
CNN_FunBar: Advanced Learning Technique for Fungi ITS Region Classification

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Barcode Datasets
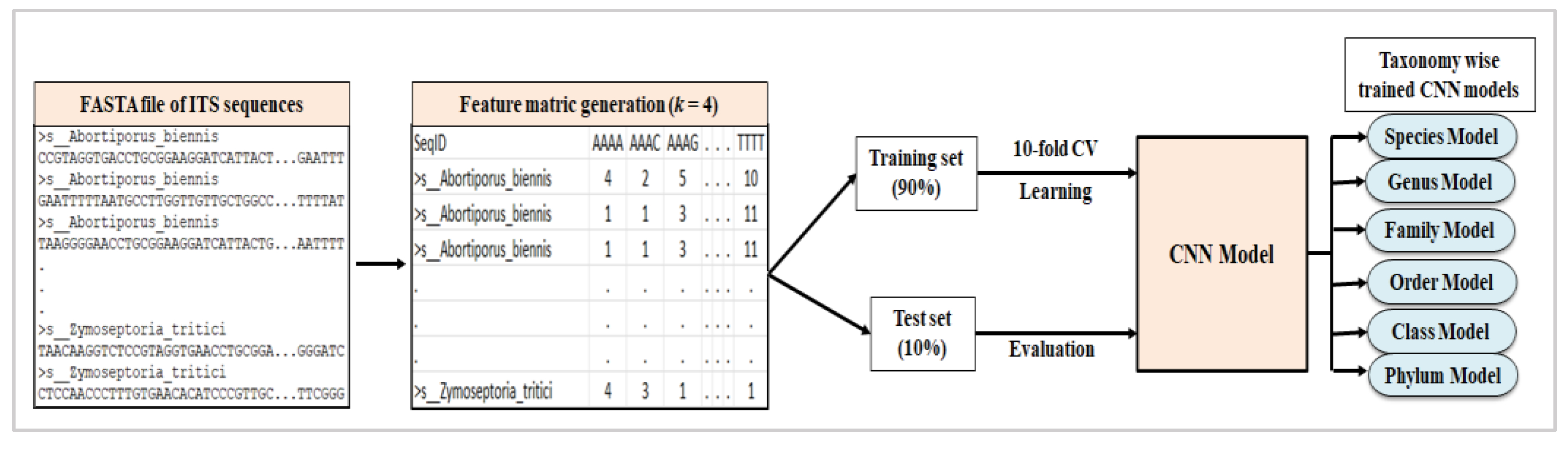
2.2. Feature Vector Generation
2.3. Supervised Classifiers
2.3.1. Convolutional Neural Network (CNN)
2.3.2. Support Vector Machine (SVM)
2.3.3. K-Nearest Neighbor (KNN)
2.3.4. Naïve-Bayes Method
2.3.5. Random Forest (RF)
2.3.6. RDP Classifier
2.4. Training and Evaluation
2.5. Comparison of CNN with Existing Fungi ITS Classification Software
2.6. Implementation Details
- CPU: 64-bit Intel(R)-Xeon(R), 2.9 GHz, 1 TB;
- RAM: 128 GB;
- OS: Linux RedHat.
3. Results and Discussion
3.1. Effect of Convolution Kernel Size and Filter Numbers on CNN Model Performance
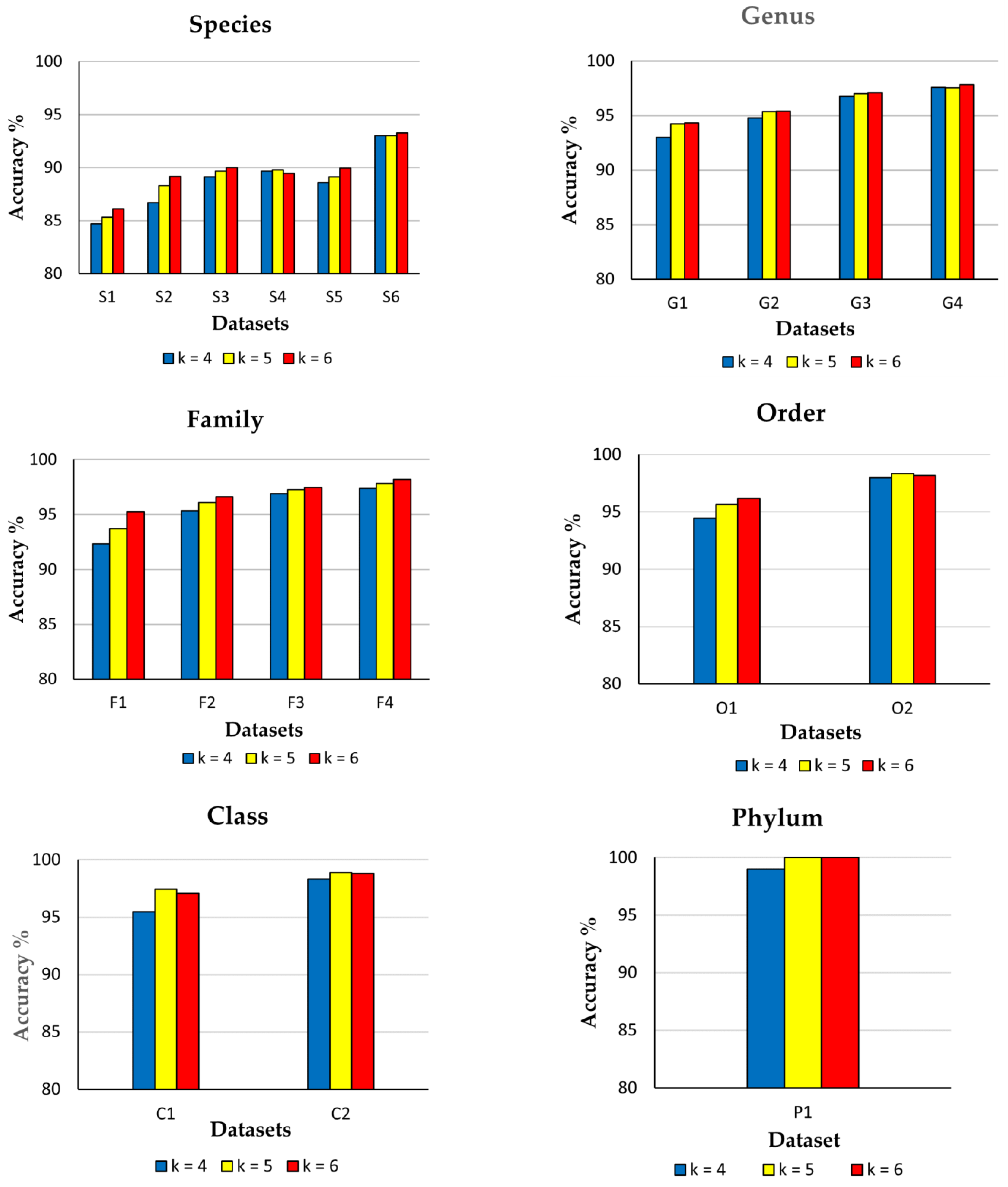
3.2. Impact of Diversity and k-mer Sizes on CNN Model Performance
3.3. Classification Performance Analysis Based on Varying Class Frequencies
3.4. Comparison among Classification Performances of CNN and Other Machine Learning Algorithms
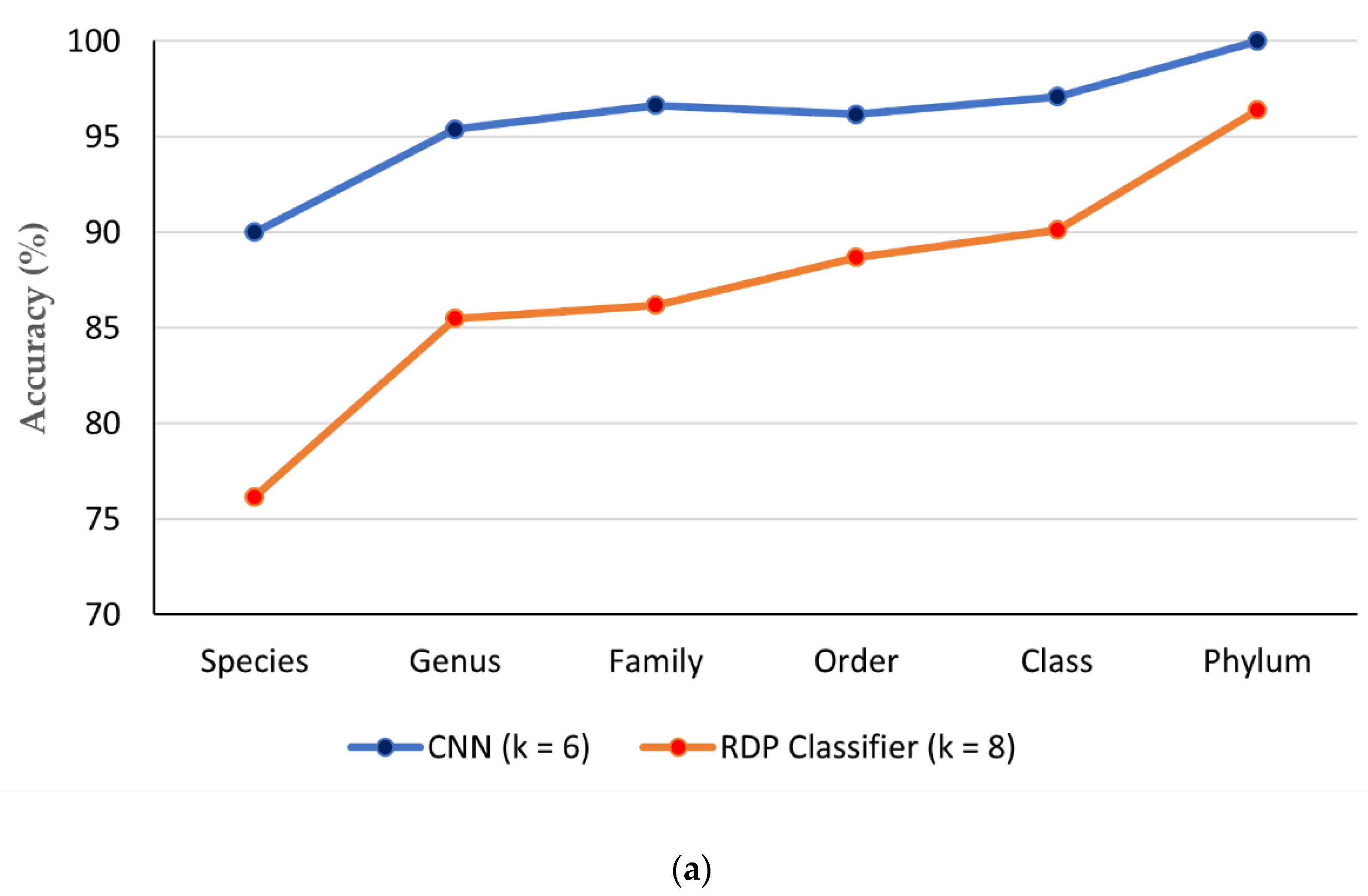
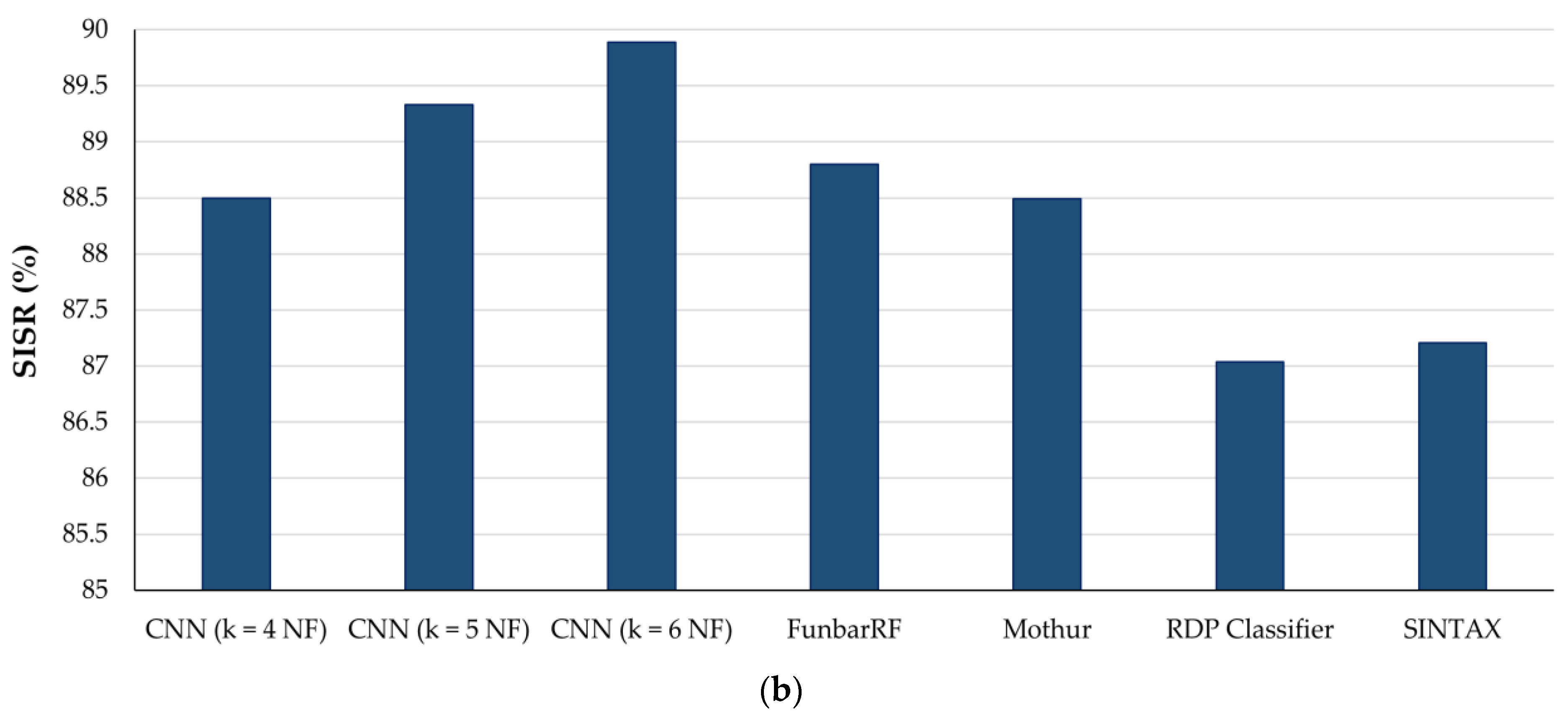
3.5. Comparison of CNN with Existing ITS Classification Software
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferreira, V.; Elosegi, A.; Tiegs, S.D.; Von Schiller, D.; Young, R. Organic Matter Decomposition and Ecosystem Metabolism as Tools to Assess the Functional Integrity of Streams and Rivers—A Systematic Review. Water 2020, 12, 3523. [Google Scholar] [CrossRef]
- Bhattacharyya, S.S.; Ros, G.H.; Furtak, K.; Iqbal, H.M.N.; Parra-Saldívar, R. Soil carbon sequestration—An interplay between soil microbial community and soil organic matter dynamics. Sci. Total. Environ. 2022, 815, 152928. [Google Scholar] [CrossRef] [PubMed]
- Chukwuneme, C.; Ayangbenro, A.; Babalola, O. Metagenomic Analyses of Plant Growth-Promoting and Carbon-Cycling Genes in Maize Rhizosphere Soils with Distinct Land-Use and Management Histories. Genes 2021, 12, 1431. [Google Scholar] [CrossRef] [PubMed]
- Enebe, M.; Babalola, O. The Influence of Soil Fertilization on the Distribution and Diversity of Phosphorus Cycling Genes and Microbes Community of Maize Rhizosphere Using Shotgun Metagenomics. Genes 2021, 12, 1022. [Google Scholar] [CrossRef] [PubMed]
- Aasfar, A.; Bargaz, A.; Yaakoubi, K.; Hilali, A.; Bennis, I.; Zeroual, Y.; Kadmiri, I.M. Nitrogen Fixing Azotobacter Species as Potential Soil Biological Enhancers for Crop Nutrition and Yield Stability. Front. Microbiol. 2021, 12, 628379. [Google Scholar] [CrossRef] [PubMed]
- Bloch, S.E.; Ryu, M.-H.; Ozaydin, B.; Broglie, R. Harnessing atmospheric nitrogen for cereal crop production. Curr. Opin. Biotechnol. 2020, 62, 181–188. [Google Scholar] [CrossRef]
- Dixit, R.; Wasiullah; Malaviya, D.; Pandiyan, K.; Singh, U.B.; Sahu, A.; Shukla, R.; Singh, B.P.; Rai, J.P.; Sharma, P.K.; et al. Bioremediation of Heavy Metals from Soil and Aquatic Environment: An Overview of Principles and Criteria of Fundamental Processes. Sustainability 2015, 7, 2189–2212. [Google Scholar] [CrossRef] [Green Version]
- Behera, B.K.; Chakraborty, H.J.; Patra, B.; Rout, A.K.; Dehury, B.; Das, B.K.; Sarkar, D.J.; Parida, P.K.; Raman, R.K.; Rao, A.R.; et al. Metagenomic Analysis Reveals Bacterial and Fungal Diversity and Their Bioremediation Potential from Sediments of River Ganga and Yamuna in India. Front. Microbiol. 2020, 11, 556136. [Google Scholar] [CrossRef]
- Behera, B.K.; Das, A.; Sarkar, D.J.; Weerathunge, P.; Parida, P.K.; Das, B.K.; Thavamani, P.; Ramanathan, R.; Bansal, V. Polycyclic Aromatic Hydrocarbons (PAHs) in inland aquatic ecosystems: Perils and remedies through biosensors and bioremediation. Environ. Pollut. 2018, 241, 212–233. [Google Scholar] [CrossRef]
- Pal, A.K.; Singh, J.; Soni, R.; Tripathi, P.; Kamle, M.; Tripathi, V.; Kumar, P. The role of microorganism in bioremediation for sustainable environment management. In Bioremediation of Pollutants; Elsevier: Amsterdam, The Netherlands, 2020; pp. 227–249. [Google Scholar] [CrossRef]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.-E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef] [Green Version]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieder, R.; Edwards, R. Insights into antibiotic resistance through metagenomic approaches. Futur. Microbiol. 2012, 7, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Igiehon, N.; Babalola, O. Rhizosphere Microbiome Modulators: Contributions of Nitrogen Fixing Bacteria towards Sustainable Agriculture. Int. J. Environ. Res. Public Health 2018, 15, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.; Chiu, C. The Role of Metagenomics and Next-Generation Sequencing in Infectious Disease Diagnosis. Clin. Chem. 2021, 68, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.-Y.; Yang, J.; McDermaid, A.; Zhao, J.; Kang, Y.; Ma, Q. Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes. Brief. Bioinform. 2017, 19, 1415–1429. [Google Scholar] [CrossRef] [Green Version]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Briefings Bioinform. 2017, 20, 1125–1136. [Google Scholar] [CrossRef]
- Navgire, G.S.; Goel, N.; Sawhney, G.; Sharma, M.; Kaushik, P.; Mohanta, Y.K.; Mohanta, T.K.; Al-Harrasi, A. Analysis and Interpretation of metagenomics data: An approach. Biol. Proced. Online 2022, 24, 18. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef]
- Tonge, D.P.; Pashley, C.H.; Gant, T.W. Amplicon –Based Metagenomic Analysis of Mixed Fungal Samples Using Proton Release Amplicon Sequencing. PLoS ONE 2014, 9, e93849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbareche, H.; Veillette, M.; Bilodeau, G.; Duchaine, C. Comparison of the performance of ITS1 and ITS2 as barcodes in amplicon-based sequencing of bioaerosols. Peerj 2020, 8, e8523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleifer, K.H. Classification of Bacteria and Archaea: Past, present and future. Syst. Appl. Microbiol. 2009, 32, 533–542. [Google Scholar] [CrossRef]
- Chakraborty, C.; Doss, C.G.P.; Patra, B.C.; Bandyopadhyay, S. DNA barcoding to map the microbial communities: Current advances and future directions. Appl. Microbiol. Biotechnol. 2014, 98, 3425–3436. [Google Scholar] [CrossRef]
- Igiehon, N.O.; Babalola, O.O. Below-ground-above-ground Plant-microbial Interactions: Focusing on Soybean, Rhizobacteria and Mycorrhizal Fungi. Open Microbiol. J. 2018, 12, 261–279. [Google Scholar] [CrossRef]
- Hol, W.H.G.; Bezemer, T.M.; Biere, A. Getting the ecology into interactions between plants and the plant growth-promoting bacterium Pseudomonas fluorescens. Front. Plant Sci. 2013, 4, 81. [Google Scholar] [CrossRef] [Green Version]
- Hartley, S.E.; Gange, A.C. Impacts of Plant Symbiotic Fungi on Insect Herbivores: Mutualism in a Multitrophic Context. Annu. Rev. Entomol. 2009, 54, 323–342. [Google Scholar] [CrossRef]
- Martin, F.M.; Uroz, S.; Barker, D.G. Ancestral alliances: Plant mutualistic symbioses with fungi and bacteria. Science 2017, 356, eaad4501. [Google Scholar] [CrossRef]
- Lindahl, B.D.; Tunlid, A. Ectomycorrhizal fungi—Potential organic matter decomposers, yet not saprotrophs. New Phytol. 2015, 205, 1443–1447. [Google Scholar] [CrossRef]
- Singh, T.; Singh, A.P. White and Brown Rot Fungi as Decomposers of Lignocellulosic Materials and Their Role in Waste and Pollution Control. In Fungal Applications in Sustainable Environmental Biotechnology; Springer: Cham, Switzerland, 2016; pp. 233–247. [Google Scholar] [CrossRef]
- Kück, U.; Bloemendal, S.; Teichert, I. Putting Fungi to Work: Harvesting a Cornucopia of Drugs, Toxins, and Antibiotics. PLoS Pathog. 2014, 10, e1003950. [Google Scholar] [CrossRef] [PubMed]
- Silber, J.; Kramer, A.; Labes, A.; Tasdemir, D. From Discovery to Production: Biotechnology of Marine Fungi for the Production of New Antibiotics. Mar. Drugs 2016, 14, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCartney, H.A.; Foster, S.J.; Fraaije, B.A.; Ward, E. Molecular diagnostics for fungal plant pathogens. Pest Manag. Sci. 2003, 59, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Thambugala, K.M.; Daranagama, D.A.; Phillips, A.J.L.; Kannangara, S.D.; Promputtha, I. Fungi vs. Fungi in Biocontrol: An Overview of Fungal Antagonists Applied Against Fungal Plant Pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 604923. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.Á.; Gabaldón, T. Fungal evolution: Diversity, taxonomy and phylogeny of the Fungi. Biol. Rev. 2019, 94, 2101–2137. [Google Scholar] [CrossRef] [PubMed]
- Grządziel, J.; Gałązka, A. Fungal Biodiversity of the Most Common Types of Polish Soil in a Long-Term Microplot Experiment. Front. Microbiol. 2019, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, L.G.; Szöllősi, G. Fungal Phylogeny in the Age of Genomics: Insights into Phylogenetic Inference from Genome-Scale Datasets. Adv. Genet. 2017, 100, 49–72. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toju, H.; Tanabe, A.; Yamamoto, S.; Sato, H. High-Coverage ITS Primers for the DNA-Based Identification of Ascomycetes and Basidiomycetes in Environmental Samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef] [Green Version]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, V.; Wang, Q.; Greenfield, P.; Charleston, M.; Porras-Alfaro, A.; Kuske, C.R.; Cole, J.R.; Midgley, D.J.; Tran-Dinh, N. Fungal identification using a Bayesian classifier and the Warcup training set of internal transcribed spacer sequences. Mycologia 2016, 108, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D.N. BARCODING: BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Serrano, L.; Restrepo, S.; Bustos, J.R.; Zambrano, M.M.; Anzola, J.M. Mycofier: A new machine learning-based classifier for fungal ITS sequences. BMC Res. Notes 2016, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. SINTAX: A Simple Non-Bayesian Taxonomy Classifier for 16S and ITS Sequences. bioRxiv 2016, 074161. [Google Scholar] [CrossRef] [Green Version]
- Meher, P.K.; Sahu, T.K.; Gahoi, S.; Tomar, R.; Rao, A.R. funbarRF: DNA barcode-based fungal species prediction using multiclass Random Forest supervised learning model. BMC Genet. 2019, 20, 2. [Google Scholar] [CrossRef]
- Jing, Y.; Bian, Y.; Hu, Z.; Wang, L.; Xie, X.-Q.S. Deep Learning for Drug Design: An Artificial Intelligence Paradigm for Drug Discovery in the Big Data Era. AAPS J. 2018, 20, 58. [Google Scholar] [CrossRef]
- Kotsiopoulos, T.; Sarigiannidis, P.; Ioannidis, D.; Tzovaras, D. Machine Learning and Deep Learning in smart manufacturing: The Smart Grid paradigm. Comput. Sci. Rev. 2021, 40, 100341. [Google Scholar] [CrossRef]
- Fiannaca, A.; La Paglia, L.; La Rosa, M.; Lo Bosco, G.; Renda, G.; Rizzo, R.; Gaglio, S.; Urso, A. Deep learning models for bacteria taxonomic classification of metagenomic data. BMC Bioinform. 2018, 19, 198. [Google Scholar] [CrossRef]
- Pagès, H.; Aboyoun, P.; Gentleman, R.; DebRoy, S. Biostrings: Efficient Manipulation of Biological Strings. 2022. Available online: https://bioconductor.org/packages/Biostrings.%0A%0A (accessed on 25 October 2022).
- Pedregosa, F.; Gaël, V.; Alexandre, G.; Vincent, M.; Bertrand, T.; Olivier, G.; Mathieu, B.; Al, E. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Zhang, W.; Peng, G.; Li, C.; Chen, Y.; Zhang, Z. A New Deep Learning Model for Fault Diagnosis with Good Anti-Noise and Domain Adaptation Ability on Raw Vibration Signals. Sensors 2017, 17, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.; Hinton, G.E. Rectified Linear Units Improve Restricted Boltzmann Machines. In Proceedings of the 27th international conference on machine learning (ICML-10), Haifa, Israel, 21–24 June 2010; pp. 807–814. [Google Scholar] [CrossRef]
- Lecun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef] [Green Version]
- Lücke, J.; Sahani, M. Generalized Softmax Networks for Non-linear Component Extraction. In Artificial Neural Networks—ICANN 2007; Springer: Berlin/Heidelberg, Germany, 2007; pp. 657–667. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Zhang, Z.; Sabuncu, M.R. Generalized Cross Entropy Loss for Training Deep Neural Networks with Noisy Labels. arXiv 2018, arXiv:1805.07836. [Google Scholar]
- Vapnik, V. The Nature of Statistical Learning Theory; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1995. [Google Scholar]
- Crisci, C.; Ghattas, B.; Perera, G. A review of supervised machine learning algorithms and their applications to ecological data. Ecol. Model. 2012, 240, 113–122. [Google Scholar] [CrossRef]
- Hota, S.; Pathak, S. KNN classifier based approach for multi-class sentiment analysis of twitter data. Int. J. Eng. Technol. 2018, 7, 1372. [Google Scholar] [CrossRef] [Green Version]
- Schulman, P. Bayes’ Theorem—A Review. Cardiol. Clin. 1984, 2, 319–328. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Yacouby, R.; Axman, D. Probabilistic Extension of Precision, Recall, and F1 Score for More Thorough Evaluation of Classification Models. In Proceedings of the First Workshop on Evaluation and Comparison of NLP Systems, Online, 20 November 2020; Association for Computational Linguistics: Stroudsburg, PA, USA, 2020; pp. 79–91. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triantaphyllou, E. Multi-criteria decision making methods. In Multi-Criteria Decision Making Methods: A Comparative Study; Springer: Boston, MA, USA, 2000; pp. 5–21. [Google Scholar] [CrossRef]
- Lai, Y.-J.; Liu, T.-Y.; Hwang, C.-L. TOPSIS for MODM. Eur. J. Oper. Res. 1994, 76, 486–500. [Google Scholar] [CrossRef]
- Yazdi, M.M. Topsis: TOPSIS Method for Multiple-Criteria Decision Making (MCDM). Available online: https://cran.r-project.org/web/packages/topsis/topsis.pdf (accessed on 16 November 2022).
- Gulli, A.; Pal, S. Deep Learning with Keras; Packt Publishing Ltd.: Birmingham, UK, 2017. [Google Scholar]
- TensorFlow. 2021. Available online: https://imagej.net/libs/tensorflow (accessed on 20 October 2022).
- Vu, D.; Groenewald, M.; Verkley, G. Convolutional neural networks improve fungal classification. Sci. Rep. 2020, 10, 12628. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Class | Order | Family | Genus | Species | Total ITS Sequences |
|---|---|---|---|---|---|---|
| 2 | 49 | 274 | 1049 | 6367 | 44,167 | 4,504,529 |
| Taxonomic Level | Datasets | No. of Unique Categories | No. of Sequence per Category | Total No. of ITS Sequences |
|---|---|---|---|---|
| Species | S1 | 2752 | 20 | 55,040 |
| S2 | 939 | 50 | 46,950 | |
| S3 | 429 | 100 | 42,900 | |
| S4 | 148 | 250 | 37,000 | |
| S5 | 117 | 300 | 35,100 | |
| S6 | 64 | 500 | 32,000 | |
| Genus | G1 | 1682 | 50 | 84,100 |
| G2 | 1293 | 100 | 129,300 | |
| G3 | 841 | 250 | 210,250 | |
| G4 | 580 | 500 | 290,000 | |
| Family | F1 | 520 | 50 | 26,000 |
| F2 | 453 | 100 | 45,300 | |
| F3 | 369 | 250 | 92,250 | |
| F4 | 301 | 500 | 150,500 | |
| Order | O1 | 149 | 100 | 14,900 |
| O2 | 106 | 500 | 53,000 | |
| Class | C1 | 31 | 100 | 3100 |
| C2 | 25 | 500 | 12,500 | |
| Phylum | P1 | 2 | 500 | 1000 |
| Taxonomic Level | No. of Unique Categories | No. of Sequence per Category | Total No. of ITS Sequences |
|---|---|---|---|
| Species | 64 | 100 | 6400 |
| 250 | 16,000 | ||
| 500 | 32,000 | ||
| Genus | 580 | 100 | 58,000 |
| 250 | 145,000 | ||
| 500 | 290,000 | ||
| Family | 301 | 100 | 30,100 |
| 250 | 75,250 | ||
| 500 | 150,500 | ||
| Order | 106 | 100 | 10,600 |
| 250 | 26,500 | ||
| 500 | 53,000 | ||
| Class | 25 | 100 | 2500 |
| 250 | 6250 | ||
| 500 | 12,500 |
| CNN Model | Accuracy (%) | Precision (%) | Recall (%) | F1 Score (%) | MCC Score (%) | ||
|---|---|---|---|---|---|---|---|
| Name | Kernel Size (m) | No. of Filters in 1st and 2nd Convolution Layers (n1, n2) | |||||
| CNN-1 | 5 | 5, 10 | 87.88 | 87.74 | 87.86 | 87.84 | 87.85 |
| CNN-2 | 3 | 16, 32 | 88.39 | 89.10 | 88.37 | 88.23 | 88.37 |
| CNN-3 | 3 | 32, 64 | 89.14 | 89.78 | 89.13 | 89.01 | 89.11 |
| CNN-4 | 3 | 64, 128 | 88.16 | 88.69 | 88.15 | 87.98 | 88.13 |
| Taxonomic Level | Datasets | Total No. of Levels | Accuracy (%) for Different k-mer Sizes | ||
|---|---|---|---|---|---|
| k = 4 | k = 5 | k = 6 | |||
| Species | S1 | 2752 | 84.70 | 85.34 | 86.12 |
| S2 | 939 | 86.69 | 88.31 | 89.18 | |
| S3 | 429 | 89.14 | 89.67 | 90.00 | |
| S4 | 148 | 89.65 | 89.81 | 89.46 | |
| S5 | 117 | 88.60 | 89.14 | 89.94 | |
| S6 | 64 | 93.03 | 93.03 | 93.28 | |
| Genus | G1 | 1682 | 92.98 | 94.23 | 94.33 |
| G2 | 1293 | 94.78 | 95.37 | 95.39 | |
| G3 | 841 | 96.74 | 97.00 | 97.10 | |
| G4 | 580 | 97.59 | 97.54 | 97.83 | |
| Family | F1 | 520 | 92.35 | 93.73 | 95.23 |
| F2 | 453 | 95.32 | 96.11 | 96.64 | |
| F3 | 369 | 96.89 | 97.25 | 97.49 | |
| F4 | 301 | 97.37 | 97.82 | 98.21 | |
| Order | O1 | 149 | 94.43 | 95.64 | 96.17 |
| O2 | 106 | 98.00 | 98.33 | 98.19 | |
| Class | C1 | 31 | 95.48 | 97.42 | 97.09 |
| C2 | 25 | 98.32 | 98.88 | 98.80 | |
| Phylum | P1 | 2 | 99.00 | 100.00 | 100.00 |
| Taxonomic Level (N = No. of Unique Categories) | Sequence per Category | Average Values of Performance Metrics (%) | ||||
|---|---|---|---|---|---|---|
| Accuracy | Precision | Recall | F1 | MCC | ||
| Species (N = 64) | 100 | 91.87 | 92.25 | 91.86 | 91.67 | 91.76 |
| 250 | 91.31 | 91.37 | 91.31 | 91.23 | 91.18 | |
| 500 | 93.28 | 93.36 | 93.28 | 93.22 | 93.18 | |
| Genus (N = 580) | 100 | 96.48 | 96.76 | 96.48 | 96.44 | 96.48 |
| 250 | 97.18 | 97.32 | 97.18 | 97.17 | 97.17 | |
| 500 | 97.83 | 97.88 | 97.83 | 97.83 | 97.83 | |
| Family (N = 301) | 100 | 96.44 | 96.69 | 96.44 | 96.43 | 96.43 |
| 250 | 97.42 | 97.55 | 97.42 | 97.42 | 97.41 | |
| 500 | 98.21 | 98.24 | 98.20 | 98.20 | 98.20 | |
| Order (N = 106) | 100 | 96.70 | 97.10 | 96.69 | 96.66 | 96.67 |
| 250 | 97.32 | 97.48 | 97.32 | 97.32 | 97.30 | |
| 500 | 98.19 | 98.25 | 98.19 | 98.19 | 98.17 | |
| Class (N = 25) | 100 | 97.60 | 97.84 | 97.60 | 97.63 | 97.51 |
| 250 | 98.40 | 98.52 | 98.41 | 98.42 | 98.34 | |
| 500 | 98.80 | 98.86 | 98.80 | 98.80 | 98.75 | |
| Taxonomic Level (N = No. of Unique Categories) | Value of k | Average Accuracy (%) | ||||
|---|---|---|---|---|---|---|
| CNN | SVM | KNN | Naïve-Bayes | Random Forest | ||
| Species (N = 429) | 4 | 89.14 | 85.8 | 86.71 | 74.68 | 89.12 |
| 5 | 89.67 | 87.35 | 88.57 | 75.17 | 89.34 | |
| 6 | 90.00 | 88.02 | 88.58 | 76.01 | 89.68 | |
| Genus (N = 1293) | 4 | 94.78 | 92.65 | 93.1 | 83.33 | 94.57 |
| 5 | 95.37 | 92.98 | 93.52 | 84.00 | 95.01 | |
| 6 | 95.39 | 93.07 | 93.64 | 84.12 | 95.17 | |
| Family (N = 453) | 4 | 95.32 | 94.13 | 93.07 | 85.29 | 94.56 |
| 5 | 96.11 | 95.22 | 94.15 | 85.79 | 95.58 | |
| 6 | 96.64 | 95.31 | 93.71 | 85.91 | 96.64 | |
| Order (N = 149) | 4 | 94.43 | 92.69 | 91.54 | 85.40 | 94.16 |
| 5 | 95.64 | 94.02 | 93.69 | 87.00 | 95.63 | |
| 6 | 96.17 | 95.27 | 93.73 | 88.54 | 96.04 | |
| Class (N = 31) | 4 | 95.48 | 93.62 | 92.52 | 88.38 | 95.45 |
| 5 | 97.42 | 95.19 | 93.44 | 89.16 | 97.06 | |
| 6 | 97.09 | 95.84 | 92.11 | 89.29 | 97.38 | |
| Phylum (N = 2) | 4 | 99.00 | 98.88 | 99.00 | 95.00 | 99.00 |
| 5 | 100.00 | 99.55 | 99.01 | 96.00 | 99.00 | |
| 6 | 100.00 | 99.55 | 100.00 | 96.52 | 100.00 | |
| Taxonomic Level | Model | Score | Rank |
|---|---|---|---|
| Species | CNN | 1.0000 | 1 |
| SVM | 0.1701 | 4 | |
| KNN | 0.2335 | 3 | |
| Random Forest | 0.7877 | 2 | |
| Genus | CNN | 0.9978 | 1 |
| SVM | 0.1201 | 4 | |
| KNN | 0.2171 | 3 | |
| Random Forest | 0.9043 | 2 | |
| Family | CNN | 1.0000 | 1 |
| SVM | 0.6319 | 3 | |
| KNN | 0.2310 | 4 | |
| Random Forest | 0.9939 | 2 | |
| Order | CNN | 1.0000 | 1 |
| SVM | 0.6253 | 3 | |
| KNN | 0.2011 | 4 | |
| Random Forest | 0.9204 | 2 | |
| Class | CNN | 0.9418 | 2 |
| SVM | 0.6928 | 3 | |
| KNN | 0.3414 | 4 | |
| Random Forest | 1.0000 | 1 | |
| Phylum | CNN | 1.0000 | 1 |
| SVM | 0.7824 | 2 | |
| KNN | 1.0000 | 1 | |
| Random Forest | 1.0000 | 1 |
| Classifier | Taxonomic Levels | |||||
|---|---|---|---|---|---|---|
| Species | Genus | Family | Order | Class | Phylum | |
| CNN (k = 6) | 90 | 95.39 | 96.64 | 96.17 | 97.09 | 100 |
| RDP Classifier (k = 8) | 76.18 | 85.50 | 86.20 | 88.69 | 90.13 | 96.41 |
| Algorithm/ Software | Feature Type | Avg. Accuracy (%) | Correct Predictions | SISR (%) |
|---|---|---|---|---|
| k = 4 NF | 89.21 | 12,063 | 88.50 | |
| CNN | k = 5 NF | 90.75 | 12,176 | 89.33 |
| k = 6 NF | 91.27 | 12,252 | 89.89 | |
| FunbarRF (Random Forest) | g-spaced di-nucleotide features (g = 1 + 2 + 3 + 4 + 5) | ~89 | 12,104 | 88.80 |
| Mothur (KNN) | k = 8 NF | ~89 | 12,062 | 88.49 |
| RDP Classifier (Naïve-Bayes) | k = 8 NF | ~87 | 11,864 | 87.04 |
| SINTAX (Non-Bayesian) | k = 8 NF | ~87 | 11,887 | 87.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, R.; Rai, A.; Mishra, D.C. CNN_FunBar: Advanced Learning Technique for Fungi ITS Region Classification. Genes 2023, 14, 634. https://doi.org/10.3390/genes14030634
Das R, Rai A, Mishra DC. CNN_FunBar: Advanced Learning Technique for Fungi ITS Region Classification. Genes. 2023; 14(3):634. https://doi.org/10.3390/genes14030634
Chicago/Turabian StyleDas, Ritwika, Anil Rai, and Dwijesh Chandra Mishra. 2023. "CNN_FunBar: Advanced Learning Technique for Fungi ITS Region Classification" Genes 14, no. 3: 634. https://doi.org/10.3390/genes14030634