ophthalmic exam findings | |
| Nystagmus | 47 (89) |
| Fundus hypopigmentation | 36 (68) |
| Foveal hypoplasia | 45 (85) |
| Iris transillumination defects | 20 (38) |
| Number of Patients with Positive Diagnostic Yield (%) | Number of Patients with Negative Diagnostic Yield (%) | p-Value | |
|---|---|---|---|
| Overall After variant reclassification | 35 (66) 37 (70) | 18 (34) 16 (30) | -- |
| Cutaneous involvement | |||
| Yes No | 31 (76) 4 (33) | 10 (24) 8 (67) | 0.007 * |
| Ocular Manifestations | |||
| Nystagmus Fundus hypopigmentation Foveal hypoplasia Iris transillumination defects | 33 (70) 25 (69) 31 (69) 16 (80) | 14 (30) 11 (31) 14 (31) 4 (20) | 0.82 |
| Race and ethnicity | |||
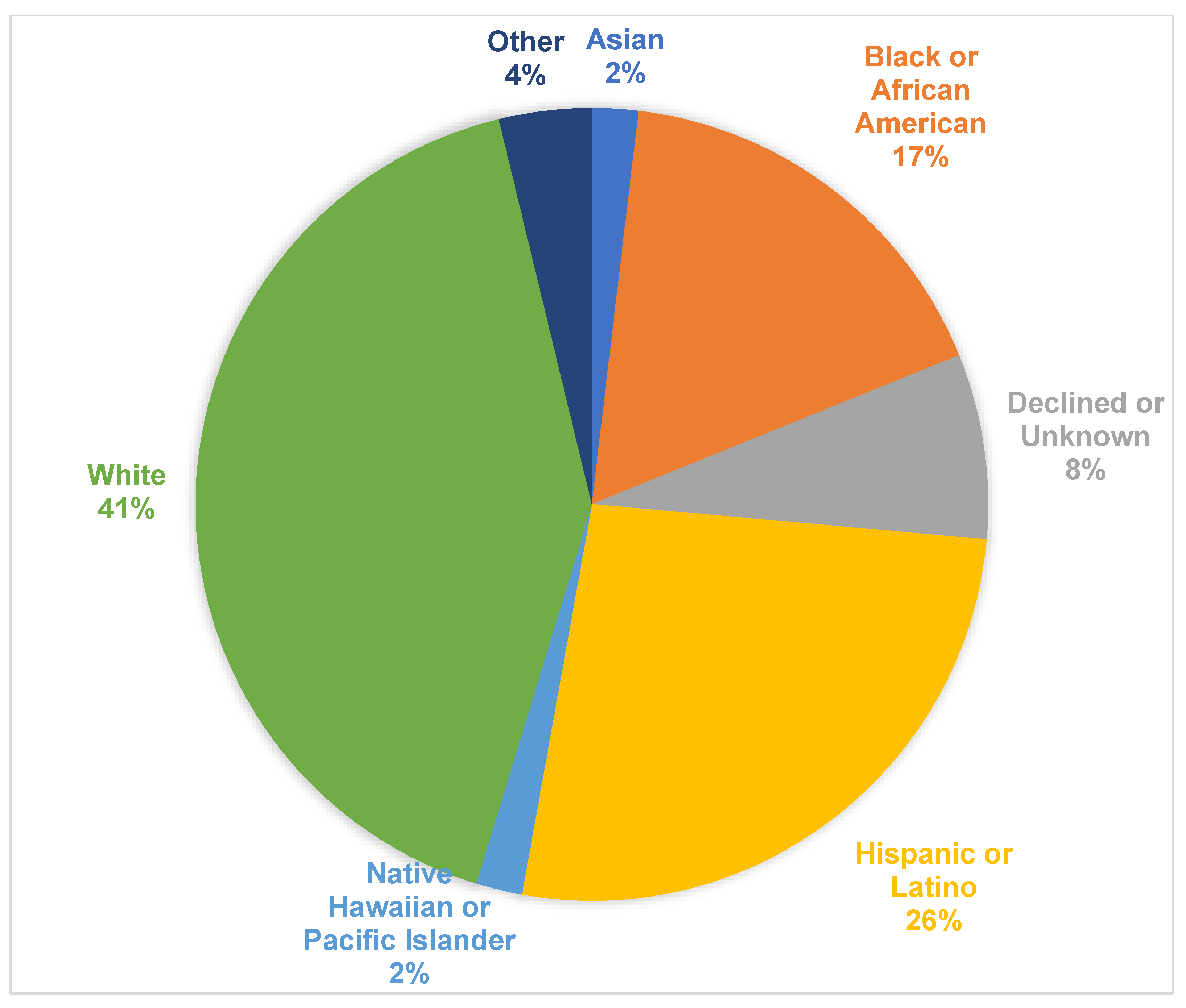
| Asian Black or African American Declined or Unknown Hispanic or Latino Native Hawaiian or Pacific Islander Other White | 0 (0) 7 (78) 3 (75) 9 (64) 1 (100) 2 (100) 13 (59) | 1 (100) 2 (22) 1 (25) 5 (36) 0 (0) 0 (0) 9 (41) | 0.59 |
| Gene | N (%) |
|---|---|
| OCA2 (%) | 15 (28) |
| TYR (%) | 11 (20) |
| HPS5 (%) | 3 (6) |
| TYRP1 (%) | 2 (4) |
| HPS1 (%) | 1 (2) |
| HPS6 (%) | 1 (2) |
| SLC45A2 (%) | 1 (2) |
| OA1/GPR143 (%) | 1 (2) |
| % Taken from total number of patients in the study (53) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, K.S.; Bohnsack, B.L.; Ing, A.; Drackley, A.; Castelluccio, V.; Zhang, K.X.; Ralay-Ranaivo, H.; Rossen, J.L. Diagnostic Yield of Genetic Testing for Ocular and Oculocutaneous Albinism in a Diverse United States Pediatric Population. Genes 2023, 14, 135. https://doi.org/10.3390/genes14010135
Chan KS, Bohnsack BL, Ing A, Drackley A, Castelluccio V, Zhang KX, Ralay-Ranaivo H, Rossen JL. Diagnostic Yield of Genetic Testing for Ocular and Oculocutaneous Albinism in a Diverse United States Pediatric Population. Genes. 2023; 14(1):135. https://doi.org/10.3390/genes14010135
Chicago/Turabian StyleChan, Kyle S., Brenda L. Bohnsack, Alexander Ing, Andy Drackley, Valerie Castelluccio, Kevin X. Zhang, Hanta Ralay-Ranaivo, and Jennifer L. Rossen. 2023. "Diagnostic Yield of Genetic Testing for Ocular and Oculocutaneous Albinism in a Diverse United States Pediatric Population" Genes 14, no. 1: 135. https://doi.org/10.3390/genes14010135