
Chromosomal Microarray Analysis Identifies a Novel SALL1 Deletion, Supporting the Association of Haploinsufficiency with a Mild Phenotype of Townes–Brocks Syndrome
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
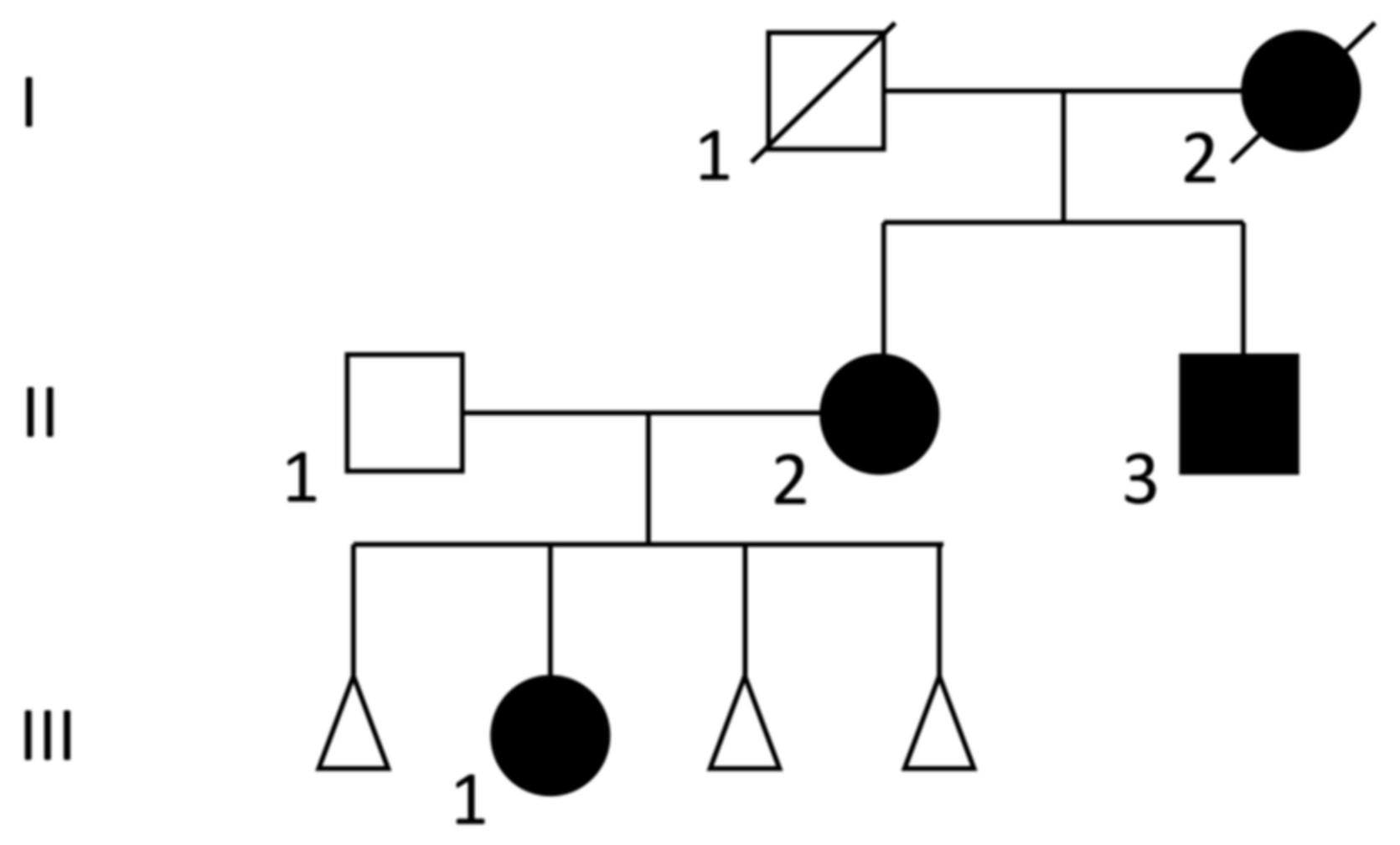
3.1. Clinical Description
3.2. Genetic Analysis
3.3. Review of Deletions in the Literature and in Databases
3.4. Review of Clinical Features in Carriers of the p.Arg276Ter Mutation and Comparison with Deletion Carriers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- South, S.T.; Lee, C.; Lamb, A.N.; Higgins, A.W.; Kearney, H.M.; Working Group for the American College of Medical Genetics and Genomics Laboratory Quality Assurance Committee. ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet. Med. 2013, 15, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Hempel, A.; Pagnamenta, A.T.; Blyth, M.; Mansour, S.; McConnell, V.; Kou, I.; Ikegawa, S.; Tsurusaki, Y.; Matsumoto, N.; Lo-Castro, A.; et al. Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin-Siris syndrome. J. Med. Genet. 2016, 53, 152–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, E.; Graziano, C.; Minopoli, F.; Bacchelli, E.; Magini, P.; Diquigiovanni, C.; Lomartire, S.; Bianco, F.; Vargiolu, M.; Parchi, P.; et al. Maternally inherited genetic variants of CADPS2 are present in autism spectrum disorders and intellectual disability patients. EMBO Mol. Med. 2014, 6, 795–809. [Google Scholar] [CrossRef]
- Kohlhase, J. Townes-Brocks Syndrome. In GeneReviews®; Adam, M.P., Ed.; University of Washington: Seattle, WA, USA, 2007. [Google Scholar]
- Botzenhart, E.M.; Green, A.; Ilyina, H.; König, R.; Lowry, R.B.; Lo, I.F.; Shohat, M.; Burke, L.; McGaughran, J.; Chafai, R.; et al. SALL1 mutation analysis in Townes-Brocks syndrome: Twelve novel mutations and expansion of the phenotype. Hum. Mutat. 2005, 26, 282. [Google Scholar] [CrossRef]
- Botzenhart, E.M.; Bartalini, G.; Blair, E.; Brady, A.F.; Elmslie, F.; Chong, K.L.; Christy, K.; Torres-Martinez, W.; Danesino, C.; Deardorff, M.A.; et al. Townes-Brocks syndrome: Twenty novel SALL1 mutations in sporadic and familial cases and refinement of the SALL1 hot spot region. Hum. Mutat. 2007, 28, 204–205. [Google Scholar] [CrossRef]
- Kohlhase, J.; Wischermann, A.; Reichenbach, H.; Froster, U.; Engel, W. Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet. 1998, 18, 81–83. [Google Scholar] [CrossRef]
- De Celis, J.F.; Barrio, R. Regulation and function of Spalt proteins during animal development. Int. J. Dev. Biol. 2009, 53, 1385–1398. [Google Scholar] [CrossRef] [Green Version]
- Bozal-Basterra, L.; Martín-Ruíz, I.; Pirone, L.; Liang, Y.; Sigurðsson, J.O.; Gonzalez-Santamarta, M.; Giordano, I.; Gabicagogeascoa, E.; de Luca, A.; Rodríguez, J.A.; et al. Truncated SALL1 Impedes Primary Cilia Function in Townes-Brocks Syndrome. Am. J. Hum. Genet. 2018, 102, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Kohlhase, J.; Liebers, M.; Backe, J.; Baumann-Müller, A.; Bembea, M.; Destrée, A.; Gattas, M.; Grüßner, S.; Müller, T.; Mortier, G.; et al. High incidence of the R276X SALL1 mutation in sporadic but not familial Townes-Brocks syndrome and report of the first familial case. J. Med. Genet. 2003, 40, e127. [Google Scholar] [CrossRef]
- Nishinakamura, R.; Matsumoto, Y.; Nakao, K.; Nakamura, K.; Sato, A.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Scully, S.; Lacey, D.L.; et al. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development 2001, 128, 3105–3115. [Google Scholar] [CrossRef]
- Kiefer, S.M.; Ohlemiller, K.K.; Yang, J.; McDill, B.W.; Kohlhase, J.; Rauchman, M. Expression of a truncated Sall1 transcriptional repressor is responsible for Townes-Brocks syndrome birth defects. Hum. Mol. Genet. 2003, 12, 2221–2227. [Google Scholar] [CrossRef]
- Kiefer, S.M.; Robbins, L.; Barina, A.; Zhang, Z.; Rauchman, M. SALL1 truncated protein expression in Townes-Brocks syndrome leads to ectopic expression of downstream genes. Hum. Mut. 2008, 29, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Borozdin, W.; Steinmann, K.; Albrecht, B.; Bottani, A.; Devriendt, K.; Leipoldt, M.; Kohlhase, J. Detection of heterozygous SALL1 deletions by quantitative real time PCR proves the contribution of a SALL1 dosage effect in the pathogenesis of Townes-Brocks syndrome. Hum. Mutat. 2006, 27, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.M.; Hopkin, R.; Bao, L.; Ware, S.M. Implications for genotype-phenotype predictions in Townes-Brocks syndrome: Case report of a novel SALL1 deletion and review of the literature. Am. J. Med. Genet. A 2012, 158A, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.A.; May, K.M. Deletion upstream of SALL1 producing Townes-Brocks syndrome. Am. J. Med. Genet. A 2016, 170A, 2476–2478. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Kohlhase, J.; Taschner, P.E.; Burfeind, P.; Pasche, B.; Newman, B.; Blanck, C.; Breuning, M.H.; Kate, L.P.T.; Maaswinkel-Mooy, P.; Mitulla, B.; et al. Molecular analysis of SALL1 mutations in Townes-Brocks syndrome. Am. J. Hum. Genet. 1999, 64, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Marlin, S.; Blanchard, S.; Slim, R.; Lacombe, D.; Denoyelle, F.; Alessandri, J.L.; Calzolari, E.; Drouin-Garraud, V.; Ferraz, F.G.; Fourmaintraux, A.; et al. Townes-Brocks syndrome: Detection of a SALL1 mutation hot spot and evidence for a position effect in one patient. Hum. Mut. 1999, 14, 377–386. [Google Scholar] [CrossRef]
- Keegan, C.E.; Mulliken, J.B.; Wu, B.L.; Korf, B.R. Townes-Brocks syndrome versus expanded spectrum hemifacial microsomia: Review of eight patients and further evidence of a “hot spot” for mutation in the SALL1 gene. Genet. Med. 2001, 3, 310–313. [Google Scholar] [CrossRef]
- Barry, J.S.; Reddy, M.A. The association of an epibulbar dermoid and Duane syndrome in a patient with a SALL1 mutation (Townes-Brocks Syndrome). Ophth. Genet. 2008, 29, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Van Bever, Y.; Gischler, S.J.; Hoeve, H.L.; Smit, L.S.; Nauta, J.; Dooijes, D. Obstructive apneas and severe dysphagia in a girl with Townes-Brocks syndrome and atypical feet involvement. Eur. J. Med. Genet. 2009, 52, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.; Braun, D.A.; Amann, K.; Tan, W.; Shril, S.; Connaughton, D.M.; Nakayama, M.; Schneider, R.; Kitzler, T.M.; van der Ven, A.T.; et al. Whole-Exome Sequencing Enables a Precision Medicine Approach for Kidney Transplant Recipients. J. Am. Soc. Nephrol. 2019, 30, 201–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valikodath, N.G.; Jain, S.; Miller, M.; Kaufman, L.M. Ocular features of Townes-Brocks syndrome. J. AAPOS 2020, 24, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Furniss, D.; Critchley, P.; Giele, H.; Wilkie, A.O. Nonsense-mediated decay and the molecular pathogenesis of mutations in SALL1 and GLI3. Am. J. Med. Genet. A. 2007, 143A, 3150–3160. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Moriniere, V.; Knuppel, T.; Charbit, M.; Dusek, J.; Ghiggeri, G.M.; Jankauskiené, A.; Mir, S.; Montini, G.; Peco-Antic, A.; et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: Results of the ESCAPE study. J. Am. Soc. Nephrol. 2006, 17, 2864–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodopiutz, J.; Zoller, H.; Fenwick, A.L.; Arnhold, R.; Schmid, M.; Prayer, D.; Müller, T.; Repa, A.; Pollak, A.; Aufricht, C.; et al. Homozygous SALL1 mutation causes a novel multiple congenital anomaly-mental retardation syndrome. J. Pediatr. 2013, 162, 612–617. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.; Lenarduzzi, S.; Cappellani, S.; Pecile, V.; Morgutti, M.; Orzan, E.; Ghiselli, S.; Ambrosetti, U.; Brumat, M.; Gajendrarao, P.; et al. Genomic Studies in a Large Cohort of Hearing Impaired Italian Patients Revealed Several New Alleles, a Rare Case of Uniparental Disomy (UPD) and the Importance to Search for Copy Number Variations. Front. Genet. 2018, 9, 681. [Google Scholar] [CrossRef]
- Levy, M.A.; McConkey, H.; Kerkhof, J.; Barat-Houari, M.; Bargiacchi, S.; Biamino, E.; Bralo, M.P.; Cappuccio, G.; Ciolfi, A.; Clarke, A.; et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Adv. 2021, 3, 100075. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Deletions | Family | Patient | IA/AS | DE | HM | FM | HI | CHD | RA | DD/ID |
|---|---|---|---|---|---|---|---|---|---|---|
| This report | 1 | 1 | VPA | + | + | + | + | − | − | + |
| 2 | − | − | − | − | + | − | − | − | ||
| 3 | + | + | + | − | + | − | − | − | ||
| [15] | 1 | 1 | VPA | + | + | + | + | NR | NR | − |
| 2 | + | + | + | − | NR | − | − | − | ||
| 3 | 1 | + | + | + | + | + | − | + | − | |
| [16] | 1 | 1 | + | + | − | + | − | − | − | + |
| 2 | + | − | + | − | − | − | − | − | ||
| [17] | 1 | 1 | + | AES | + | NR | NR | − | + | + |
| DECIPHER | 1 | 1 | + | NR | + | NR | + | NR | NR | + |
| TOT | 6 | 10 | 7/10 | 6/9 | 8/10 | 4/8 | 6/8 | 0/8 | 2/8 | 4/10 |
| p.Arg276Ter | Patient | IA/AS | DE | HM | FM | HI | CHD | RA | DD/ID |
|---|---|---|---|---|---|---|---|---|---|
| [19] | 3-1 | + | + | + | + | + | − | − | + |
| 4-1 | + | + | + | + | + | − | + | − | |
| 5-1 | − | + | + | + | + | + | + | − | |
| [20] | TB6 | + | + | + | − | + | + | − | NR |
| TB7 | + | + | + | − | + | − | + | NR | |
| TB8 | + | + | + | + | + | − | + | NR | |
| [21] | 2 | AT | + | + | + | NR | + | + | NR |
| [11] | 103/1 | AT | + | + | + | + | + | + | − |
| 119/1 | + | + | + | + | + | − | − | NR | |
| 149/1 | + | + | + | − | + | + | + | NR | |
| 182/1 | + | + | + | + | − | − | − | + | |
| 186/1 | + | + | + | + | + | + | − | − | |
| 188/1 | + | + | − | − | − | − | + | − | |
| 205/1 | AT | + | + | + | + | NR | + | − | |
| 224/1 | VPA | + | + | + | + | + | − | − | |
| 224/2 | + | + | + | + | − | + | − | NR | |
| [22] | AT | + | + | + | + | NR | − | NR | |
| [23] | + | + | + | + | + | − | + | + | |
| [24] | B934 | NR | + | + | NR | + | + | NR | NR |
| [25] | + | + | + | NR | + | + | + | NR | |
| TOT | 20 | 13/19 | 20/20 | 19/20 | 14/18 | 16/19 | 10/18 | 11/19 | 3/10 |
| IA/AS | DE | HM | FM | HI | CHD | RA | DD/ID | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yes/Tot | % | Yes/Tot | % | Yes/Tot | % | Yes/Tot | % | Yes/Tot | % | Yes/Tot | % | Yes/Tot | % | Yes/Tot | % | |
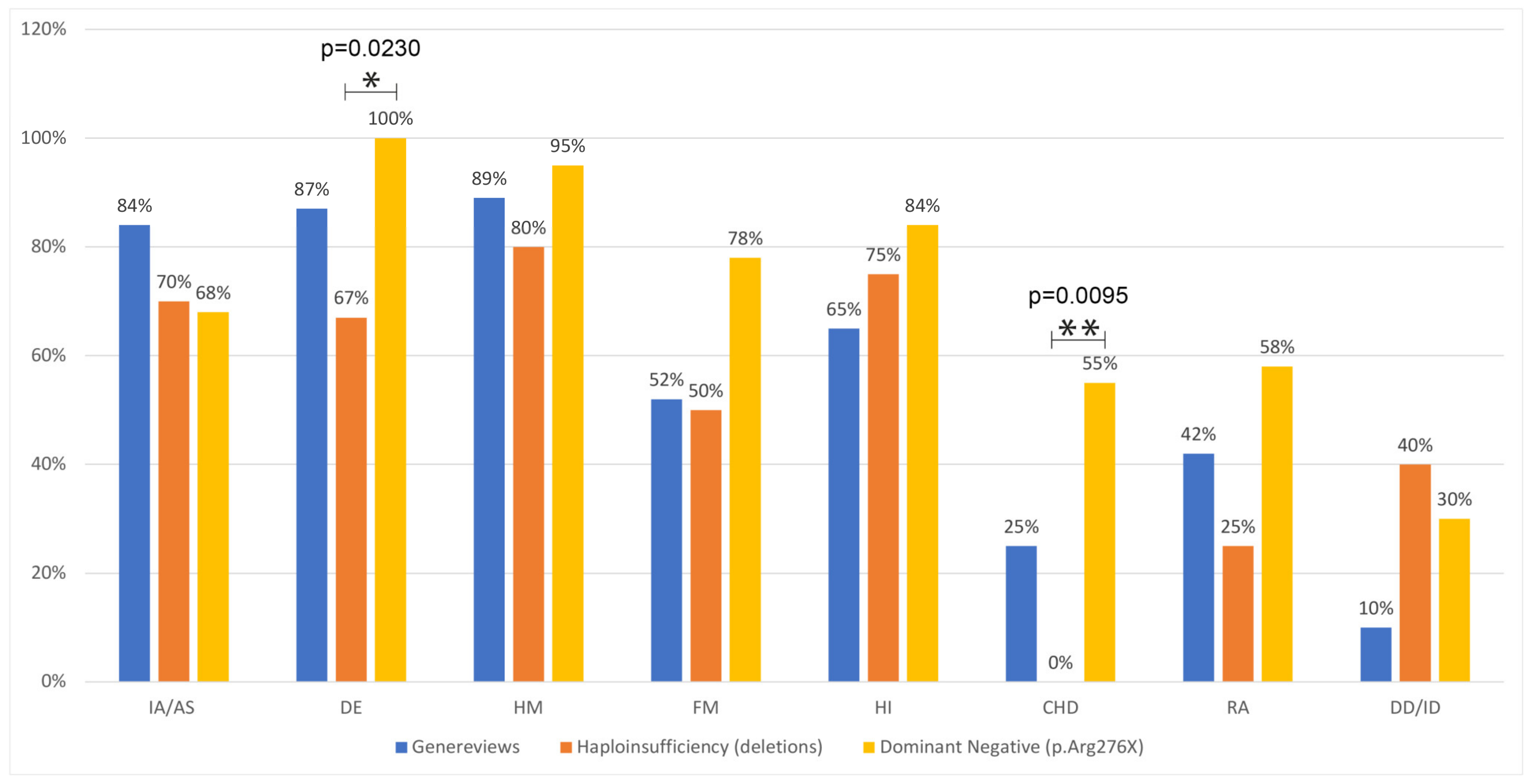
| Group 1: haploinsufficiency (deletions) | 7/10 | 70% | 6/9 | 67% | 8/10 | 80% | 4/8 | 50% | 6/8 | 75% | 0/8 | 0% | 2/8 | 25% | 4/10 | 40% |
| Group 2: dominant negative (p.Arg276Ter) | 13/19 | 68% | 20/20 | 100% | 19/20 | 95% | 14/18 | 78% | 16/19 | 84% | 10/18 | 55% | 11/19 | 58% | 3/10 | 30% |
| p-value | 1.0000 | 0.0230 | 0.2512 | 0.1972 | 0.6159 | 0.0095 | 0.2087 | 1.0000 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Innoceta, A.M.; Olivucci, G.; Parmeggiani, G.; Scarano, E.; Pragliola, A.; Graziano, C. Chromosomal Microarray Analysis Identifies a Novel SALL1 Deletion, Supporting the Association of Haploinsufficiency with a Mild Phenotype of Townes–Brocks Syndrome. Genes 2023, 14, 258. https://doi.org/10.3390/genes14020258
Innoceta AM, Olivucci G, Parmeggiani G, Scarano E, Pragliola A, Graziano C. Chromosomal Microarray Analysis Identifies a Novel SALL1 Deletion, Supporting the Association of Haploinsufficiency with a Mild Phenotype of Townes–Brocks Syndrome. Genes. 2023; 14(2):258. https://doi.org/10.3390/genes14020258
Chicago/Turabian StyleInnoceta, Anna Maria, Giulia Olivucci, Giulia Parmeggiani, Emanuela Scarano, Antonella Pragliola, and Claudio Graziano. 2023. "Chromosomal Microarray Analysis Identifies a Novel SALL1 Deletion, Supporting the Association of Haploinsufficiency with a Mild Phenotype of Townes–Brocks Syndrome" Genes 14, no. 2: 258. https://doi.org/10.3390/genes14020258