
Transcriptomic Profiling Reveals an Enhancer RNA Signature for Recurrence Prediction in Colorectal Cancer


Abstract
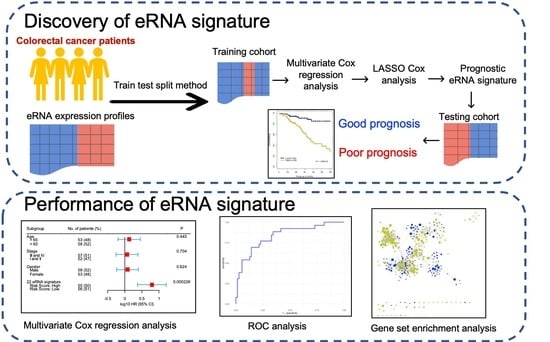
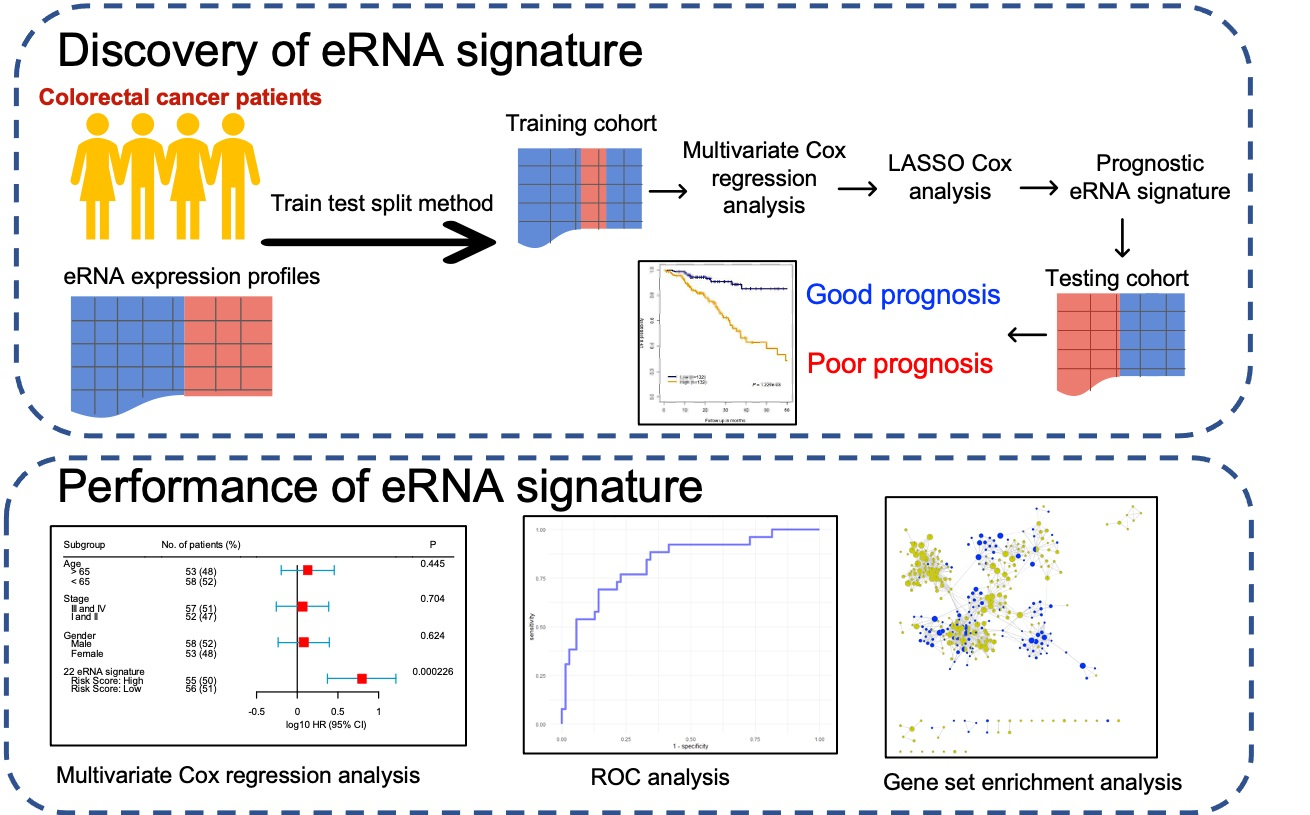
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
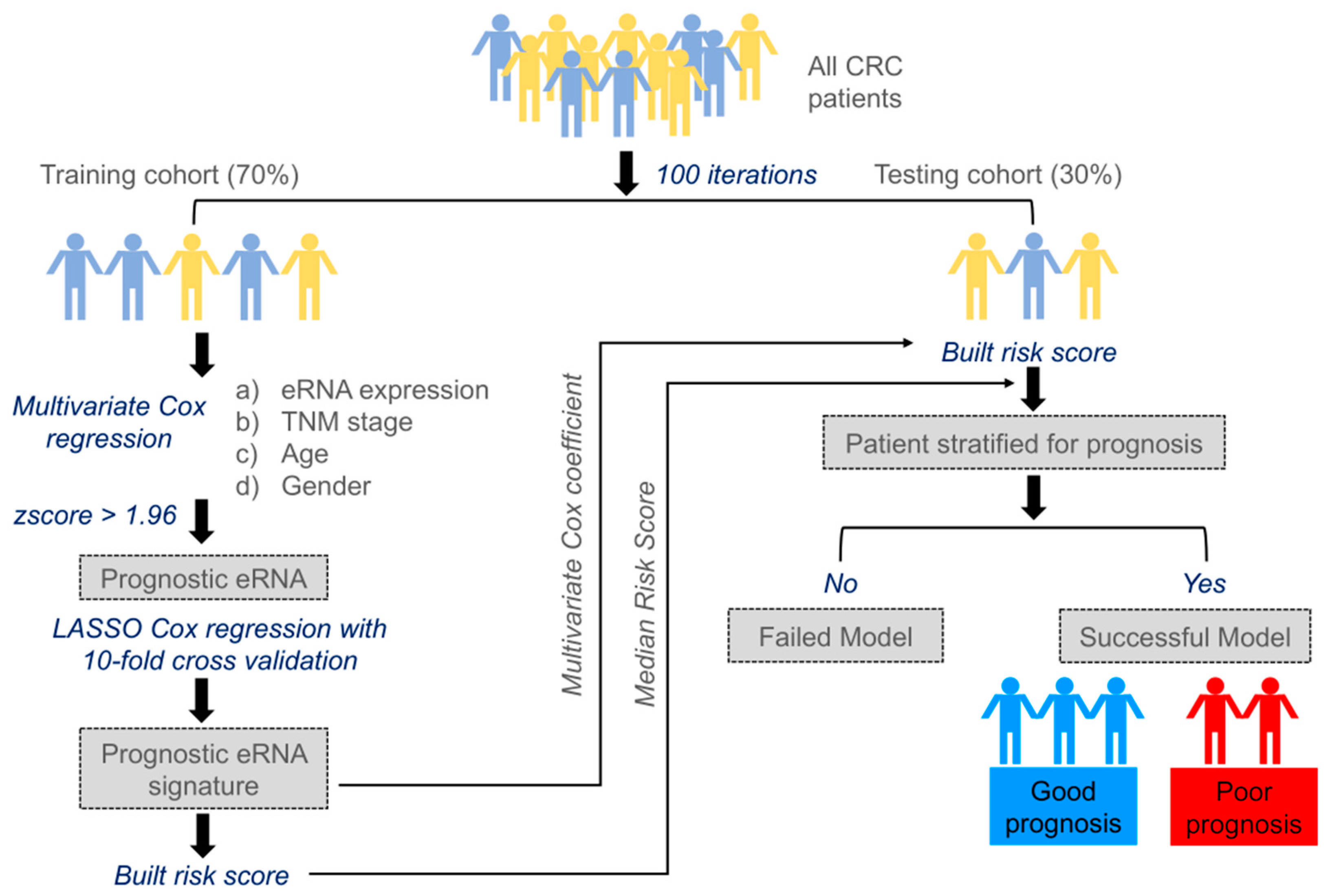
2.1. Building Survival Model to Identify Clinically Relevant eRNAs
- We randomly selected 70% of the entire cohort from The Cancer Genome Atlas (TCGA) as the training set and reserved the remaining 30% of the cohort as the testing set. To avoid deviation affecting the stability of the model, we maintained the distribution of disease-free and relapsed patients from the entire cohort in both training and testing sets.
- Multivariate Cox regression analysis was carried out training on 477 eRNAs for CC and 460 eRNAs for RC, along with controlling the effects from other clinical risk factors, including patient age, gender, and TNM stage.
- eRNAs significantly associated (p < 0.05 & z-score > 1.96) for predicting patient disease-free survival (DFS) were retained and termed as prognostic eRNAs and were further selected by LASSO Cox regularization with 10-fold cross-validation.
- Following LASSO Cox regularization and eRNA selection, a risk score formula was established. The risk score for each patient was calculated by a linear combination of expression and multivariate Cox coefficient of eRNAs.
- Patients were classified into low-risk or high-risk groups using the median risk score as the cut-off threshold from the training set. The coefficient for each eRNA and the cut-off value of the risk score from the training set was used to calculate the risk score and to stratify patients into individual risk groups in the testing set.
- Survival differences between the two groups were estimated using Kaplan–Meier curve and compared using the log-rank test.
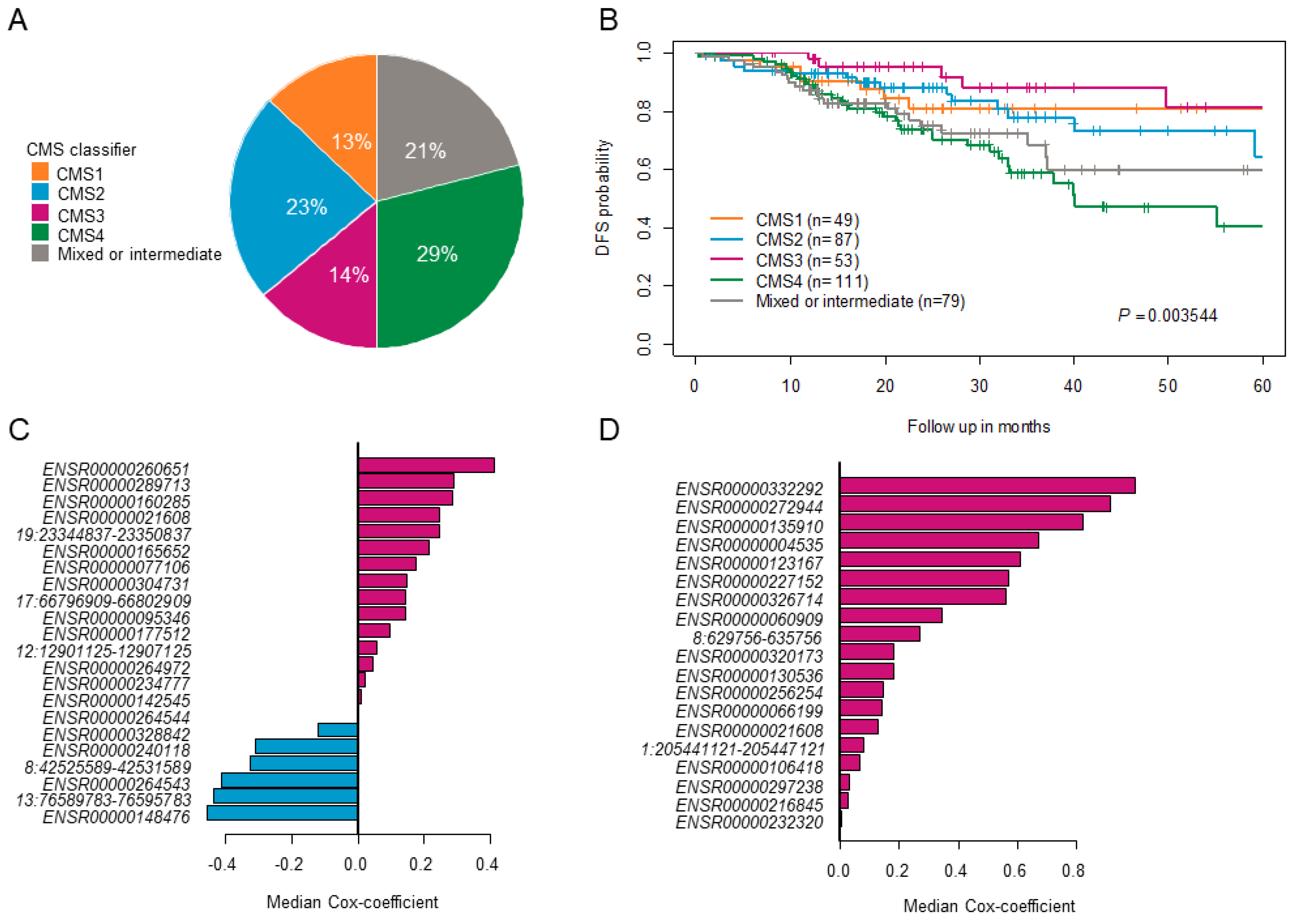
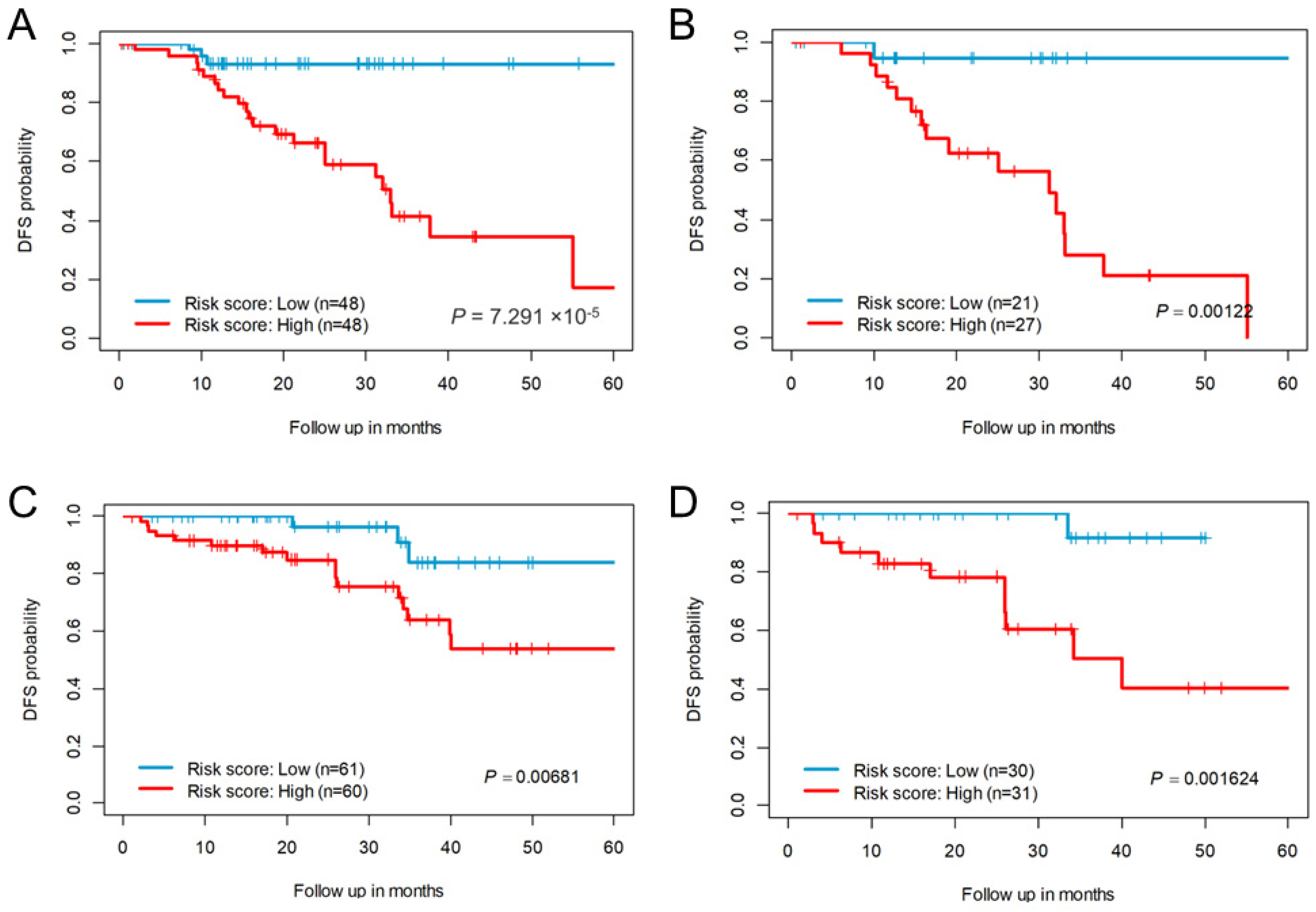
2.2. Prognostic Association of eRNA Signature Risk Score with Disease-Free Survival in Patients
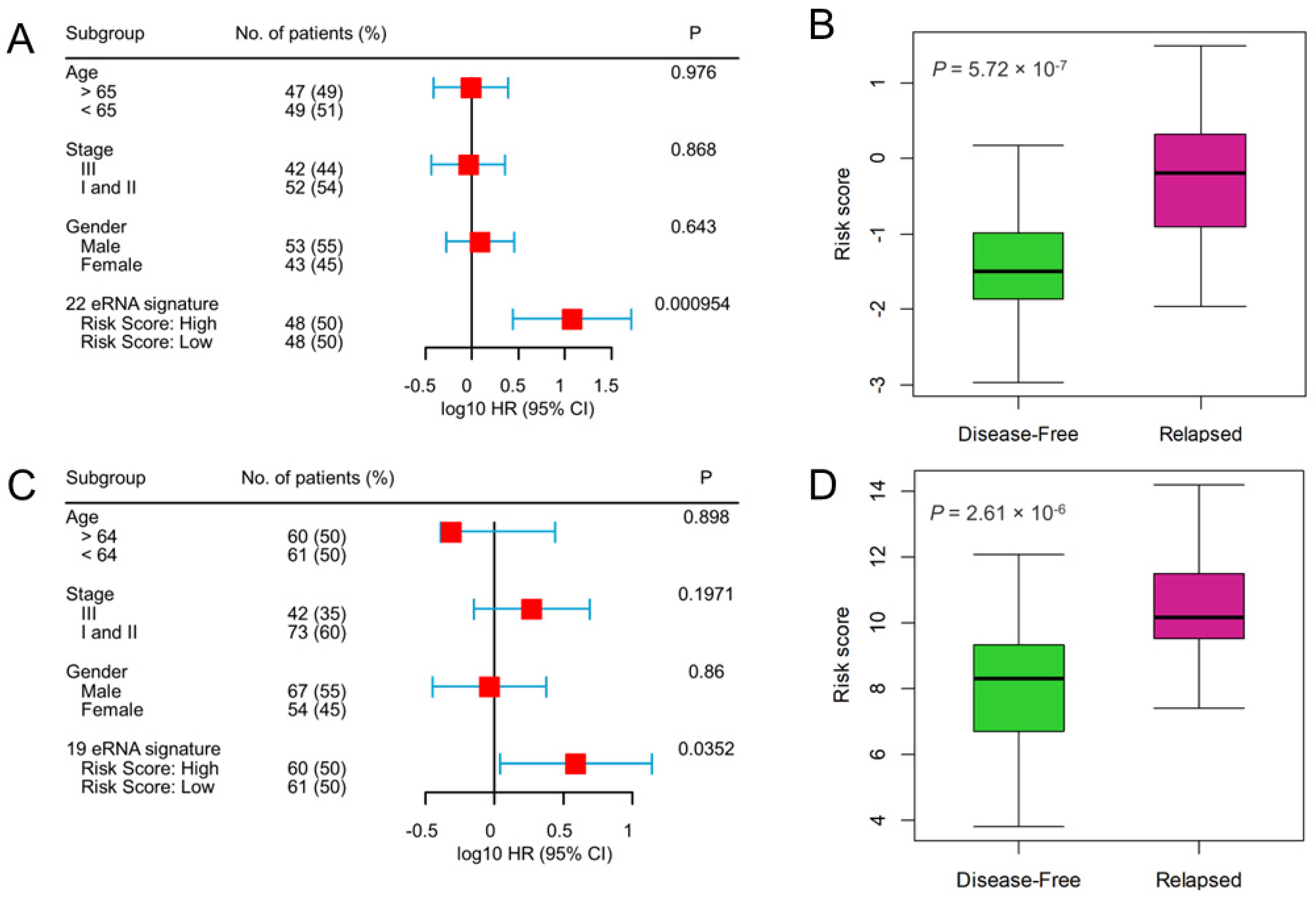
2.3. Recurrence Prediction by eRNA Signature Is Independent of Clinical Risk Factors
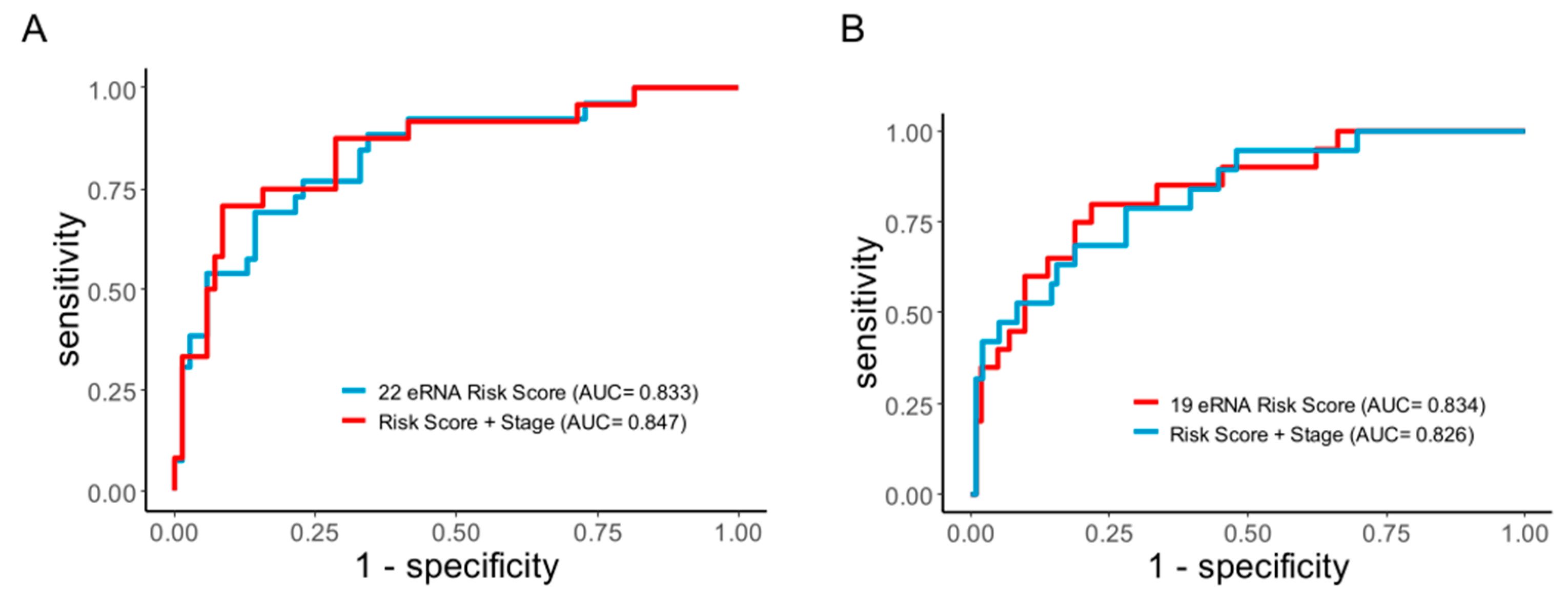
2.4. The eRNA Signature Is a Better Predictor of Recurrence with High Sensitivity and Specificity
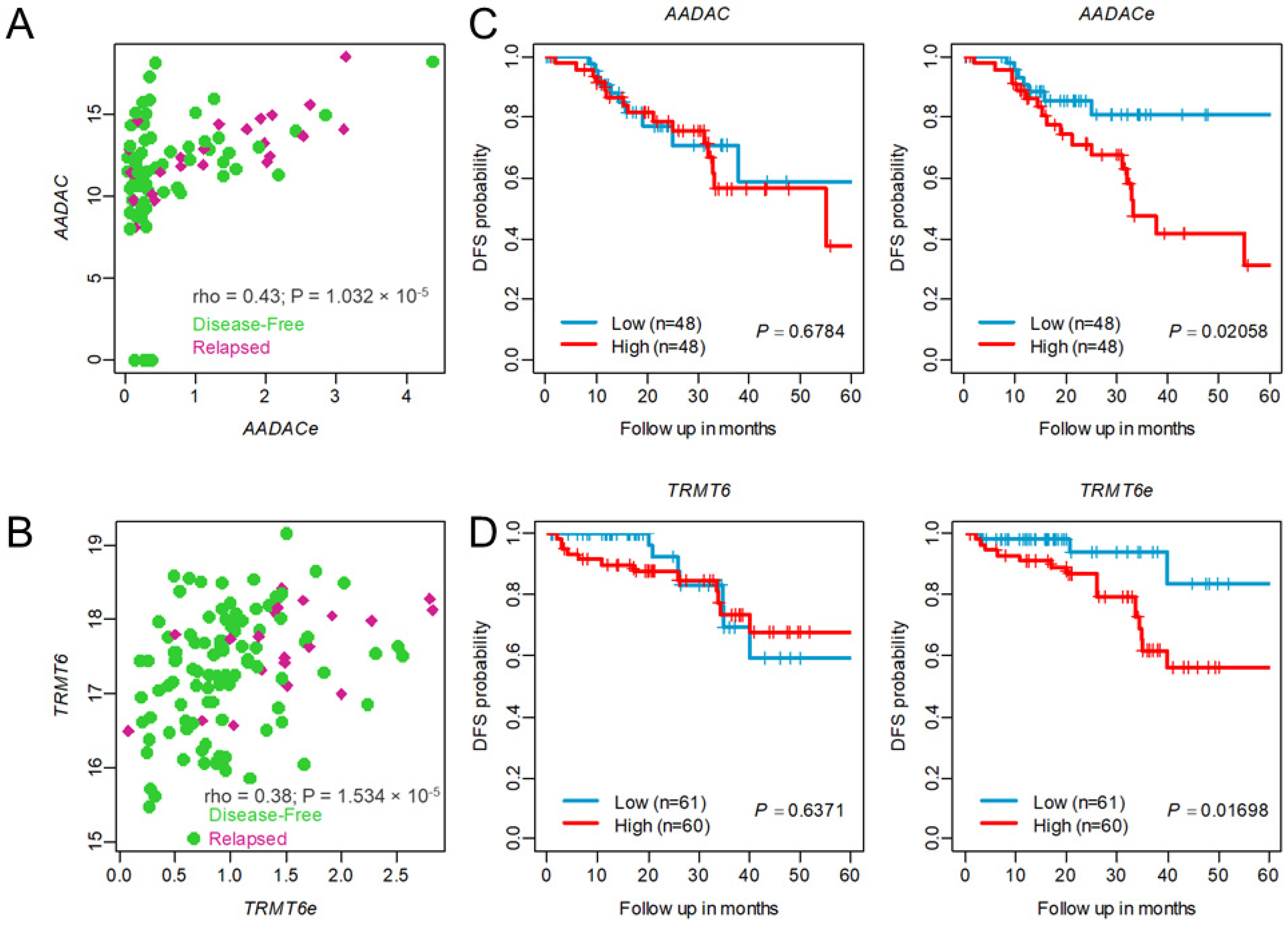
2.5. Higher Expression of eRNAs Associated with Tumor Recurrence Compared to Its Target Genes
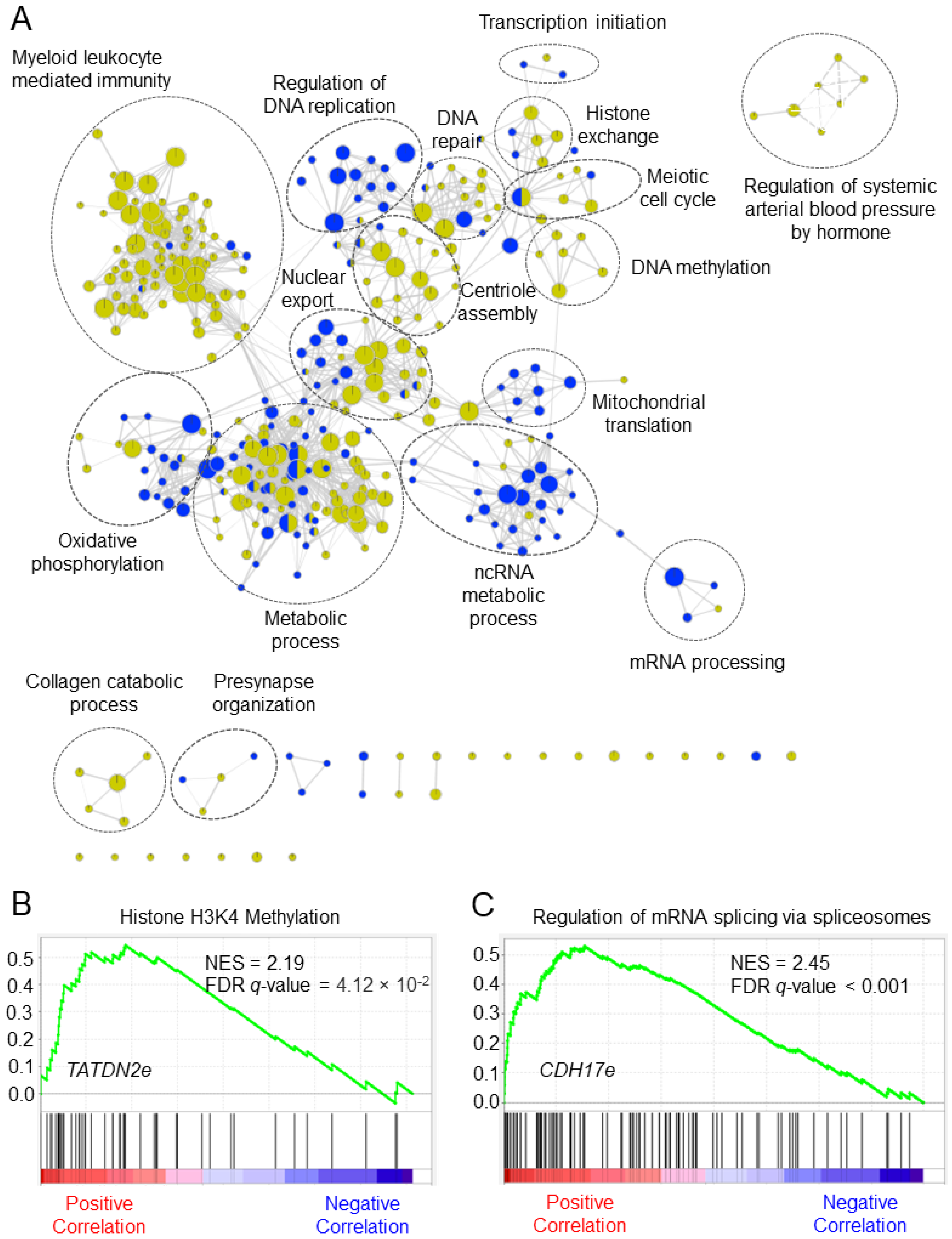
2.6. Putative Biological Functions of eRNA Signature in Colorectal Cancer
3. Discussion
4. Conclusions
5. Methods
5.1. CRC Patients Datasets
5.2. Consensus Molecular Subtypes (CMS) Classification
5.3. Identification of Prognostic eRNAs Associated with Tumor Relapse
5.4. Risk Score Calculation
5.5. Statistical Analysis
5.6. Gene Set Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2022; American Cancer Society: Atlanta, GA, USA, 2022. [Google Scholar]
- Blank, A.; Roberts, D.E.I.; Dawson, H.; Zlobec, I.; Lugli, A. Tumor heterogeneity in primary colorectal cancer and corresponding metastases. Does the apple fall far from the tree? Front. Med. 2018, 5, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, T.S.; Clark, J.W.; Haigis, K.M. Cancers of the colon and rectum: Identical or fraternal twins? Cancer Discov. AACR 2012, 2, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Marks, K.; West, N.; Morris, E.; Quirke, P. Clinicopathological, genomic and immunological factors in colorectal cancer prognosis. J. Br. Surg. 2018, 105, e99–e109. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Mason, M.J.; Sinicrope, F.A.; Phipps, A.I.; Tejpar, S.; Nesbakken, A.; Danielsen, S.A.; Sveen, A.; Buchanan, D.D.; Clendenning, M.; et al. Prediction of overall survival in stage II and III colon cancer beyond TNM system: A retrospective, pooled biomarker study. Ann. Oncol. 2017, 28, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Tolone, S.; Allaria, A.; Napolitano, V.; Ilaria, F.; Ottavia, M.; Cesaro, E.; Docimo, L.; Fei, L. Lymph node ratio versus TNM system as a prognostic factor in colorectal cancer staging. A Single Center experience. Open Med. 2019, 14, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.C.; Park, Y.-Y.; Park, E.S.; Lim, J.Y.; Kim, S.M.; Kim, S.-B.; Kim, J.; Kim, S.C.; Chu, I.-S.; Smith, J.J. Prognostic gene expression signature associated with two molecularly distinct subtypes of colorectal cancer. Gut 2012, 61, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, R.; Ozawa, T.; Gao, F.; Wang, X.; Goel, A.; Nozawa, H.; Hata, K.; Nagata, H.; Okada, S.; Watanabe, T. Gene expression signature in surgical tissues and endoscopic biopsies identifies high-risk T1 colorectal cancers. Gastroenterology 2019, 156, 2338–2341.e2333. [Google Scholar] [CrossRef]
- Ye, S.-B.; Cheng, Y.-K.; Hu, J.-C.; Gao, F.; Lan, P. Development and validation of an individualized gene expression-based signature to predict overall survival in metastatic colorectal cancer. Ann. Transl. Med. 2020, 8, 96. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, J.; Wang, J.; Xu, M.; Zhang, Y.; Sun, P.; Liang, L. Development and clinical validation of a novel 4-gene prognostic signature predicting survival in colorectal cancer. Front. Oncol. 2020, 10, 595. [Google Scholar] [CrossRef]
- Brouwer, N.P.; Bos, A.C.; Lemmens, V.E.; Tanis, P.J.; Hugen, N.; Nagtegaal, I.D.; de Wilt, J.H.; Verhoeven, R.H. An overview of 25 years of incidence, treatment and outcome of colorectal cancer patients. Int. J. Cancer 2018, 143, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Xiong, F.; Li, W. Enhancer RNAs in cancer: Regulation, mechanisms and therapeutic potential. RNA Biol. 2020, 17, 1550–1559. [Google Scholar] [CrossRef]
- Xiang, J.F.; Yin, Q.F.; Chen, T.; Zhang, Y.; Zhang, X.O.; Wu, Z.; Zhang, S.; Wang, H.B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014, 24, 513–531. [Google Scholar] [CrossRef] [Green Version]
- McCleland, M.L.; Mesh, K.; Lorenzana, E.; Chopra, V.S.; Segal, E.; Watanabe, C.; Haley, B.; Mayba, O.; Yaylaoglu, M.; Gnad, F.; et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Investig. 2016, 126, 639–652. [Google Scholar] [CrossRef] [Green Version]
- Melo, C.A.; Drost, J.; Wijchers, P.J.; van de Werken, H.; de Wit, E.; Oude Vrielink, J.A.; Elkon, R.; Melo, S.A.; Léveillé, N.; Kalluri, R.; et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 2013, 49, 524–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Wang, L.; Boldrup, L.; Coates, P.J.; Fahraeus, R.; Sgaramella, N.; Wilms, T.; Nylander, K. AP001056. 1, a prognosis-related enhancer RNA in squamous cell carcinoma of the head and neck. Cancers 2019, 11, 347. [Google Scholar] [CrossRef]
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-L.; Fei, T.; Chen, Y.; Li, T.; Gao, Y.; Wang, X.; Sun, T.; Sweeney, C.J.; Lee, G.-S.M.; Chen, S.; et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. USA 2014, 111, 7319–7324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.H.; Ruan, H.; Ye, Y.; Krakowiak, J.; Hu, Q.; Xiang, Y.; Gong, J.; Zhou, B.; Wang, L.; et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 2019, 10, 4562. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zhu, X.; Yan, T.; Yu, C.; Shen, C.; Hu, Y.; Hong, J.; Chen, H.; Fang, J.Y. Recurrence-associated gene signature optimizes recurrence-free survival prediction of colorectal cancer. Mol. Oncol. 2017, 11, 1544–1560. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Zhang, M.; Zhu, H.; Xu, J. A 15-gene signature for prediction of colon cancer recurrence and prognosis based on SVM. Gene 2017, 604, 33–40. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. AACR 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Therneau, T. A package for survival analysis in S. R Package Version 2015, 2, 38. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Hadley, W. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahu, D.; Lin, C.-C.; Goel, A. Transcriptomic Profiling Reveals an Enhancer RNA Signature for Recurrence Prediction in Colorectal Cancer. Genes 2023, 14, 137. https://doi.org/10.3390/genes14010137
Sahu D, Lin C-C, Goel A. Transcriptomic Profiling Reveals an Enhancer RNA Signature for Recurrence Prediction in Colorectal Cancer. Genes. 2023; 14(1):137. https://doi.org/10.3390/genes14010137
Chicago/Turabian StyleSahu, Divya, Chen-Ching Lin, and Ajay Goel. 2023. "Transcriptomic Profiling Reveals an Enhancer RNA Signature for Recurrence Prediction in Colorectal Cancer" Genes 14, no. 1: 137. https://doi.org/10.3390/genes14010137