
Unravelling the Evolutionary Dynamics of High-Risk Klebsiella pneumoniae ST147 Clones: Insights from Comparative Pangenome Analysis
 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Reconstruction, Finishing and Quality Assessment of KP_SDL79
2.2. Acquisition of Genomic Sequences from Public Databases and Data Analysis
2.3. Genome Similarity Estimation
2.4. Pangenome Analysis and Functional Annotation
2.5. SNP Analysis and Phylogeny Reconstruction
2.6. BEAST Analysis
2.7. Association of AMR, Virulence Determinants and Plasmids
2.8. Genome Mining and Identification of Essential Genomic Elements
2.9. Statistical Analysis
3. Results
3.1. Improvement of Draft Assembly of KP_SDL79
3.2. Dataset Description and Genome Statistics
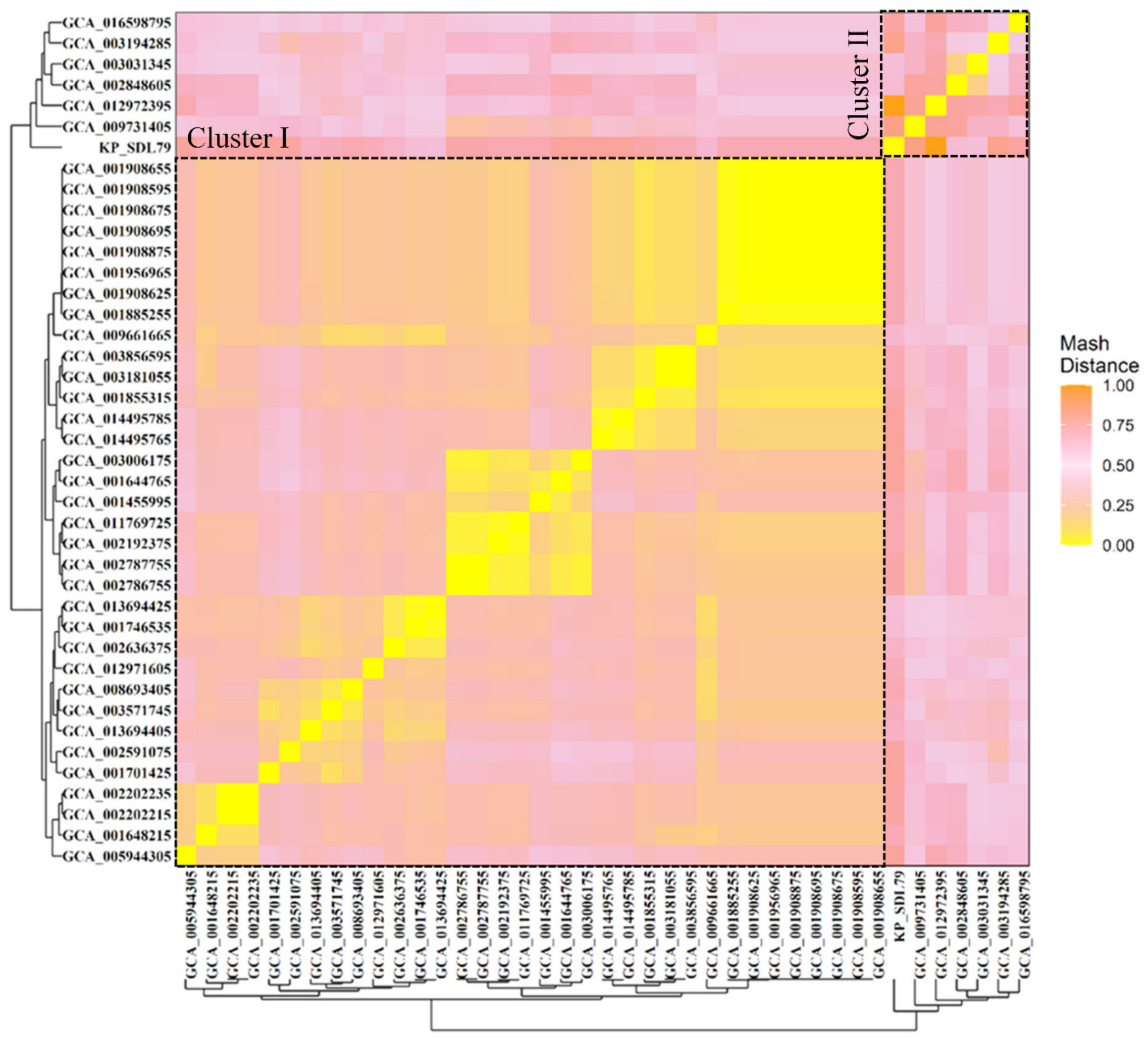
3.3. Genomic Relatedness Analysis Using Mash Distance
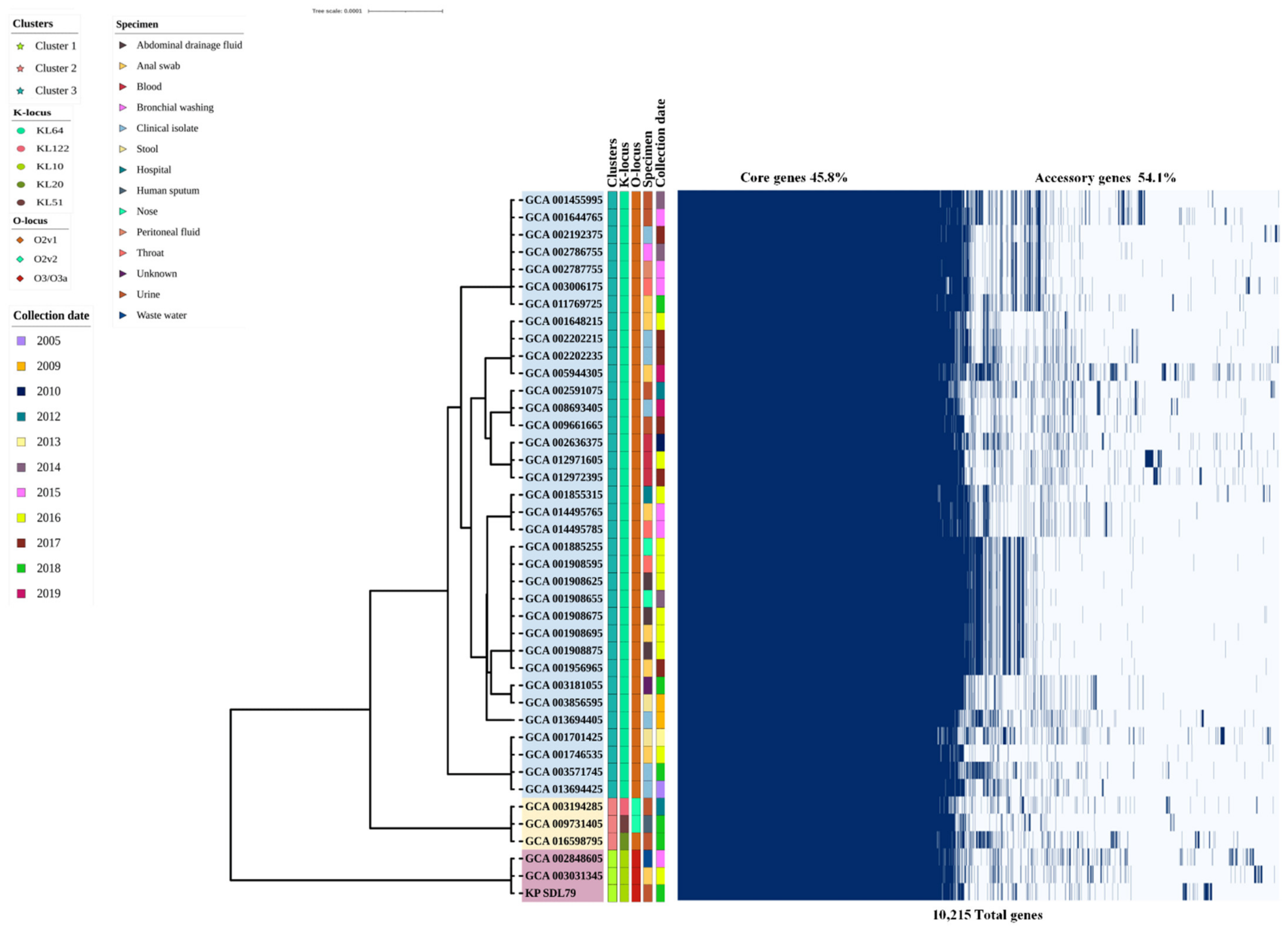
3.4. Insights into Pangenome Structure, Core Phylogenetic Relationships and Functional Characterization
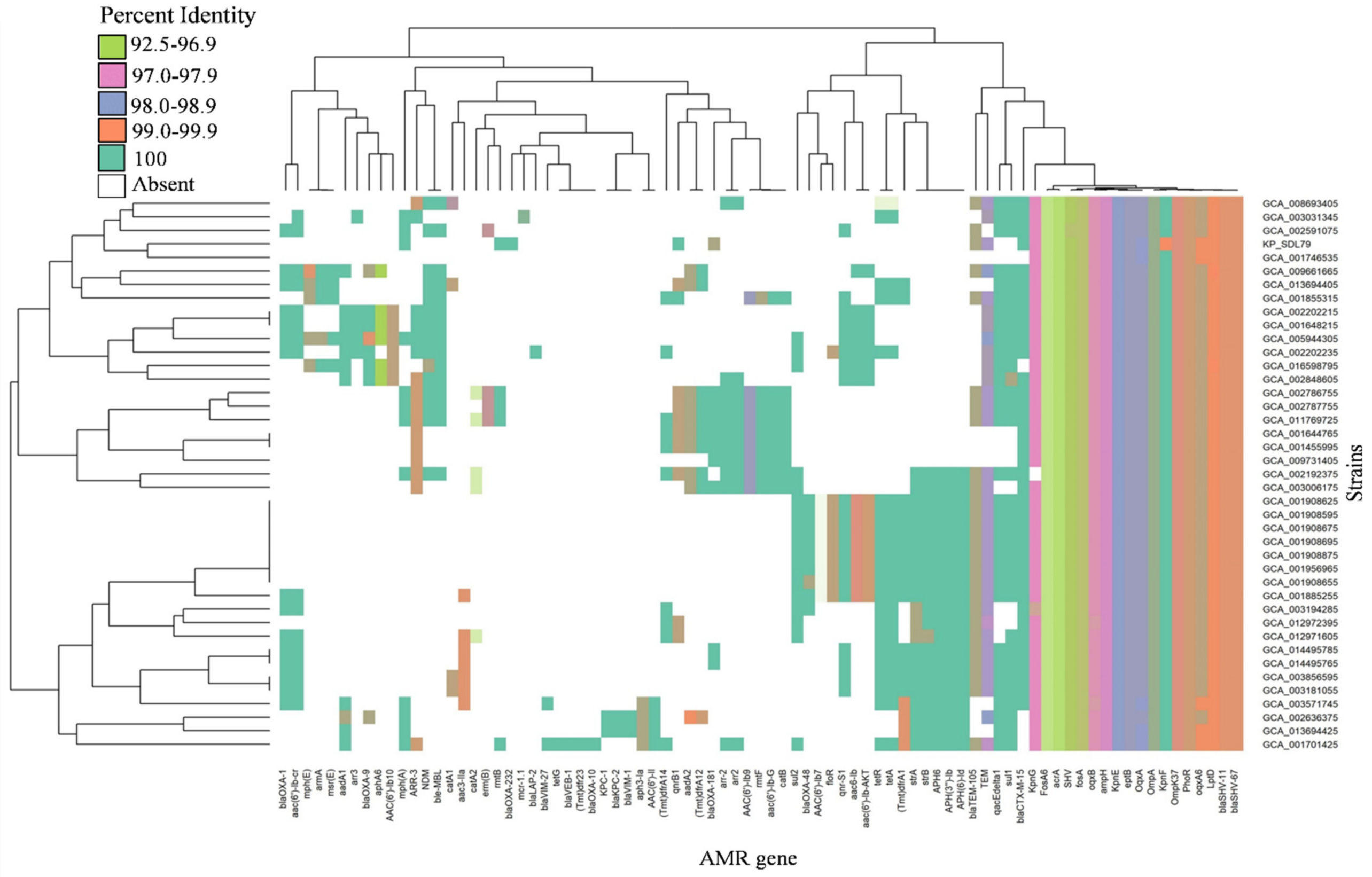
3.5. Distribution of Antibiotic Resistance and Virulence Genes
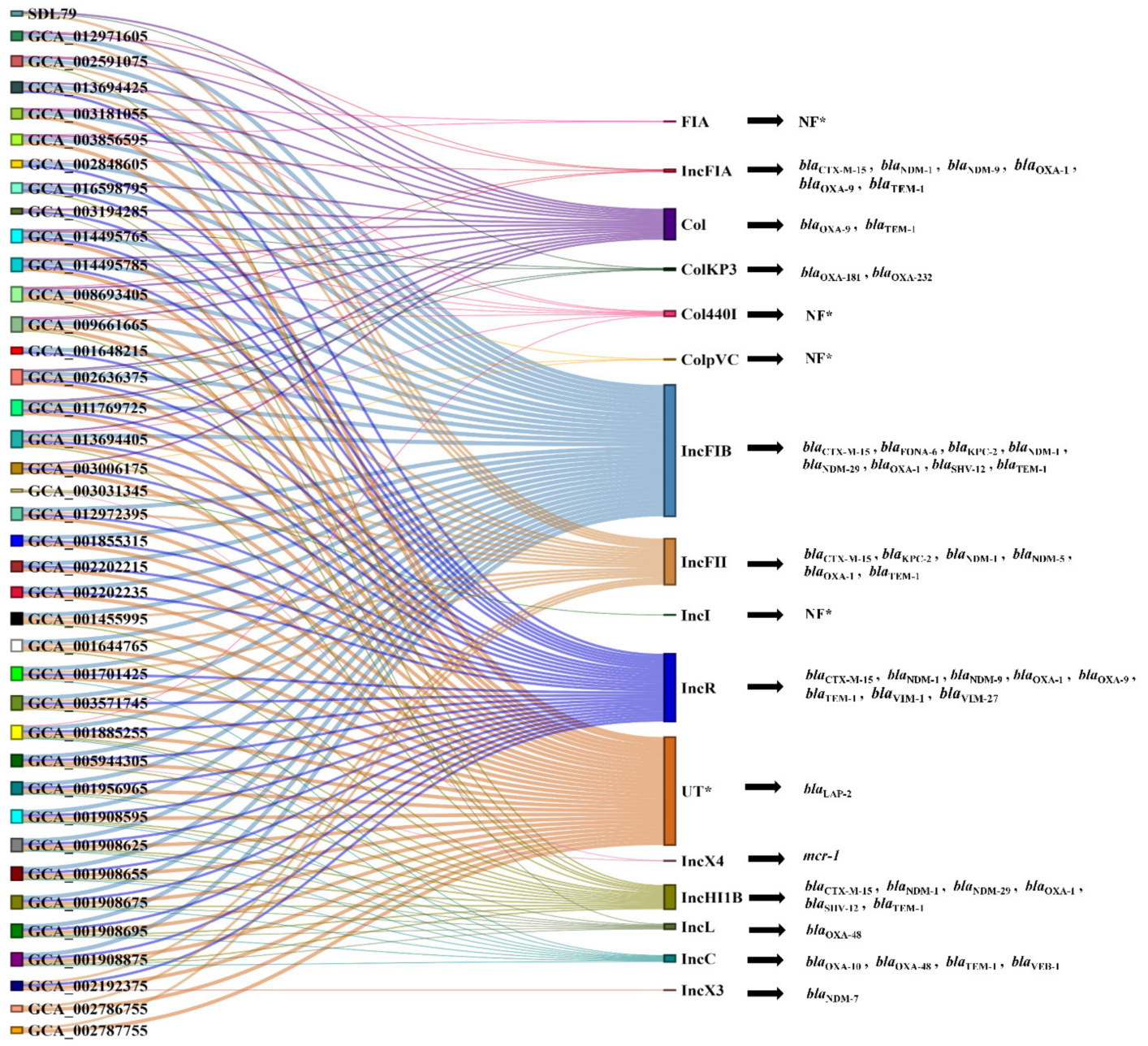
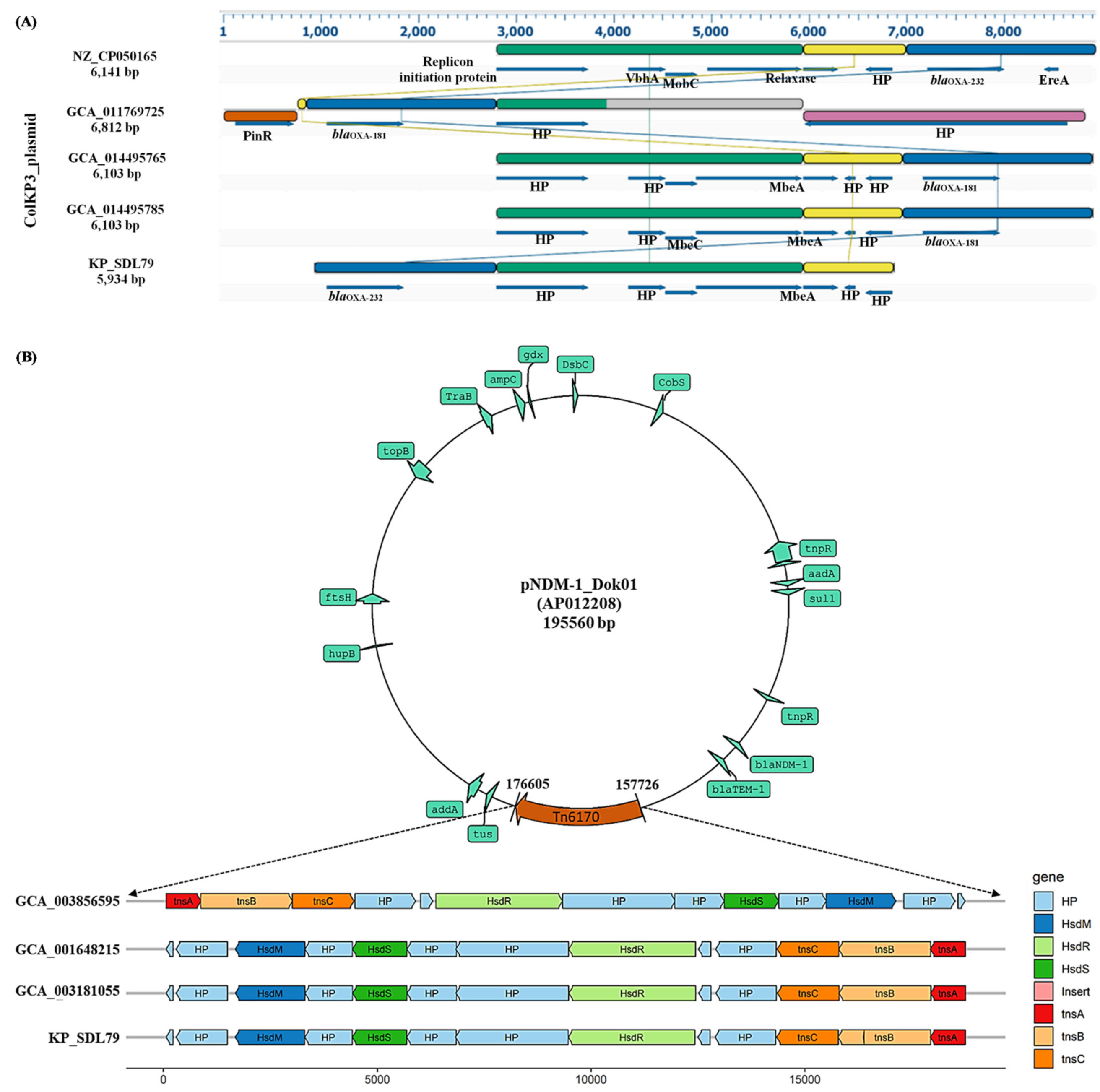
3.6. Plasmid Prediction and Synteny Analysis
3.7. Identification of Insertion Elements and Characterization of Tn6170
3.8. Prediction of Serotypes, Prophages and CRISPR-Types
3.9. ST147 Isolates Have Significant and Diverse Biosynthetic Potential
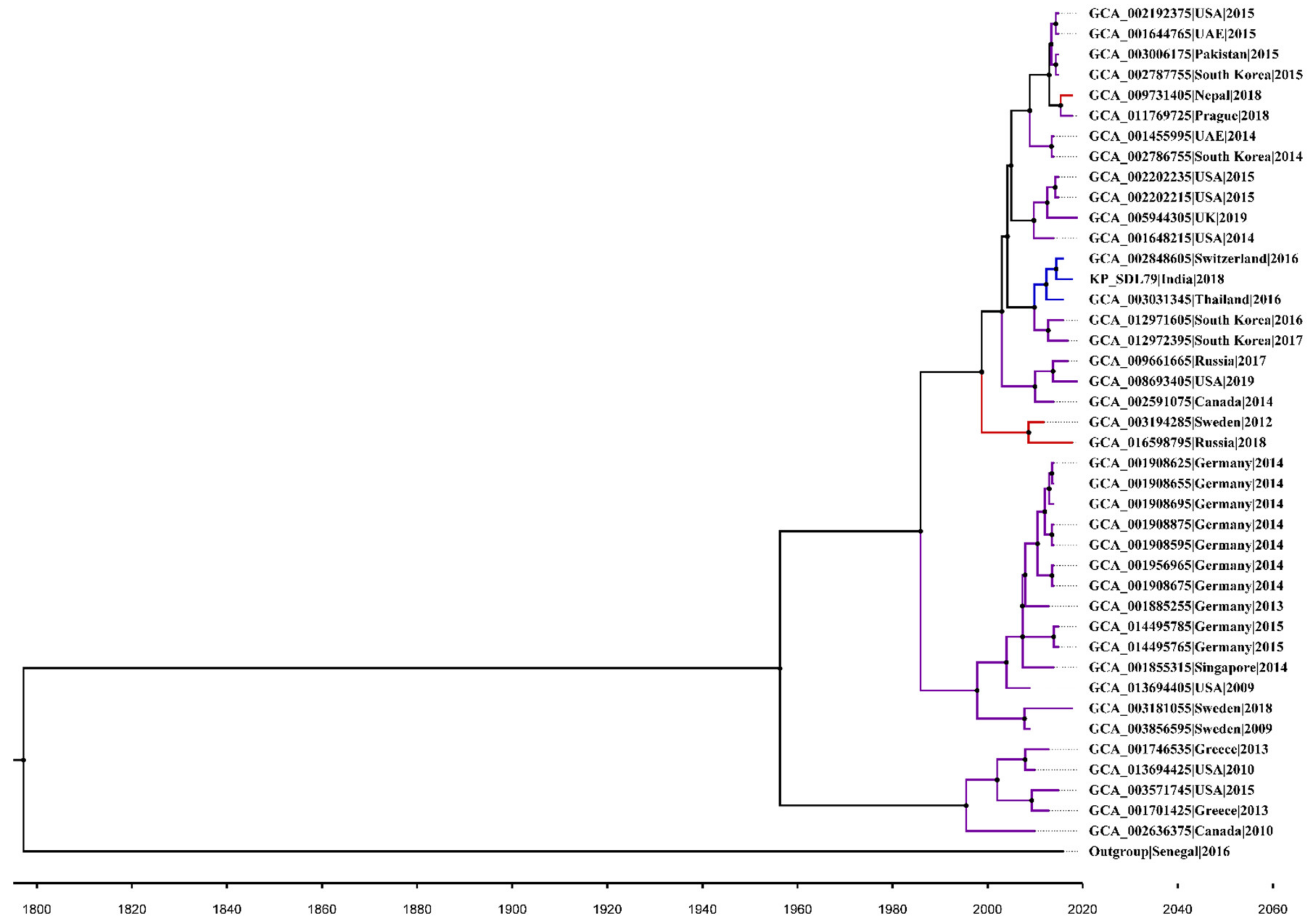
3.10. Core SNP Identification and Phylogenetic Reconstruction
3.11. Bayesian Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Temkin, E.; Fallach, N.; Almagor, J.; Gladstone, B.P.; Tacconelli, E.; Carmeli, Y. Estimating the number of infections caused by antibiotic-resistant Escherichia coli and Klebsiella pneumoniae in 2014: A modelling study. Lancet Glob. Health 2018, 6, e969–e979. [Google Scholar] [CrossRef]
- Navon-Venezia, S.; Kondratyeva, K.; Carattoli, A. Klebsiella pneumoniae: A major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol. Rev. 2017, 41, 252–275. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Peirano, G.; Chen, L.; Kreiswirth, B.N.; Pitout, J.D.D. Emerging Antimicrobial-Resistant High-Risk Klebsiella pneumoniae Clones ST307 and ST147. Antimicrob. Agents Chemother. 2020, 64, e01148-20. [Google Scholar] [CrossRef]
- Protonotariou, E.; Poulou, A.; Politi, L.; Sgouropoulos, I.; Metallidis, S.; Kachrimanidou, M.; Pournaras, S.; Tsakris, A.; Skoura, L. Hospital outbreak due to a Klebsiella pneumoniae ST147 clonal strain co-producing KPC-2 and VIM-1 carbapenemases in a tertiary teaching hospital in Northern Greece. Int. J. Antimicrob. Agents 2018, 52, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.; Desai, S.; Passet, V.; Gajjar, D.; Brisse, S. Genomic evolution of the globally disseminated multidrug-resistant Klebsiella pneumoniae clonal group 147. Microb. Genom. 2022, 8, 000737. [Google Scholar] [CrossRef]
- Uelze, L.; Grützke, J.; Borowiak, M.; Hammerl, J.A.; Juraschek, K.; Deneke, C.; Tausch, S.H.; Malorny, B. Typing methods based on whole genome sequencing data. One Health Outlook 2020, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Carlos Guimaraes, L.; Benevides de Jesus, L.; Vinicius Canario Viana, M.; Silva, A.; Thiago Juca Ramos, R.; de Castro Soares, S.; Azevedo, V. Inside the Pan-genome—Methods and Software Overview. Curr. Genom. 2015, 16, 245–252. [Google Scholar] [CrossRef]
- Dey, S.; Gaur, M.; Sahoo, R.K.; Das, A.; Jain, B.; Pati, S.; Subudhi, E. Genomic characterization of XDR Klebsiella pneumoniae ST147 co-resistant to carbapenem and colistin—The first report in India. J. Glob. Antimicrob. Resist. 2020, 22, 54–56. [Google Scholar] [CrossRef]
- Andrews, S.; Krueger, F.; Seconds-Pichon, A.; Biggins, F.; Wingett, S. FastQC. A quality control tool for high throughput sequence data. Babraham Inst. 2015, 1, 1. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 January 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: {A} parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D. RepeatMasker. Biotech Softw. Internet Rep. 2000, 1, 36–39. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Raney, B.; Paten, B.; Pham, S. Ragout—A reference-assisted assembly tool for bacterial genomes. Bioinformatics 2014, 30, i302–i309. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Mohite, O.S.; Lloyd, C.J.; Monk, J.M.; Weber, T.; Palsson, B.O. Pangenome analysis of Enterobacteria reveals richness of secondary metabolite gene clusters and their associated gene sets. Synth. Syst. Biotechnol. 2022, 7, 900–910. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Yuan, C.; Yang, H. Research on K-Value Selection Method of K-Means Clustering Algorithm. J. Multidiscip. Sci. J. 2019, 2, 16. [Google Scholar] [CrossRef]
- Sitto, F.; Battistuzzi, F.U. Estimating Pangenomes with Roary. Mol. Biol. Evol. 2020, 37, 933–939. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; Lees, J.A.; Bentley, S.D.; Frost, S.D.W.; Corander, J. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 2019, 47, 5539–5549. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.N.; Slezak, T.; Hall, B.G. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 2015, 31, 2877–2878. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Kleinheinz, K.A.; Joensen, K.G.; Larsen, M.V. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 2014, 4, e27943. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.; Lund, O.; Villa, L.; Aarestrup, F.; Hasman, H. PlasmidFinder and pMLST: In silico detection and typing of plasmids. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34 (Suppl. 1), D32–D36. [Google Scholar] [CrossRef]
- David, C.; Aude, B.; Claire, T.-N.; Marie, T.; Juraj, M.; Bertrand, N.; Eduardo, P.C.R.; Gilles, V.; Daniel, G.; Christine, P. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar]
- Song, W.; Sun, H.-X.; Zhang, C.; Cheng, L.; Peng, Y.; Deng, Z.; Wang, D.; Wang, Y.; Hu, M.; Liu, W.; et al. Prophage Hunter: An integrative hunting tool for active prophages. Nucleic Acids Res. 2019, 47, W74–W80. [Google Scholar] [CrossRef]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Kurittu, P.; Khakipoor, B.; Aarnio, M.; Nykäsenoja, S.; Brouwer, M.; Myllyniemi, A.-L.; Vatunen, E.; Heikinheimo, A. Plasmid-Borne and Chromosomal ESBL/AmpC Genes in Escherichia coli and Klebsiella pneumoniae in Global Food Products. Front. Microbiol. 2021, 12, 592291. [Google Scholar] [CrossRef]
- Peters, J.E. Tn7. Microbiol. Spectr. 2014, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Harmer, C.J.; Hall, R.M. pRMH760, a Precursor of A/C 2 Plasmids Carrying bla CMY and bla NDM Genes. Microb. Drug Resist. 2014, 20, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Kryazhimskiy, S.; Plotkin, J.B. The Population Genetics of dN/dS. PLoS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef] [PubMed]
- Denton, J.F.; Lugo-Martinez, J.; Tucker, A.E.; Schrider, D.R.; Warren, W.C.; Hahn, M.W. Extensive Error in the Number of Genes Inferred from Draft Genome Assemblies. PLoS Comput. Biol. 2014, 10, e1003998. [Google Scholar] [CrossRef] [PubMed]
- Passarelli-Araujo, H.; Palmeiro, J.K.; Moharana, K.C.; Pedrosa-Silva, F.; Dalla-Costa, L.M.; Venancio, T.M. Genomic analysis unveils important aspects of population structure, virulence, and antimicrobial resistance in Klebsiella aerogenes. FEBS J. 2019, 286, 3797–3810. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, C.; Liu, Y.; Zhang, Y.; Xiong, Z.; Fan, Y.; Zou, X.; Lu, B.; Cao, B. Genomic characteristics of clinically important ST11 Klebsiella pneumoniae strains worldwide. J. Glob. Antimicrob. Resist. 2020, 22, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Zhang, R.; Liu, L.; Li, R.; Lin, D.; Chan, E.W.-C.; Chen, S. Genome analysis of clinical multilocus sequence Type 11 Klebsiella pneumoniae from China. Microb. Genom. 2018, 4, e000149. [Google Scholar] [CrossRef]
- Nüesch-Inderbinen, M.; Zurfluh, K.; Stevens, M.J.A.; Stephan, R. Complete and assembled genome sequence of an NDM-9- and CTX-M-15-producing Klebsiella pneumoniae ST147 wastewater isolate from Switzerland. J. Glob. Antimicrob. Resist. 2018, 13, 53–54. [Google Scholar] [CrossRef]
- Sundaresan, A.K.; Vincent, K.; Mohan, G.B.M.; Ramakrishnan, J. Association of sequence types, antimicrobial resistance and virulence genes in Indian isolates of Klebsiella pneumoniae: A comparative genomics study. J. Glob. Antimicrob. Resist. 2022, 30, 431–441. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef]
- Patel, P.K.; Russo, T.A.; Karchmer, A.W. Hypervirulent Klebsiella pneumoniae. Open Forum Infect. Dis. 2014, 1, ofu028. [Google Scholar] [CrossRef]
- Turton, J.F.; Payne, Z.; Coward, A.; Hopkins, K.L.; Turton, J.A.; Doumith, M.; Woodford, N. Virulence genes in isolates of Klebsiella pneumoniae from the UK during 2016, including among carbapenemase gene-positive hypervirulent K1-st23 and ‘non-hypervirulent’ types ST147, ST15 and ST383. J. Med. Microbiol. 2018, 67, 118–128. [Google Scholar] [CrossRef]
- Rodrigues, C.; Machado, E.; Ramos, H.; Peixe, L.; Novais, Â. Expansion of ESBL-producing Klebsiella pneumoniae in hospitalized patients: A successful story of international clones (ST15, ST147, ST336) and epidemic plasmids (IncR, IncFIIK). Int. J. Med. Microbiol. 2014, 304, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Shankar, C.; Mathur, P.; Venkatesan, M.; Pragasam, A.K.; Anandan, S.; Khurana, S.; Veeraraghavan, B. Rapidly disseminating blaOXA-232 carrying Klebsiella pneumoniae belonging to ST231 in India: Multiple and varied mobile genetic elements. BMC Microbiol. 2019, 19, 137. [Google Scholar] [CrossRef]
- Comandatore, F.; Sassera, D.; Bayliss, S.C.; Scaltriti, E.; Gaiarsa, S.; Cao, X.; Gales, A.C.; Saito, R.; Pongolini, S.; Brisse, S.; et al. Gene Composition as a Potential Barrier to Large Recombinations in the Bacterial Pathogen Klebsiella pneumoniae. Genome Biol. Evol. 2019, 11, 3240–3251. [Google Scholar] [CrossRef] [PubMed]
- Di Pilato, V.; De Angelis, L.H.; Aiezza, N.; Baccani, I.; Niccolai, C.; Parisio, E.M.; Giordano, C.; Camarlinghi, G.; Barnini, S.; Forni, S.; et al. Resistome and virulome accretion in an NDM-1-producing ST147 sublineage of Klebsiella pneumoniae associated with an outbreak in Tuscany, Italy: A genotypic and phenotypic characterisation. Lancet Microbe 2022, 3, e224–e234. [Google Scholar] [CrossRef] [PubMed]
- Artyszuk, D.; Izdebski, R.; Maciejewska, A.; Kaszowska, M.; Herud, A.; Szijártó, V.; Gniadkowski, M.; Lukasiewicz, J. The Impact of Insertion Sequences on O-Serotype Phenotype and Its O-Locus-Based Prediction in Klebsiella pneumoniae O2 and O1. Int. J. Mol. Sci. 2020, 21, 6572. [Google Scholar] [CrossRef]
- Bobay, L.-M.; Touchon, M.; Rocha, E.P.C. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 12127–12132. [Google Scholar] [CrossRef]
- Sekizuka, T.; Matsui, M.; Yamane, K.; Takeuchi, F.; Ohnishi, M.; Hishinuma, A.; Arakawa, Y.; Kuroda, M. Complete Sequencing of the blaNDM-1-Positive IncA/C Plasmid from Escherichia coli ST38 Isolate Suggests a Possible Origin from Plant Pathogens. PLoS ONE 2011, 6, e25334. [Google Scholar] [CrossRef]
- Belknap, K.C.; Park, C.J.; Barth, B.M.; Andam, C.P. Genome mining of biosynthetic and chemotherapeutic gene clusters in Streptomyces bacteria. Sci. Rep. 2020, 10, 2003. [Google Scholar] [CrossRef]
- Guerra, M.E.S.; Destro, G.; Vieira, B.; Lima, A.S.; Ferraz, L.F.C.; Hakansson, A.P.; Darrieux, M.; Converso, T.R. Klebsiella pneumoniae Biofilms and Their Role in Disease Pathogenesis. Front. Cell. Infect. Microbiol. 2022, 12. [Google Scholar] [CrossRef]
- Li, H.-Y.; Kao, C.-Y.; Lin, W.-H.; Zheng, P.-X.; Yan, J.-J.; Wang, M.-C.; Teng, C.-H.; Tseng, C.-C.; Wu, J.-J. Characterization of CRISPR-Cas Systems in Clinical Klebsiella pneumoniae Isolates Uncovers Its Potential Association With Antibiotic Susceptibility. Front. Microbiol. 2018, 9, 1595. [Google Scholar] [CrossRef] [PubMed]
- Zemmour, A.; Dali-Yahia, R.; Maatallah, M.; Saidi-Ouahrani, N.; Rahmani, B.; Benhamouche, N.; Al-Farsi, H.M.; Giske, C.G. High-risk clones of extended-spectrumβ- lactamase-producing Klebsiella pneumoniae isolated from the University Hospital Establishment of Oran, Algeria (2011-2012). PLoS ONE 2021, 16, e0254805. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Fournier, P.-E.; Raoult, D. Genome and pan-genome analysis to classify emerging bacteria. Biol. Direct 2019, 14, 5. [Google Scholar] [CrossRef]
- Rahman, S.; Pond, S.L.K.; Webb, A.; Hey, J. Weak selection on synonymous codons substantially inflates dN/dS estimates in bacteria. Proc. Natl. Acad. Sci. USA 2021, 118, e2023575118. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ma, Y.; Chen, H.; Dai, J.; Luo, H.; Jia, M.; Song, Z. Complete genome sequencing and evolutionary analysis of HCV subtype 6xg from IDUs in Yunnan, China. PLoS ONE 2019, 14, e0217010. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Country | Host | Isolation Source | Collection Year | Chromosome | Genome Size (bp) | GC Content (%) | CDS | rRNA | tRNA | misc_RNA | tmRNA | K-Locus | O-Locus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GCA_001455995 | United Arab Emirates | Human | Urine | 2014 | 7 | 5980203 | 57.33 | 5631 | 25 | 89 | 130 | 1 | KL64 | O2v1 |
| GCA_001644765 | United Arab Emirates | Human | Urine sample | 2015 | 6 | 6088457 | 57.29 | 5736 | 25 | 89 | 132 | 1 | KL64 | O2v1 |
| GCA_001648215 | USA | Human | Perirectal swab | 2014 | 4 | 5566588 | 57.43 | 5101 | 25 | 87 | 112 | 1 | KL64 | O2v1 |
| GCA_001701425 | Greece | Human | Stool | 2013 | 6 | 5760711 | 57.58 | 5403 | 25 | 88 | 122 | 1 | KL64 | O2v1 |
| GCA_001746535 | Greece | Human | Rectal swab | 2013 | 1 | 5344576 | 57.42 | 4946 | 31 | 103 | 116 | 1 | KL64 | O2v1 |
| GCA_001855315 | Singapore | Human | Hospital | 2014 | 5 | 5718854 | 57.3 | 5297 | 25 | 90 | 110 | 1 | KL64 | O2v1 |
| GCA_001885255 | Germany | Human | Nose | 2013 | 7 | 5920942 | 57.36 | 5559 | 25 | 88 | 120 | 1 | KL64 | O2v1 |
| GCA_001908595 | Germany | Human | Tracheal secretion | 2014 | 6 | 5916317 | 57.36 | 5550 | 25 | 88 | 119 | 1 | KL64 | O2v1 |
| GCA_001908625 | Germany | Human | Abdominal drainage fluid | 2014 | 6 | 5907691 | 57.36 | 5541 | 25 | 88 | 118 | 1 | KL64 | O2v1 |
| GCA_001908655 | Germany | Human | Nasal swab | 2014 | 6 | 5913496 | 57.36 | 5551 | 25 | 88 | 119 | 1 | KL64 | O2v1 |
| GCA_001908675 | Germany | Human | Abdominal drainage fluid | 2014 | 6 | 5921376 | 57.36 | 5553 | 25 | 88 | 119 | 1 | KL64 | O2v1 |
| GCA_001908695 | Germany | Human | Anal swab | 2014 | 6 | 5914083 | 57.36 | 5543 | 25 | 88 | 119 | 1 | KL64 | O2v1 |
| GCA_001908875 | Germany | Human | Abdominal drain fluid | 2014 | 6 | 5920288 | 57.36 | 5551 | 25 | 88 | 119 | 1 | KL64 | O2v1 |
| GCA_001956965 | Germany | Human | Anal swab | 2014 | 5 | 5869161 | 57.36 | 5497 | 25 | 88 | 118 | 1 | KL64 | O2v1 |
| GCA_002192375 | USA | Human | Medical | 2015 | 5 | 5708112 | 57.3 | 5320 | 25 | 89 | 120 | 1 | KL64 | O2v1 |
| GCA_002202215 | USA | Human | Medical | 2015 | 5 | 5656270 | 57.41 | 5238 | 25 | 88 | 116 | 1 | KL64 | O2v1 |
| GCA_002202235 | USA | Human | Medical | 2015 | 6 | 5782129 | 57.42 | 5383 | 25 | 88 | 120 | 1 | KL64 | O2v1 |
| GCA_002591075 | Canada | Human | Urine | 2014 | 6 | 5752865 | 57.56 | 5369 | 25 | 86 | 121 | 1 | KL64 | O2v1 |
| GCA_002636375 | Canada | Human | Blood | 2010 | 7 | 5727717 | 57.45 | 5397 | 25 | 90 | 118 | 1 | KL64 | O2v1 |
| GCA_002786755 | South Korea | Human | Bronchial washing | 2014 | 4 | 5720338 | 57.32 | 5346 | 25 | 89 | 129 | 1 | KL64 | O2v1 |
| GCA_002787755 | South Korea | Human | Peritoneal fluid | 2015 | 4 | 5707921 | 57.32 | 5317 | 25 | 89 | 128 | 1 | KL64 | O2v1 |
| GCA_002848605 | Switzerland | Environment | Waste water | 2016 | 3 | 5630984 | 57.4 | 5428 | 25 | 87 | 122 | 1 | KL10 | O3/O3a |
| GCA_003006175 | Pakistan | Human | Trachaeal secretion | 2015 | 8 | 5752275 | 57.29 | 5368 | 25 | 89 | 128 | 1 | KL64 | O2v1 |
| GCA_003031345 | Thailand | Human | Rectal swab | 2016 | 4 | 5640638 | 57.42 | 5292 | 25 | 86 | 123 | 1 | KL10 | O3/O3a |
| GCA_003181055 | Sweden | Human | Unknown | 2018 | 5 | 5657209 | 57.27 | 5220 | 25 | 88 | 116 | 1 | KL64 | O2v1 |
| GCA_003194285 | Sweden | Human | Urine | 2012 | 5 | 5391141 | 57.54 | 5005 | 28 | 87 | 117 | 1 | KL122 | O2v2 |
| GCA_003571745 | USA | Human | Medical | 2015 | 6 | 5768716 | 57.54 | 5375 | 25 | 89 | 124 | 1 | KL64 | O2v1 |
| GCA_003856595 | Sweden | Human | Faeces | 2009 | 5 | 5657200 | 57.27 | 5219 | 25 | 88 | 116 | 1 | KL64 | O2v1 |
| GCA_005944305 | United Kingdom | Human | Rectal swab | 2019 | 8 | 6109775 | 57.34 | 5828 | 25 | 88 | 119 | 1 | KL64 | O2v1 |
| GCA_008693405 | USA | Human | Clinical isolate | 2019 | 8 | 5642810 | 57.48 | 5249 | 24 | 87 | 120 | 1 | KL64 | O2v1 |
| GCA_009661665 | Russia | Human | Urine | 2017 | 6 | 5645610 | 57.46 | 5244 | 25 | 88 | 115 | 1 | KL64 | O2v1 |
| GCA_009731405 | Nepal | Human | Human sputum | 2018 | 1 | 5422388 | 57.28 | 5006 | 25 | 88 | 110 | 1 | KL51 | O2v2 |
| GCA_011769725 | Czech Republic | Human | Rectal swab | 2018 | 11 | 5919785 | 57.33 | 5514 | 25 | 90 | 136 | 1 | KL64 | O2v1 |
| GCA_012971605 | South Korea | Human | Blood | 2016 | 6 | 5820688 | 57.35 | 5432 | 25 | 88 | 123 | 1 | KL64 | O2v1 |
| GCA_012972395 | South Korea | Human | Blood | 2017 | 9 | 5688247 | 57.36 | 5308 | 25 | 86 | 113 | 1 | KL64 | O2v1 |
| GCA_013694405 | USA | Human | Clinical isolate | 2009 | 8 | 5793006 | 57.46 | 5412 | 25 | 86 | 123 | 1 | KL64 | O2v1 |
| GCA_013694425 | USA | Human | Clinical isolate | 2010 | 5 | 5610747 | 57.44 | 5237 | 25 | 88 | 115 | 1 | KL64 | O2v1 |
| GCA_014495765 | Germany | Human | Rectal swab | 2015 | 9 | 5730837 | 57.24 | 5315 | 25 | 89 | 120 | 1 | KL64 | O2v1 |
| GCA_014495785 | Germany | Human | Throat | 2015 | 9 | 5729180 | 57.24 | 5303 | 25 | 89 | 120 | 1 | KL64 | O2v1 |
| GCA_016598795 | Russia | Human | Urine | 2018 | 4 | 5797747 | 57.41 | 5434 | 25 | 88 | 117 | 1 | KL20 | O2v1 |
| GCA_024124405 | India | Human | Urine | 2018 | 43 * | 5622734 | 57.21 | 5266 | 3 | 84 | 112 | 1 | KL10 | O3/O3a |
| Features | K. pneumoniae HS11286 | Unicycler | AbySS | SOAPdenovo | SPAdes |
|---|---|---|---|---|---|
| Contigs (≥1000 bp) | 1 | 65 | 119 | 129 | 132 |
| Contigs (≥50,000 bp) | 1 | 23 | 40 | 41 | 24 |
| Largest contig size (bp) | 5,333,942 | 714,080 | 323,583 | 390,642 | 1,005,571 |
| Total length (bp) | 5,333,942 | 5,562,754 | 5,833,980 | 5,815,984 | 5,988,665 |
| GC Content (%) | 57.48 | 57.21 | 56.82 | 56.78 | 56.52 |
| N50 (bp) | 5,333,942 | 218,233 | 113,012 | 105,915 | 218,806 |
| N75 (bp) | 5,333,942 | 147,654 | 66,133 | 61,400 | 129,870 |
| L50 (bp) | 1 | 7 | 17 | 18 | 8 |
| L75 (bp) | 1 | 15 | 33 | 35 | 17 |
| Genome fraction (%) | 100 | 91.557 | 91.018 | 91.133 | 91.022 |
| Ns per 100 kbp | 0 | 325.46 | 1462.79 | 1892.94 | 1879.7 |
| Mismatches per 100 kbp | 0 | 675.84 | 681.03 | 679.32 | 677.46 |
| Complete Gene | 5404 | 4736 | 4702 | 4726 | 4727 |
| Partial Gene | 0 | 84 | 87 | 94 | 95 |
| Contamination | 0 | 1.46 | 2.23 | 2.22 | 2.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, S.; Gaur, M.; Sykes, E.M.E.; Prusty, M.; Elangovan, S.; Dixit, S.; Pati, S.; Kumar, A.; Subudhi, E. Unravelling the Evolutionary Dynamics of High-Risk Klebsiella pneumoniae ST147 Clones: Insights from Comparative Pangenome Analysis. Genes 2023, 14, 1037. https://doi.org/10.3390/genes14051037
Dey S, Gaur M, Sykes EME, Prusty M, Elangovan S, Dixit S, Pati S, Kumar A, Subudhi E. Unravelling the Evolutionary Dynamics of High-Risk Klebsiella pneumoniae ST147 Clones: Insights from Comparative Pangenome Analysis. Genes. 2023; 14(5):1037. https://doi.org/10.3390/genes14051037
Chicago/Turabian StyleDey, Suchanda, Mahendra Gaur, Ellen M. E. Sykes, Monica Prusty, Selvakumar Elangovan, Sangita Dixit, Sanghamitra Pati, Ayush Kumar, and Enketeswara Subudhi. 2023. "Unravelling the Evolutionary Dynamics of High-Risk Klebsiella pneumoniae ST147 Clones: Insights from Comparative Pangenome Analysis" Genes 14, no. 5: 1037. https://doi.org/10.3390/genes14051037