

Molecular Heterogeneity of Pediatric AML with Atypical Promyelocytes Accumulation in Children—A Single Center Experience
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Morphological Examination
2.3. Flow Cytometry
2.4. Cytogenetics and Molecular Genetics
2.5. Statistics
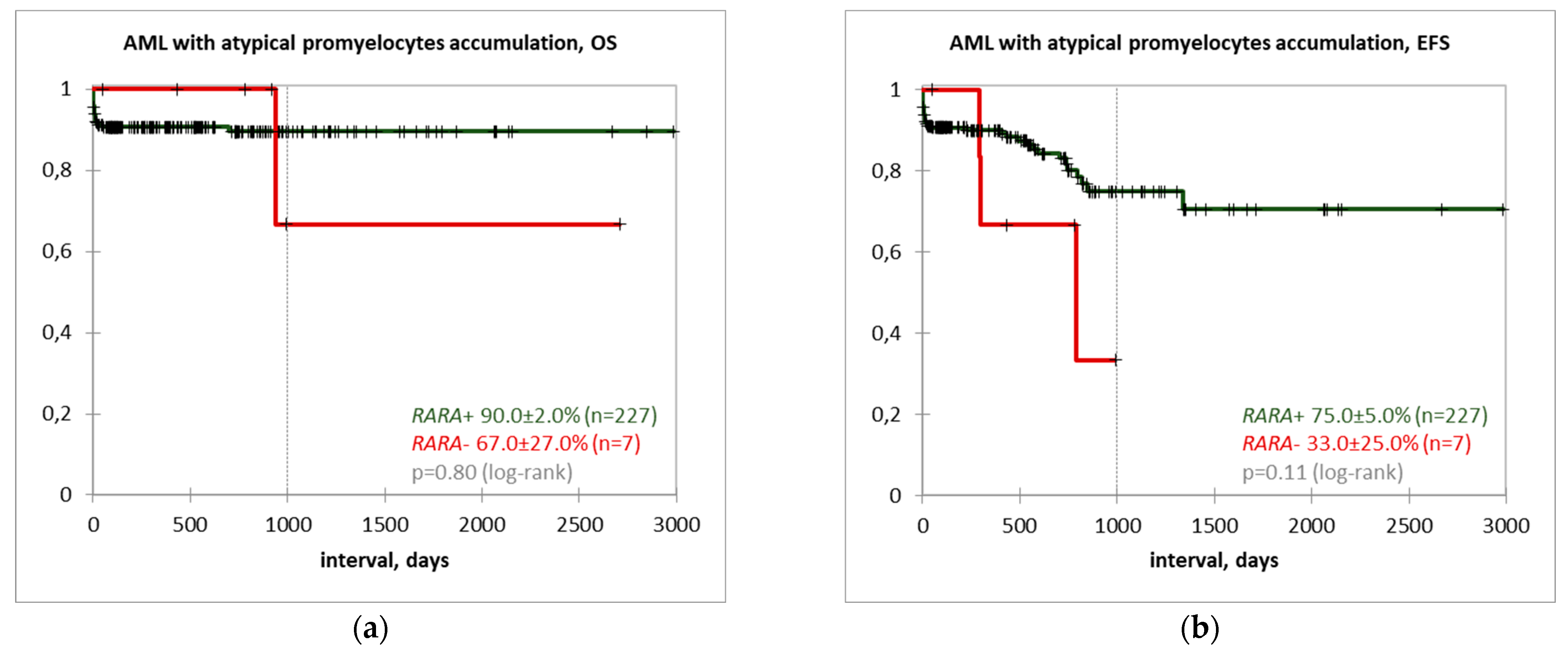
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guarnera, L.; Ottone, T.; Fabiani, E.; Divona, M.; Savi, A.; Travaglini, S.; Falconi, G.; Panetta, P.; Rapanotti, M.C.; Voso, M.T. Atypical Rearrangements in APL-Like Acute Myeloid Leukemias: Molecular Characterization and Prognosis. Front. Oncol. 2022, 12, 871590. [Google Scholar] [CrossRef]
- Marinelli, A.; Bossi, D.; Pelicci, P.G.; Minucci, S. A redundant oncogenic potential of the retinoic receptor (RAR) alpha, beta and gamma isoforms in acute promyelocytic leukemia. Leukemia 2007, 21, 647–650. [Google Scholar] [CrossRef]
- Melnick, A.; Licht, J.D. Deconstructing a disease: RARα, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 1999, 93, 3167–3215. [Google Scholar] [CrossRef]
- Sternsdorf, T.; Phan, V.T.; Maunakea, M.L.; Ocampo, C.B.; Sohal, J.; Silletto, A.; Galimi, F.; Le Beau, M.M.; Evans, R.M.; Kogan, S.C. Forced retinoic acid receptor alpha homodimers prime mice for APL-like leukemia. Cancer Cell 2006, 9, 81–94. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, J.; Yu, W.; Jin, J. Current views on the genetic landscape and management of variant acute promyelocytic leukemia. Biomark. Res. 2021, 9, 33. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann. Intern. Med. 1985, 103, 620–625. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. A variant form of hypergranular promyelocytic leukaemia (M3). Br. J. Haematol. 1980, 44, 169–170. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.Á.; Barragán, E.; Cervera, J. Acute promyelocytic leukemia: A constellation of molecular events around a single PML-RARA fusion gene. Cancers 2020, 12, 624. [Google Scholar] [CrossRef]
- Pandolfi, P.P.; Alcalay, M.; Fagioli, M.; Zangrilli, D.; Mencarelli, A.; Diverio, D.; Biondi, A.; Lo Coco, F.; Rambaldi, A.; Grignani, F.; et al. Genomic variability and alternative splicing generate multiple PML/RAR alpha transcripts that encode aberrant PML proteins and PML/RAR alpha isoforms in acute promyelocytic leukaemia. EMBO J. 1992, 11, 1397–1407. [Google Scholar] [CrossRef]
- Reiter, A.; Saussele, S.; Grimwade, D.; Wiemels, J.L.; Segal, M.R.; Lafage-Pochitaloff, M.; Walz, C.; Weisser, A.; Hochhaus, A.; Willer, A.; et al. Genomic anatomy of the specific reciprocal translocation t(15;17) in acute promyelocytic leukemia. Genes Chromosomes Cancer 2003, 36, 175–188. [Google Scholar] [CrossRef]
- Grimwade, D.; Gorman, P.; Duprez, E.; Howe, K.; Langabeer, S.; Oliver, F.; Walker, H.; Culligan, D.; Waters, J.; Pomfret, M.; et al. Characterization of cryptic rearrangements and variant translocations in acute promyelocytic leukemia. Blood 1997, 90, 4876–4885. [Google Scholar]
- Adams, J.; Nassiri, M. Acute promyelocytic leukemia a review and discussion of variant translocations. Arch. Pathol. Lab. Med. 2015, 139, 1308–1313. [Google Scholar] [CrossRef]
- Grimwade, D.; Biondi, A.; Mozziconacci, M.J.; Hagemeijer, A.; Berger, R.; Neat, M.; Howe, K.; Dastugue, N.; Jansen, J.; Radford-Weiss, I.; et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): Results of the European Working Party. Groupe Francais de Cytogenetique Hematologique, Groupe de Francais d’Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies. Blood 2000, 96, 1297–1308. [Google Scholar]
- Arana Rosainz, M.J.; Nguyen, N.; Wahed, A.; Lelenwa, L.C.; Aakash, N.; Schaefer, K.; Rios, A.; Kanaan, Z.; Chen, L. Acute myeloid leukemia with mutated NPM1 mimics acute promyelocytic leukemia presentation. Int. J. Lab. Hematol. 2021, 43, 218–226. [Google Scholar] [CrossRef]
- Chen, X.; Wang, F.; Zhang, Y.; Teng, W.; Cao, P.; Ma, X.; Liu, M.; Tian, Y.; Wang, T.; Nie, D.; et al. A novel NPM1-RARG-NPM1 chimeric fusion in acute myeloid leukaemia resembling acute promyelocytic leukaemia but resistant to all-trans retinoic acid and arsenic trioxide. Br. J. Cancer 2019, 120, 1023–1025. [Google Scholar] [CrossRef]
- Mikhailova, E.V.; Kashpor, S.A.; Zerkalenkova, E.A.; Semchenkova, A.A.; Dubrovina, M.E.; Plyasunova, S.A.; Olshanskaya, Y.V.; Kalinina, I.I.; Maschan, M.A.; Maschan, A.A.; et al. Immunophenotypic characterization of pediatric acute myeloid leukemia with inv(16)(p13.1q22)/t(16;16)(p13.1;q22)/CBFb-MYH11. Pediatr. Hematol. Oncol. Immunopathol. 2021, 20, 46–53. [Google Scholar] [CrossRef]
- Popov, A.M.; Verzhbitskaya, T.Y.; Movchan, L.V.; Demina, I.A.; Mikhailova, E.V.; Semchenkova, A.A.; Permikin, Z.V.; Shman, T.V.; Karachunskiy, A.I.; Novichkova, G.A. Flow cytometry in acute leukemia diagnostics. Guidelines of Russian-Belarusian multicenter group for pediatric leukemia studies. Pediatr. Hematol. Oncol. Immunopathol. 2023, 22, 165–177. [Google Scholar] [CrossRef]
- Kalina, T.; Flores-Montero, J.; van der Velden, V.H.; Martin-Ayuso, M.; Bottcher, S.; Ritgen, M.; Almeida, J.; Lhermitte, L.; Asnafi, V.; Mendonca, A.; et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 2012, 26, 1986–2010. [Google Scholar] [CrossRef]
- Den Nijs, J.I.; Gonggrijp, H.S.; Augustinus, E.; Leeksma, C.H.W. Hot bands: A simple G-banding method for leukemic metaphases. Cancer Genet. Cytogenet. 1985, 15, 373–374. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. An International System for Human Cytogenomic Nomenclature (2020). Cytogenet. Genome Res. 2020, 160. [Google Scholar] [CrossRef]
- Gabert, J.; Beillard, E.; van der Velden, V.H.J.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and quality control studies of ‘real time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer Program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef]
- Downing, J.R.; Shurtleff, S.A.; Zielenska, M.; Curcio-Brint, A.M.; Behm, F.G.; Head, D.R.; Sandlund, J.T.; Weisenburger, D.D.; Kossakowska, A.E.; Thorner, P.; et al. Molecular detection of the (2;5) translocation of non-Hodgkin’s lymphoma by reverse transcriptase-polymerase chain reaction. Blood 1995, 85, 3416–3422. [Google Scholar] [CrossRef]
- Van Dongen, J.J.M.; Macintyre, E.A.; Gabert, J.A.; Delabesse, E.; Rossi, V.; Saglio, G.; Gottardi, E.; Rambaldi, A.; Dotti, G.; Griesinger, F.; et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: Investigation of minimal residual disease in acute leukemia. Leukemia 1999, 13, 1901–1928. [Google Scholar] [CrossRef]
- Genome_Reference_Consortium. GRCh38 UCSC Analysis Set Files. Available online: https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/analysisSet/ (accessed on 5 November 2022).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Frohlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Dunn, T.; Berry, G.; Emig-Agius, D.; Jiang, Y.; Lei, S.; Iyer, A.; Udar, N.; Chuang, H.Y.; Hegarty, J.; Dickover, M.; et al. Pisces: An accurate and versatile variant caller for somatic and germline next-generation sequencing data. Bioinformatics 2019, 35, 1579–1581. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Meyer, C.; Schneider, B.; Reichel, M.; Angermueller, S.; Strehl, S.; Schnittger, S.; Schoch, C.; Jansen, M.W.; van Dongen, J.J.; Pieters, R.; et al. Diagnostic tool for the identification of MLL rearrangements including unknown partner genes. Proc. Natl. Acad. Sci. USA 2005, 102, 449–454. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, J.W.; Xue, H.L.; Shen, S.H.; Chen, J.; Tang, Y.J.; Yu, L.S.; Liang, H.H.; Gu, L.J.; Tang, J.Y.; et al. The genetics and clinical characteristics of children morphologically diagnosed as acute promyelocytic leukemia. Leukemia 2019, 33, 1387–1399. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Bene, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef]
- Garzon, R.; Garofalo, M.; Martelli, M.P.; Briesewitz, R.; Wang, L.; Fernandez-Cymering, C.; Volinia, S.; Liu, C.G.; Schnittger, S.; Haferlach, T.; et al. Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc. Natl. Acad. Sci. USA 2008, 105, 3945–3950. [Google Scholar] [CrossRef]
- Zarka, J.; Short, N.J.; Kanagal-Shamanna, R.; Issa, G.C. Nucleophosmin 1 Mutations in Acute Myeloid Leukemia. Genes 2020, 11, 649. [Google Scholar] [CrossRef]
- Ha, J.S.; Do, Y.R.; Ki, C.S.; Lee, C.; Kim, D.H.; Lee, W.; Ryoo, N.H.; Jeon, D.S. Identification of a novel PML-RARG fusion in acute promyelocytic leukemia. Leukemia 2017, 31, 1992–1995. [Google Scholar] [CrossRef]
- Liu, T.; Wen, L.; Yuan, H.; Wang, Y.; Yao, L.; Xu, Y.; Cen, J.; Ruan, C.; Wu, D.; Chen, S. Identification of novel recurrent CPSF6-RARG fusions in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood 2018, 131, 1870–1873. [Google Scholar] [CrossRef]
- Luo, H.; Zhang, S.; Li, K.; Chen, X.H.; Li, Y.C.; Sun, Y.; Liu, L.F.; Yu, H.Y.; Zhu, H.H. A novel entity of acute myeloid leukaemia with recurrent RARG-rearrangement resembling acute promyelocytic leukaemia. Leuk. Res. 2019, 77, 14–16. [Google Scholar] [CrossRef]
- Miller, C.A.; Tricarico, C.; Skidmore, Z.L.; Uy, G.L.; Lee, Y.S.; Hassan, A.; O’Laughlin, M.D.; Schmidt, H.; Tian, L.; Duncavage, E.J.; et al. A case of acute myeloid leukemia with promyelocytic features characterized by expression of a novel RARG-CPSF6 fusion. Blood Adv. 2018, 2, 1295–1299. [Google Scholar] [CrossRef]
- Osumi, T.; Tsujimoto, S.I.; Tamura, M.; Uchiyama, M.; Nakabayashi, K.; Okamura, K.; Yoshida, M.; Tomizawa, D.; Watanabe, A.; Takahashi, H.; et al. Recurrent RARB Translocations in Acute Promyelocytic Leukemia Lacking RARA Translocation. Cancer Res. 2018, 78, 4452–4458. [Google Scholar] [CrossRef]
- Qin, Y.Z.; Huang, X.J.; Zhu, H.H. Identification of a novel CPSF6-RARG fusion transcript in acute myeloid leukemia resembling acute promyelocytic leukemia. Leukemia 2018, 32, 2285–2287. [Google Scholar] [CrossRef]
- Shiba, N.; Yoshida, K.; Hara, Y.; Yamato, G.; Shiraishi, Y.; Matsuo, H.; Okuno, Y.; Chiba, K.; Tanaka, H.; Kaburagi, T.; et al. Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv. 2019, 3, 3157–3169. [Google Scholar] [CrossRef]
- Song, Y.; Hou, J.; Wan, L.; Liu, K.; Zhou, C.; Wei, S.; Zhang, G.; Lin, D.; Li, Y.; Fang, Q.; et al. A short report of novel RARG-HNRNPM fusion gene in resembling acute promyelocytic leukemia. Hematology 2022, 27, 518–522. [Google Scholar] [CrossRef]
- Su, Z.; Liu, X.; Xu, Y.; Hu, W.; Zhao, C.; Zhao, H.; Feng, X.; Zhang, S.; Yang, J.; Shi, X.; et al. Novel reciprocal fusion genes involving HNRNPC and RARG in acute promyelocytic leukemia lacking RARA rearrangement. Haematologica 2020, 105, e376–e378. [Google Scholar] [CrossRef]
- Such, E.; Cervera, J.; Valencia, A.; Barragan, E.; Ibanez, M.; Luna, I.; Fuster, O.; Perez-Sirvent, M.L.; Senent, L.; Sempere, A.; et al. A novel NUP98/RARG gene fusion in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood 2011, 117, 242–245. [Google Scholar] [CrossRef]
- Wang, M.; Lin, H.; Chu, X.; Wang, Z.; Yang, X.; Cen, J.; Shen, H.; Pan, J.; Wang, Y.; Shen, H.; et al. Identification of a recurrent fusion NUP98-RARG in acute myeloid leukaemia resembling acute promyelocytic leukaemia. Br. J. Haematol. 2022, 197, e73–e78. [Google Scholar] [CrossRef]
- Kang, L.C.; Smith, S.V.; Kaiser-Rogers, K.; Rao, K.; Dunphy, C.H. Two cases of acute myeloid leukemia with t(11;17) associated with varying morphology and immunophenotype: Rearrangement of the MLL gene and a region proximal to the RARalpha gene. Cancer Genet. Cytogenet. 2005, 159, 168–173. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, X.; Xu, H.; Li, J.; Yu, W. MLL-rearrangement can resemble acute promyelocytic leukemia. Leuk. Lymphoma 2019, 60, 2841–2843. [Google Scholar] [CrossRef]
- Bruyere, H.; Sutherland, H.; Chipperfield, K.; Hudoba, M. Concomitant and successive amplifications of MYC in APL-like leukemia. Cancer Genet. Cytogenet. 2010, 197, 75–80. [Google Scholar] [CrossRef]
- Sun, J.; Ning, S.; Feng, R.; Li, J.; Wang, T.; Xing, B.; Zhu, X.; Zhao, Y.; Pei, L.; Liu, H. Acute myeloid leukemia with cup-like blasts and FLT3-ITD and NPM1 mutations mimics features of acute promyelocytic leukemia: A case of durable remission after sorafenib and low-dose cytarabine. Anticancer. Drugs 2022, 33, e813–e817. [Google Scholar] [CrossRef]
- Frater, J.L.; Hoover, R.G.; Bernreuter, K.; Batanian, J.R. Deletion of MYC and presence of double minutes with MYC amplification in a morphologic acute promyelocytic leukemia-like case lacking RARA rearrangement: Could early exclusion of double-minute chromosomes be a prognostic factor? Cancer Genet. Cytogenet 2006, 166, 139–145. [Google Scholar] [CrossRef]
- Zhang, X.M.; Chang, Q.; Zeng, L.; Gu, J.; Brown, S.; Basch, R.S. TBLR1 regulates the expression of nuclear hormone receptor co-repressors. BMC Cell Biol. 2006, 7, 31. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Neubauer, K.; Zieger, B. The Mammalian Septin Interactome. Front. Cell Dev. Biol. 2017, 5, 3. [Google Scholar] [CrossRef]
- Cerveira, N.; Micci, F.; Santos, J.; Pinheiro, M.; Correia, C.; Lisboa, S.; Bizarro, S.; Norton, L.; Glomstein, A.; Asberg, A.E.; et al. Molecular characterization of the MLL-SEPT6 fusion gene in acute myeloid leukemia: Identification of novel fusion transcripts and cloning of genomic breakpoint junctions. Haematologica 2008, 93, 1076–1080. [Google Scholar] [CrossRef]
- Wuchter, C.; Harbott, J.; Schoch, C.; Schnittger, S.; Borkhardt, A.; Karawajew, L.; Ratei, R.; Ruppert, V.; Haferlach, T.; Creutzig, U.; et al. Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000, 14, 1232–1238. [Google Scholar] [CrossRef]
- Chen, X.; Cherian, S. Acute Myeloid Leukemia Immunophenotyping by Flow Cytometric Analysis. Clin. Lab. Med. 2017, 37, 753–769. [Google Scholar] [CrossRef]
- Zerkalenkova, E.; Mikhaylova, E.; Lebedeva, S.; Illarionova, O.; Baidun, L.; Kashpor, S.; Osipova, E.; Maschan, M.; Maschan, A.; Novichkova, G.; et al. Quantification of NG2-positivity for the precise prediction of KMT2A gene rearrangements in childhood acute leukemia. Genes Chromosomes Cancer 2021, 60, 88–99. [Google Scholar] [CrossRef]
- Gorczyca, W. Acute promyelocytic leukemia: Four distinct patterns by flow cytometry immunophenotyping. Pol. J. Pathol. 2012, 63, 8–17. [Google Scholar]
- Mikhailova, E.V.; Mochalova, N.S.; Kashpor, S.A.; Zerkalenkova, E.A.; Konyukhova, T.V.; Plyasunova, S.A.; Olshanskaya, Y.V.; Kalinina, I.I.; Maschan, M.A.; Maschan, A.A.; et al. Low specificity of HLA-DR expression for diagnosis of acute promyelocytic leukemia. Pediatr. Hematol. Oncol. Immunopathol. 2022, 21, 42–48. [Google Scholar] [CrossRef]
- Fang, H.; Wang, S.A.; Hu, S.; Konoplev, S.N.; Mo, H.; Liu, W.; Zuo, Z.; Xu, J.; Jorgensen, J.L.; Yin, C.C.; et al. Acute promyelocytic leukemia: Immunophenotype and differential diagnosis by flow cytometry. Cytom. B Clin. Cytom. 2022, 102, 283–291. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Cicconi, L.; Voso, M.T. Progress and criticalities in the management of acute promyelocytic leukemia. Oncotarget 2017, 8, 99221–99222. [Google Scholar] [CrossRef]
- Sobas, M.; Talarn-Forcadell, M.C.; Martinez-Cuadron, D.; Escoda, L.; Garcia-Perez, M.J.; Mariz, J.; Mela-Osorio, M.J.; Fernandez, I.; Alonso-Dominguez, J.M.; Cornago-Navascues, J.; et al. PLZF-RARalpha, NPM1-RARalpha, and Other Acute Promyelocytic Leukemia Variants: The PETHEMA Registry Experience and Systematic Literature Review. Cancers 2020, 12, 1313. [Google Scholar] [CrossRef]
- Testi, A.M.; Biondi, A.; Lo Coco, F.; Moleti, M.L.; Giona, F.; Vignetti, M.; Menna, G.; Locatelli, F.; Pession, A.; Barisone, E.; et al. GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 2005, 106, 447–453. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Hood, B.L.; Conrads, T.P.; Redner, R.L. Extrinsic apoptosis is impeded by direct binding of the APL fusion protein NPM-RAR to TRADD. Mol. Cancer Res. 2014, 12, 1283–1291. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Wen, L.; Xu, Y.; Yao, L.; Wang, N.; Wang, Q.; Liu, T.; Pan, J.; Cen, J.; Zhou, H.; Miao, M.; et al. Clinical and molecular features of acute promyelocytic leukemia with variant retinoid acid receptor fusions. Haematologica 2019, 104, e195. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Zhou, C.; Li, C.; Ru, K.; Rao, Q.; Xing, H.; Tian, Z.; Tang, K.; Mi, Y.; et al. TBLR1 fuses to retinoid acid receptor alpha in a variant t(3;17)(q26;q21) translocation of acute promyelocytic leukemia. Blood 2014, 124, 936–945. [Google Scholar] [CrossRef]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Emerenciano, M.; Meyer, C.; Mansur, M.B.; Marschalek, R.; Pombo-de-Oliveira, M.S.; Brazilian Collaborative Study Group of Infant Acute, L. The distribution of MLL breakpoints correlates with outcome in infant acute leukaemia. Br. J. Haematol. 2013, 161, 224–236. [Google Scholar] [CrossRef]
- Gregory, J.; Kim, H.; Alonzo, T.; Gerbing, R.; Woods, W.; Weinstein, H.; Shepherd, L.; Schiffer, C.; Appelbaum, F.; Willman, C.; et al. Treatment of children with acute promyelocytic leukemia: Results of the first North American Intergroup trial INT0129. Pediatr. Blood Cancer 2009, 53, 1005–1010. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | 57 | 93 | 94 | 172 | 194 | 235 | 242 |
|---|---|---|---|---|---|---|---|
| Age, years | 8 | 14 | 3 | 8 | 0 | 1 | 1 |
| Sex | F | M | F | M | M | F | F |
| WBC * | 79 | 3.4 | 7.1 | 2.2 | 113 | 55 | 60 |
| BM morphology | M3v | M3v | M3v | M3 | M3 | M3 | M3 |
| Atypical promyelocytes | 54.5% | 78% | 94.5% | 89% | 85.5% | 55% | 98.5% |
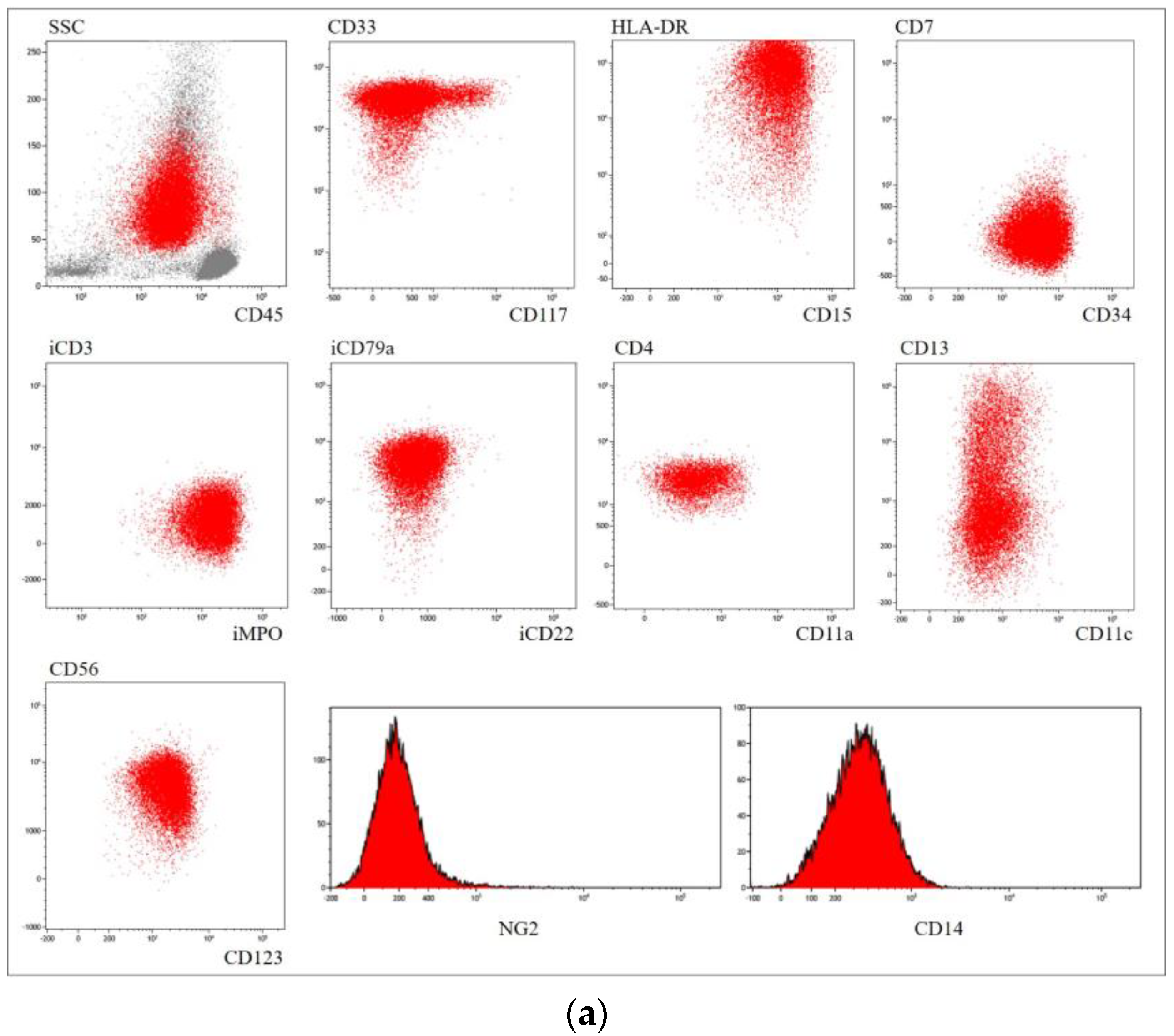
| Immunophenotype | CD33+ CD13+/− CD15+/− CD64+ CD117+ MPO+ CD123+ CD11c+/− | CD33+ CD13+ CD34+ CD2+ CD117+ MPO+ CD123+/− | CD33+ CD34+ CD15+ CD13+/− CD123+ CD56+ MPO+ iCD79a+ HLA−DR+ | CD33+ CD13+ CD117+/− MPO+ CD123+ Lysozyme+/− | CD33+/− CD13+ CD15+/− CD99+ MPO+ CD123+/− Lysozyme+/− | CD33+/− CD13+ CD15+/− CD99+ MPO+ Lysozyme+/− | CD33+ CD13+ CD15+ CD99+ MPO+ |
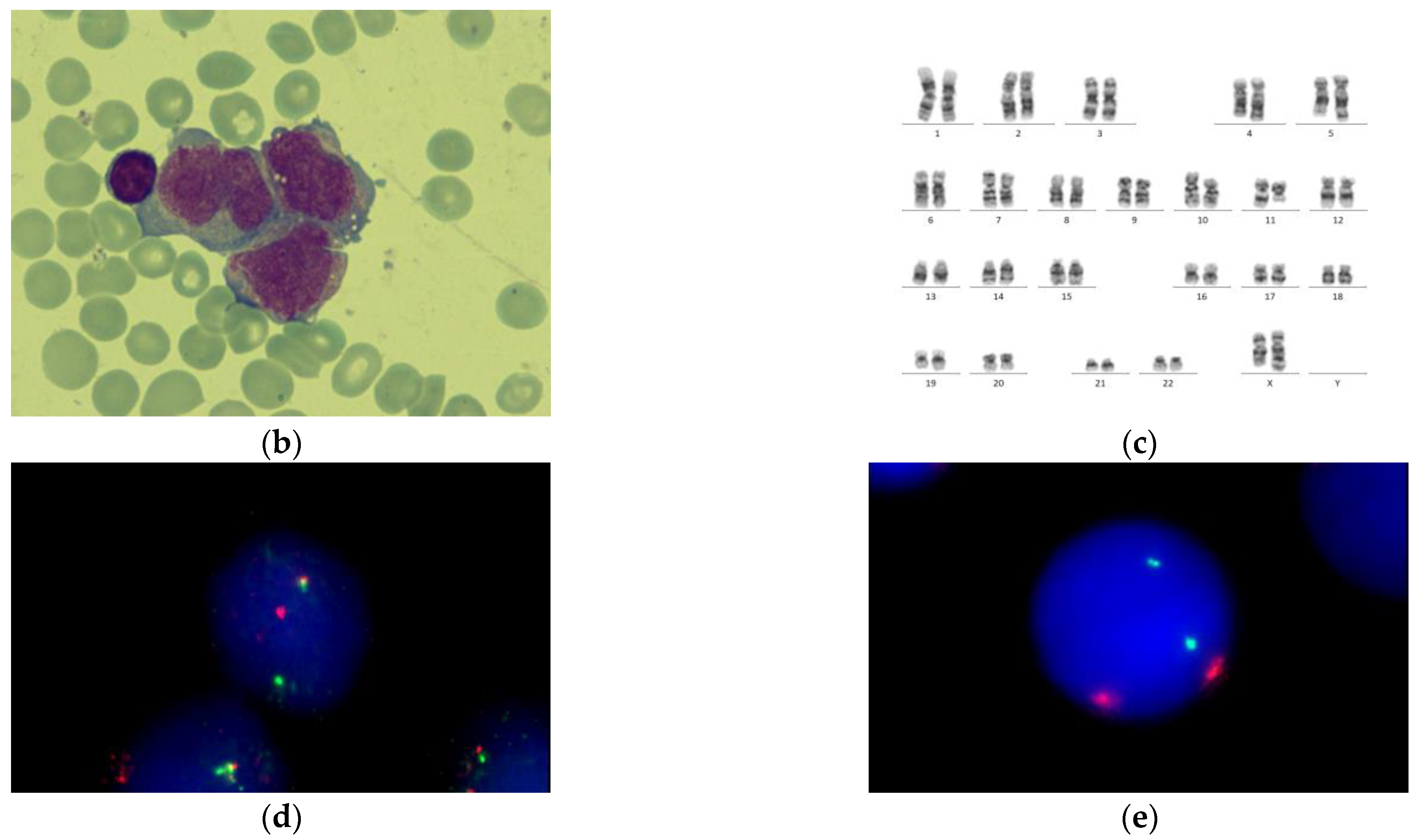
| Cytogenetics | NK ** | der(8)t(8;8)(q24;q12), der(11)t(8;11)(q12;p15) | ins(X;11) (q24;q14q25) | i(17)(q10), del TP53 | NK | NK | NK |
| FISH for t(15;17) | negative | negative | negative | negative | negative | negative | negative |
| RT-PCR for PML::RARA | negative | negative | negative | negative | negative | negative | negative |
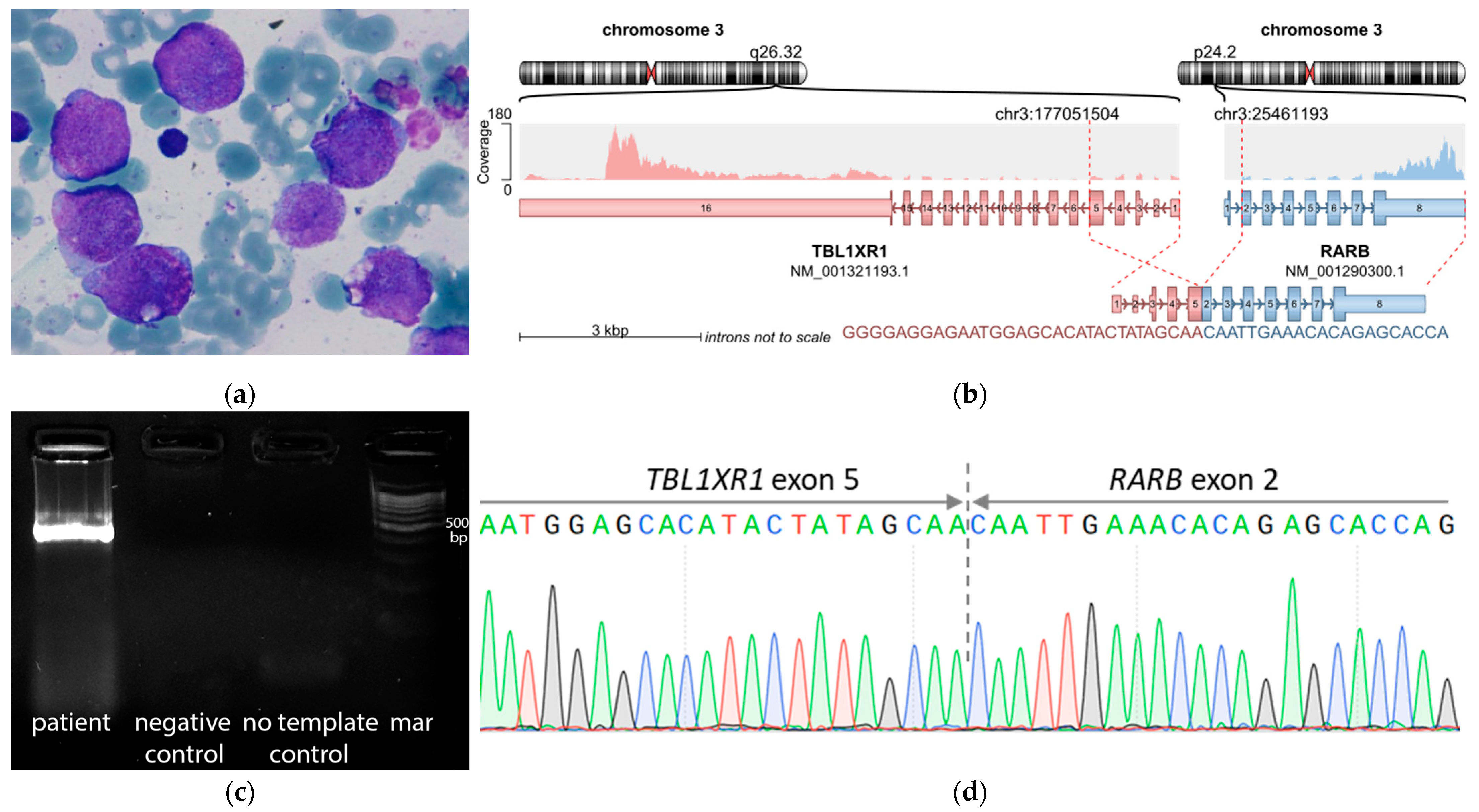
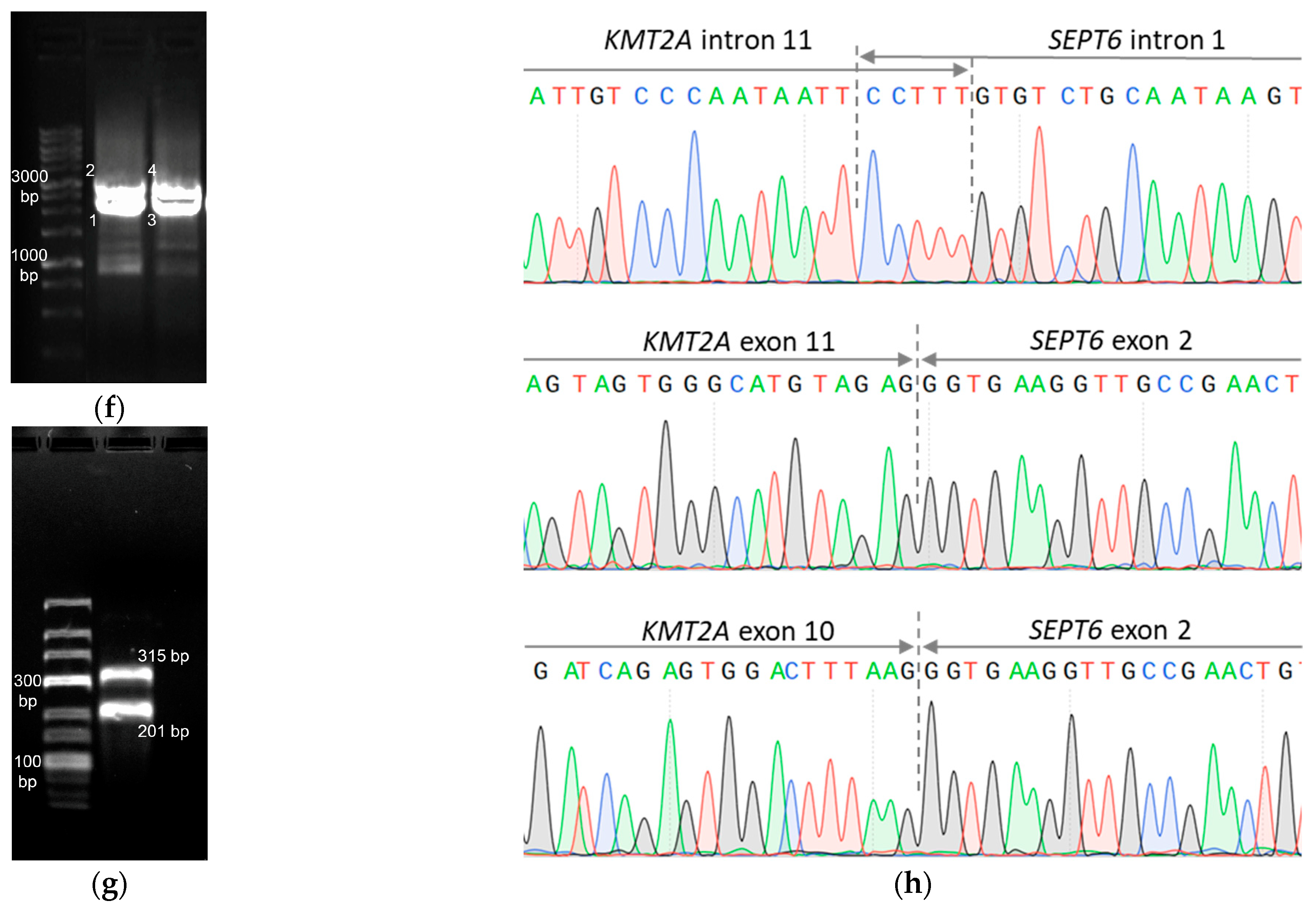
| Fusion gene | no fusions | no fusions | KMT2A::SEPT6 | no fusions | no fusions | no fusions | TBL1XR1::RARB |
| Detected mutations | NPM1 p.W288Cfs*12 FLT3 p.F594_K602dup | - | - | NRAS p.G12R | - | - | - |
| First-line therapy | cytarabine, mitoxantrone, etoposide | ATRA, cytarabine, daunorubicin | ATRA, cytarabine, daunorubicin | ATRA, cytarabine, daunorubicin | ATRA, cytarabine, daunorubicin | ATRA, cytarabine, daunorubicin | ATRA, cytarabine, daunorubicin |
| Follow-up time | 88 months | 30 months | 30 months | 25 months | 31 months | 14 months | 2 months |
| Outcome | Alive in 3rd CR *** | Alive in 3rd CR | Reached 1st CR, died of relapse | Alive in 1st CR | Alive in 1st CR | Alive in 1st CR | Reached 1st CR, lost for follow-up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borkovskaia, A.; Bogacheva, S.; Konyukhova, T.; Dadakhanova, E.; Gaskova, M.; Soldatkina, O.; Dubrovina, M.; Popov, A.; Mikhailova, E.; Inushkina, E.; et al. Molecular Heterogeneity of Pediatric AML with Atypical Promyelocytes Accumulation in Children—A Single Center Experience. Genes 2023, 14, 675. https://doi.org/10.3390/genes14030675
Borkovskaia A, Bogacheva S, Konyukhova T, Dadakhanova E, Gaskova M, Soldatkina O, Dubrovina M, Popov A, Mikhailova E, Inushkina E, et al. Molecular Heterogeneity of Pediatric AML with Atypical Promyelocytes Accumulation in Children—A Single Center Experience. Genes. 2023; 14(3):675. https://doi.org/10.3390/genes14030675
Chicago/Turabian StyleBorkovskaia, Aleksandra, Sofia Bogacheva, Tatiana Konyukhova, Elina Dadakhanova, Marina Gaskova, Olga Soldatkina, Maria Dubrovina, Alexander Popov, Ekaterina Mikhailova, Evgenia Inushkina, and et al. 2023. "Molecular Heterogeneity of Pediatric AML with Atypical Promyelocytes Accumulation in Children—A Single Center Experience" Genes 14, no. 3: 675. https://doi.org/10.3390/genes14030675