
Genome Survey Sequencing of the Mole Cricket Gryllotalpa orientalis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection and DNA Extraction
2.2. Library Preparation and Sequencing
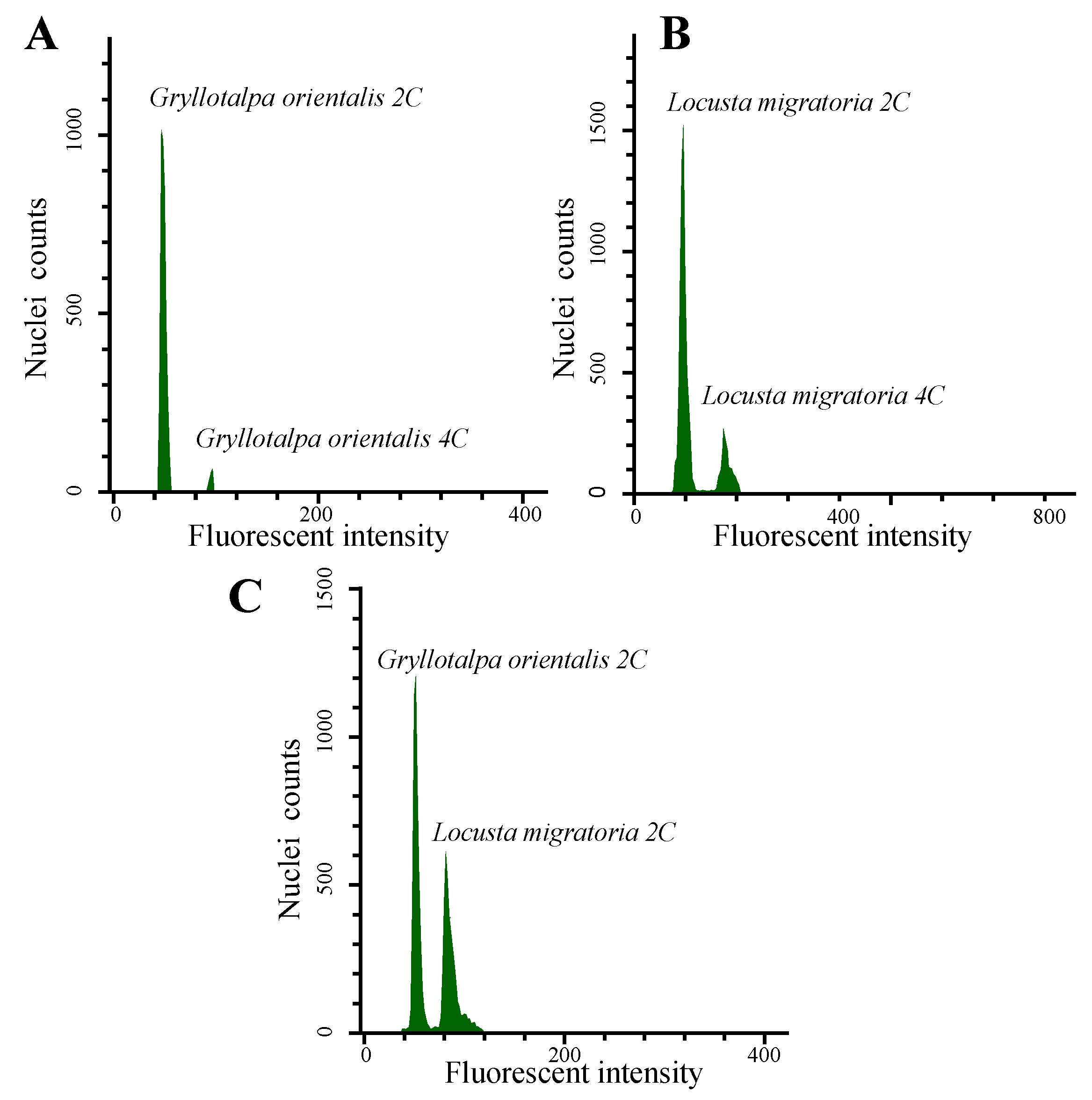
2.3. Measuring Genome Size Using Flow Cytometry (FCM)
2.4. Estimating Genome Size Based on k-mer in G. orientalis
2.5. Repetitive Elements in G. orientalis
2.6. Discovering Microsatellite in G. orientalis
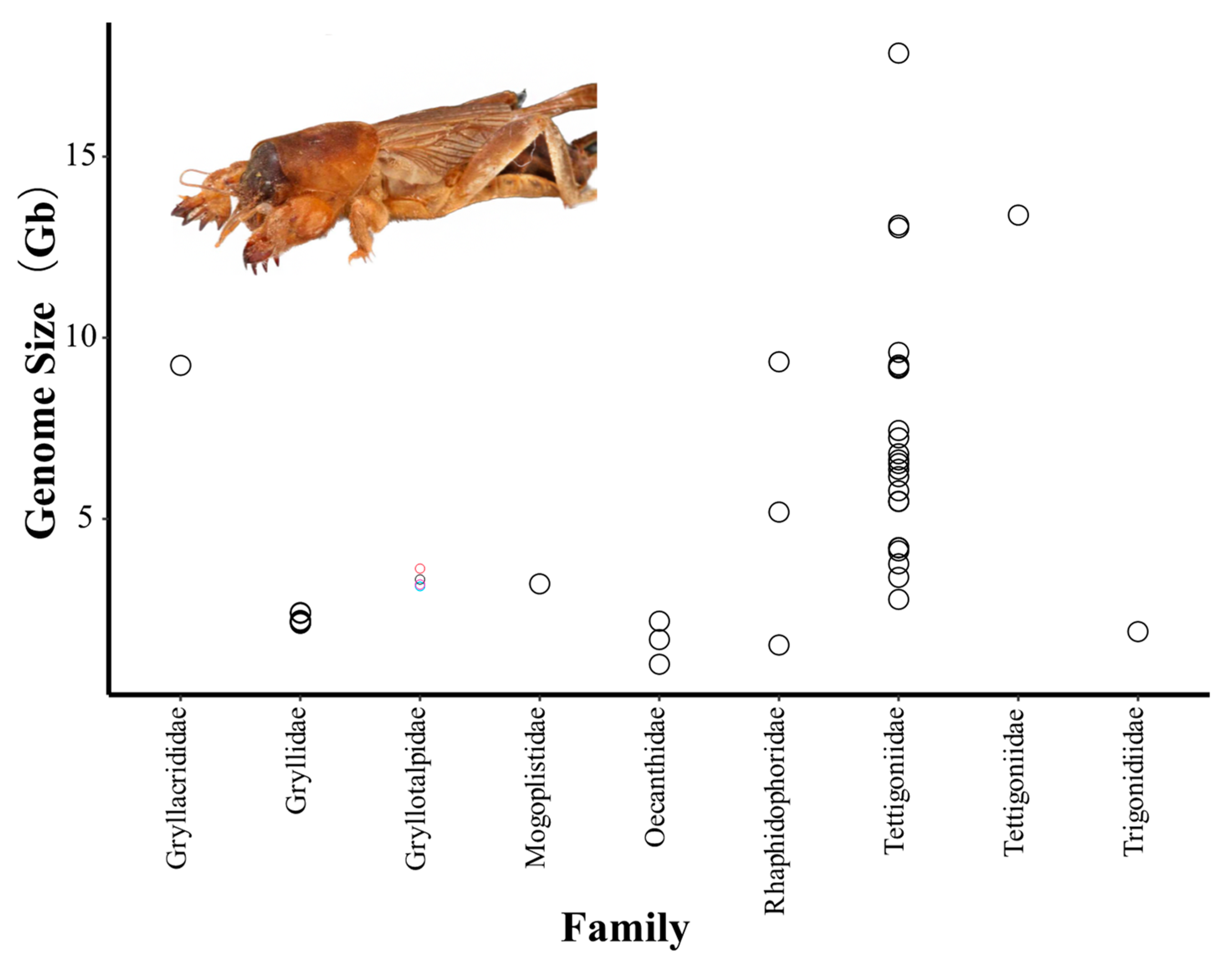
3. Results and Discussion
3.1. Genome Size Measuring Using FCM in G. orientalis
3.2. Genome Size Estimation Using k-mer in G. orientalis
3.3. Repetitive Elements in the Nuclear Genome of G. orientalis
3.4. Microsatellite Discovery in G. orientalis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cadena-Castañeda, O.J. The phylogeny of mole crickets (Orthoptera: Gryllotalpoidea: Gryllotalpidae). Zootaxa 2015, 3985, 451–490. [Google Scholar] [CrossRef] [Green Version]
- Cigliano, M.M.; Braun, H.; Eades, D.C.; Otte, D. Orthoptera Species File. Version 5.0/5.0. 2018. Available online: https://orthoptera.speciesfile.org/ (accessed on 10 November 2022).
- Leach, W.E. The Edinburgh Encyclopaedia; Blackwood: Edinburgh, UK, 1815; Volume 9, pp. 57–172. [Google Scholar]
- Cadena-Castaneda, O.J. Two new species of mole crickets (Orthoptera: Gryllotalpidae: Scapteriscinae) from the Colombian Amazon and Orinoquia rainforests. Zootaxa 2011, 3126, 62–68. [Google Scholar] [CrossRef]
- Kim, I.; Cha, S.Y.; Yoon, M.H.; Hwang, J.S.; Lee, S.M.; Sohn, H.D.; Jin, B.R. The complete nucleotide sequence and gene organization of the mitochondrial genome of the oriental mole cricket, Gryllotalpa orientalis (Orthoptera: Gryllotalpidae). Gene 2005, 353, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, K.; Tsutsumi, M.; Hotta, Y. Potato pest insect, mole cricket and the control method. Hokunou 1960, 22, 157–167. [Google Scholar]
- Matsuura, H.; Oda, H.; Ishizaki, H. Damage to Chinese yam by the African mole cricket, Gryllotalpa africana Palisot de Beauvois, and its control by chemicals. Jpn. J. Appl. Entomol. Zool. 1985, 29, 36–40. [Google Scholar] [CrossRef]
- Semlitsch, R.D. Life history of the northern mole cricket, Neocurtilla hexadactyla (Orthoptera: Gryllotalpidae), utilizing Carolina-bay habitats. Ann. Entomol. Soc. Am. 1986, 79, 256–261. [Google Scholar] [CrossRef]
- Hayslip, N.C. Notes on biological studies of mole crickets at Plant City, Florida. Fla. Entomol. 1943, 26, 33–46. [Google Scholar] [CrossRef]
- Kang, L.; Liu, C.; Liu, X. Fauna Sinica Insecta Volume 57: Orthoptera, Gryllotalpidae; Science Press: Beijing, China, 2014. [Google Scholar]
- Endo, C. Seasonal wing dimorphism and life cycle of the mole cricket Gryllotalpa orientalis (Orthoptera: Gryllotalpidae). Eur. J. Entomol. 2006, 103, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Endo, C. The underground life of the oriental mole cricket: An analysis of burrow morphology. J. Zool. 2007, 273, 414–420. [Google Scholar] [CrossRef]
- Yuan, H.; Huang, Y.; Mao, Y.; Zhang, N.; Nie, Y.; Zhang, X.; Zhou, Y.; Mao, S. The Evolutionary Patterns of Genome Size in Ensifera (Insecta: Orthoptera). Front. Genet. 2021, 12, 693541. [Google Scholar] [CrossRef]
- Gregory, T.R.; Johnston, J.S. Genome size diversity in the family Drosophilidae. Heredity 2008, 101, 228–238. [Google Scholar] [CrossRef]
- Hare, E.E.; Johnston, J.S. Genome size determination using flow cytometry of propidium iodide-stained nuclei. In Molecular Methods for Evolutionary Genetics; Springer: Berlin/Heidelberg, Germany, 2012; pp. 3–12. [Google Scholar]
- Wang, X.; Fang, X.; Yang, P.; Jiang, X.; Jiang, F.; Zhao, D.; Li, B.; Cui, F.; Wei, J.; Ma, C.; et al. The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 2014, 5, 2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbraith, D.W.; Harkins, K.R.; Maddox, J.M.; Ayres, N.M.; Sharma, D.P.; Firoozabady, E. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 1983, 220, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. 2010. [Google Scholar]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- Baeza, J.A.; Neo, M.L.; Huang, D. Genomic Survey and Resources for the Boring Giant Clam Tridacna crocea. Genes 2022, 13, 903. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [Green Version]
- Rexdb Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mobile DNA 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Castoe, T.A.; Poole, A.W.; de Koning, A.P.J.; Jones, K.L.; Tomback, D.F.; Oyler-McCance, S.J.; Fike, J.A.; Lance, S.L.; Streicher, J.W.; Smith, E.N.; et al. Rapid microsatellite identification from Illumina paired-end genomic sequencing in two birds and a snake. PLoS ONE 2012, 7, e30953. [Google Scholar] [CrossRef] [Green Version]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. In Bioinformatics Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Gregory, T.R. Animal Genome Size Database. 2015. Available online: http//www.genomesize.com (accessed on 14 November 2022).
- Dolezel, J. Nuclear DNA content and genome size of trout and human. Cytom. Part A 2003, 51, 127–128. [Google Scholar]
- Mao, Y.; Zhang, N.; Nie, Y.; Zhang, X.; Li, X.; Huang, Y. Genome Size of 17 Species from Caelifera (Orthoptera) and Determination of Internal Standards With Very Large Genome Size in Insecta. Front. Physiol. 2020, 11, 1321. [Google Scholar] [CrossRef]
- Kataoka, K.; Togawa, Y.; Sanno, R.; Asahi, T.; Yura, K. Dissecting cricket genomes for the advancement of entomology and entomophagy. Biophys. Rev. 2020, 14, 75–97. [Google Scholar] [CrossRef]
- Liu, X.; Majid, M.; Yuan, H.; Chang, H.; Zhao, L.; Nie, Y.; He, L.; Liu, X.; He, X.; Huang, Y. Transposable element expansion and low-level piRNA silencing in grasshoppers may cause genome gigantism. BMC Biol. 2022, 20, 243. [Google Scholar] [CrossRef] [PubMed]
- Ylla, G.; Nakamura, T.; Itoh, T.; Kajitani, R.; Toyoda, A.; Tomonari, S.; Bando, T.; Ishimaru, Y.; Watanabe, T.; Fuketa, M.; et al. Insights into the genomic evolution of insects from cricket genomes. Commun. Biology. 2021, 4, 733. [Google Scholar] [CrossRef]
- Vieira, M.L.C.; Santini, L.; Diniz, A.L.; Munhoz, C.D.F. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biology. 2016, 39, 312–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, K.T.; Hill, P.S.M.; Booth, W. The kin selection hypothesis in a lekking mole cricket: Assessing nested patterns of relatedness. Biol. J. Linn. Soc. 2016, 118, 382–393. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, K.; Guan, D.-L.; Huang, H.-T.; Xu, S.-Q. Genome Survey Sequencing of the Mole Cricket Gryllotalpa orientalis. Genes 2023, 14, 255. https://doi.org/10.3390/genes14020255
Sun K, Guan D-L, Huang H-T, Xu S-Q. Genome Survey Sequencing of the Mole Cricket Gryllotalpa orientalis. Genes. 2023; 14(2):255. https://doi.org/10.3390/genes14020255
Chicago/Turabian StyleSun, Kuo, De-Long Guan, Hua-Teng Huang, and Sheng-Quan Xu. 2023. "Genome Survey Sequencing of the Mole Cricket Gryllotalpa orientalis" Genes 14, no. 2: 255. https://doi.org/10.3390/genes14020255