
The Genetic Diversity of White-Backed Planthoppers (Sogatella furcifera) between Myanmar and Yunnan Province of China
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. Mitochondrial COI Gene Amplification and Sequencing
2.3. Population Genetic Diversity and Structure Analysis
2.4. Demographic History Analysis
3. Results
3.1. Genetic Diversity Analysis
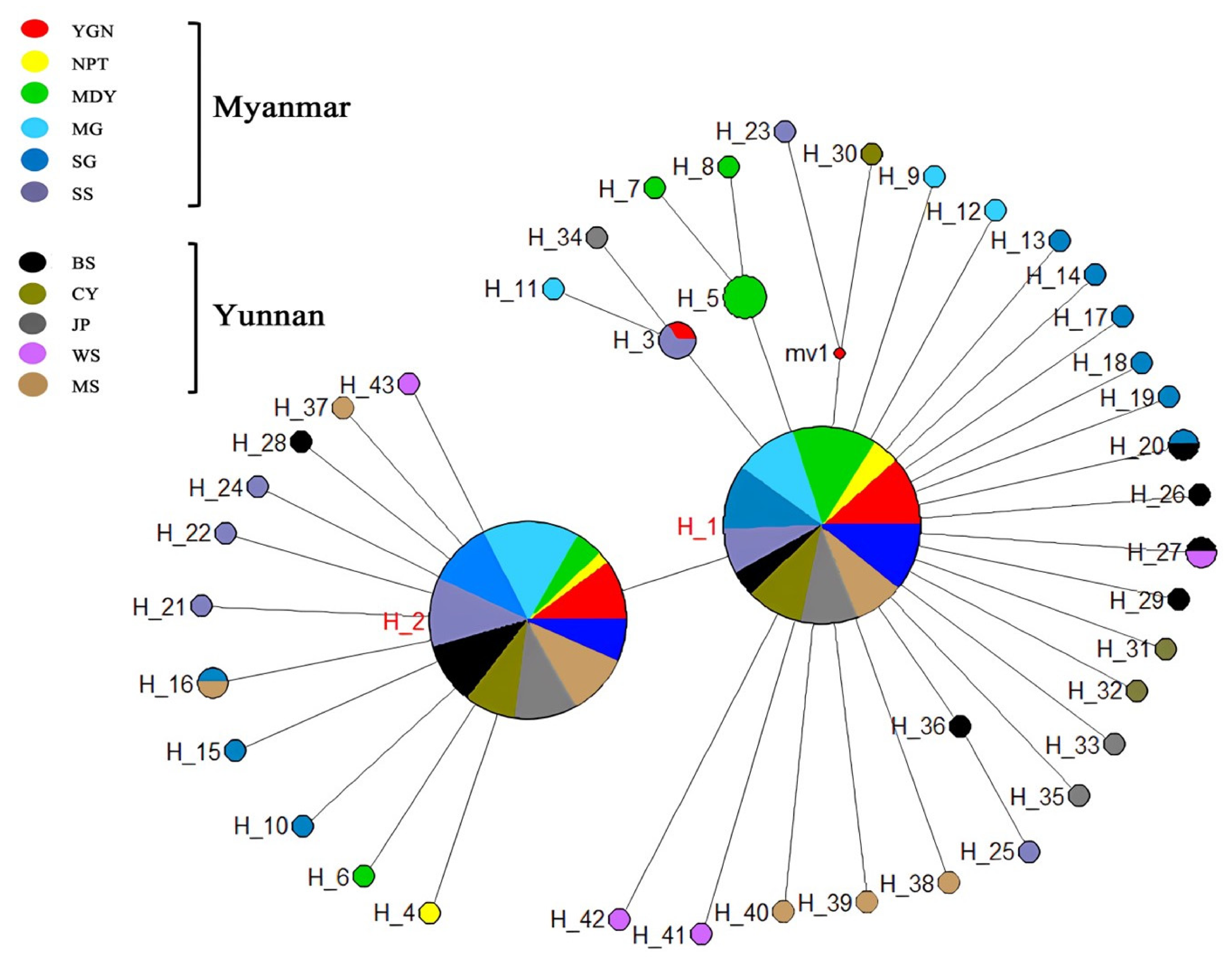
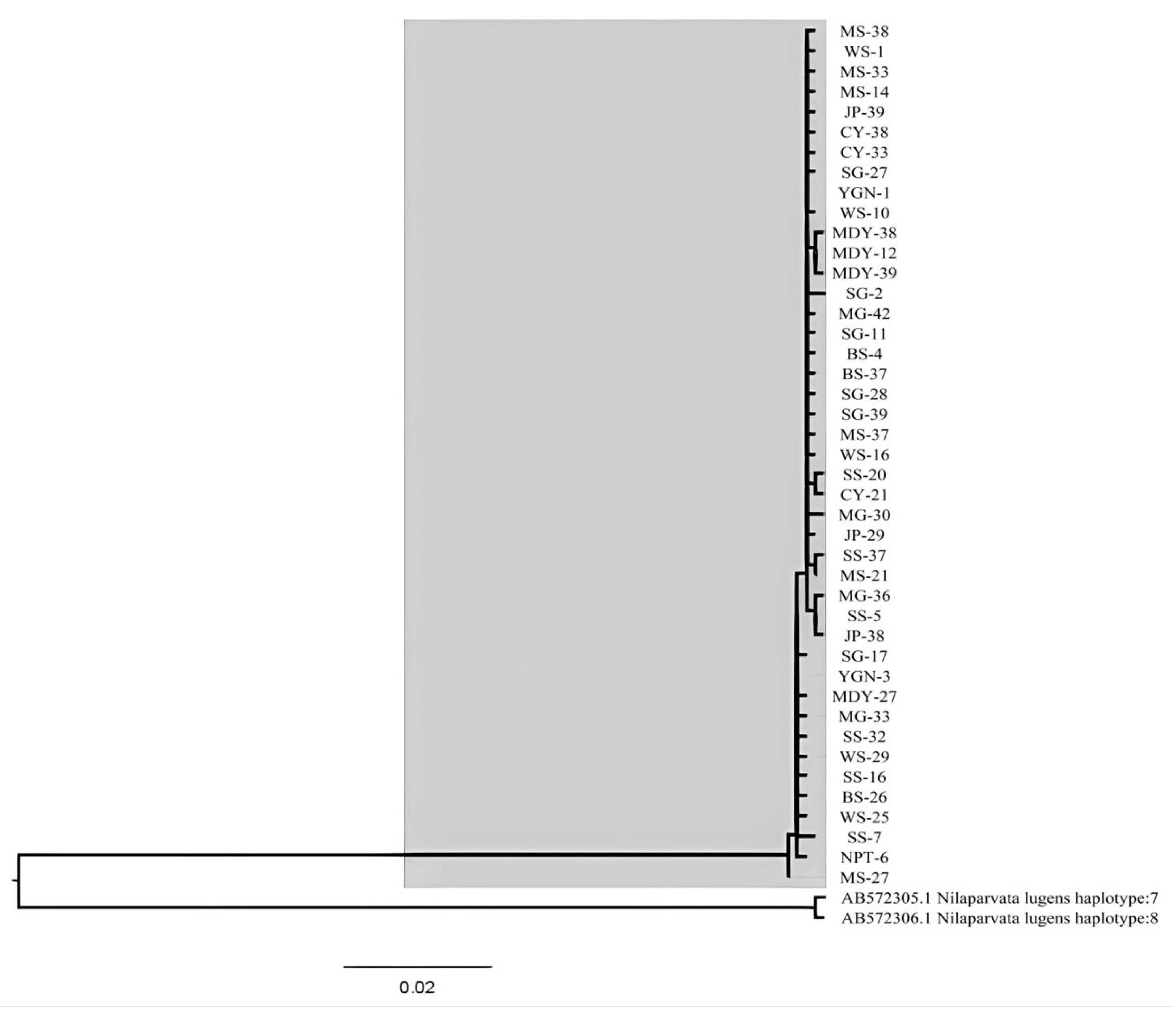
3.2. Population Genetic Structure and Haplotype Distribution
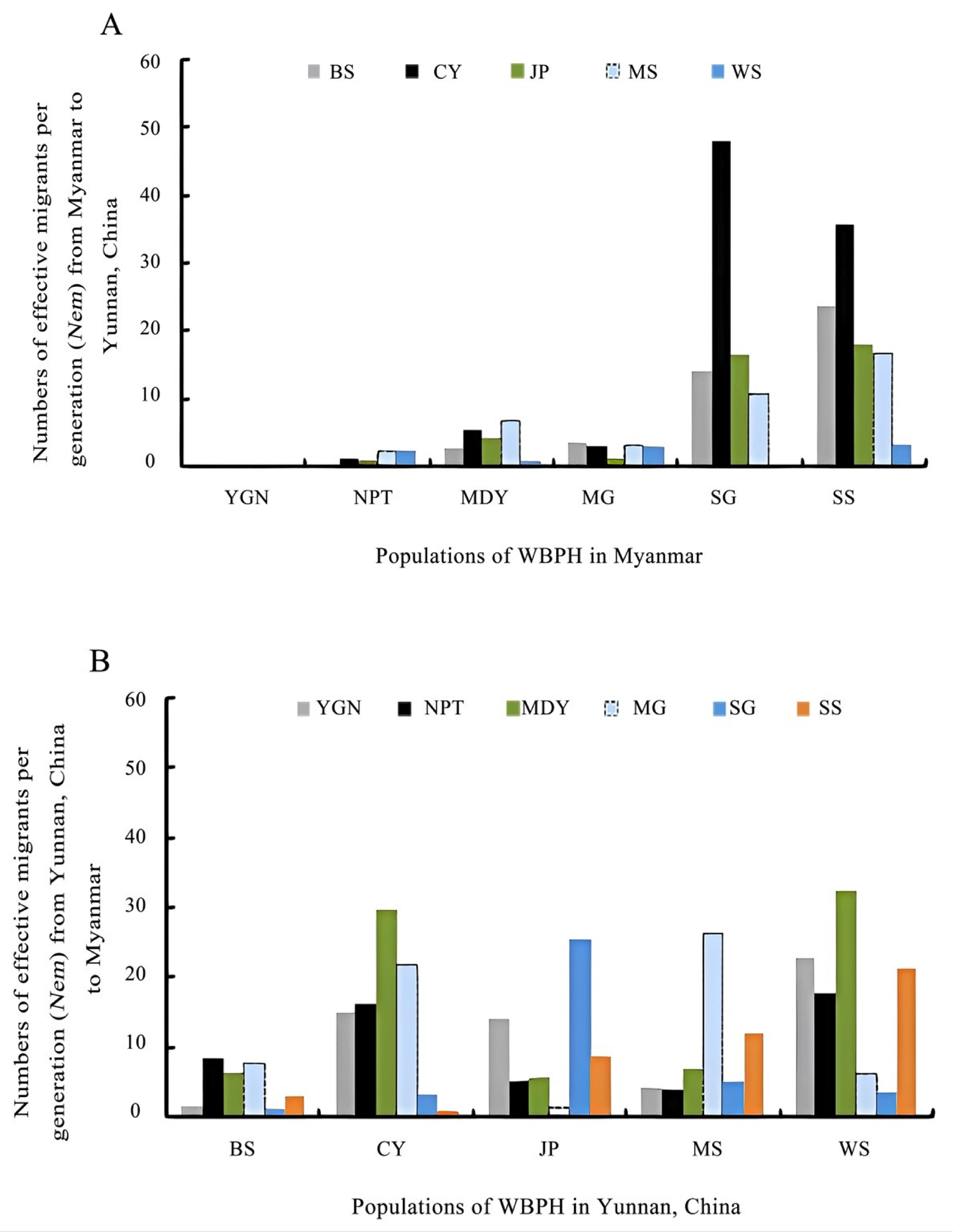
3.3. Gene Flow Based on Mitochondrial DNA
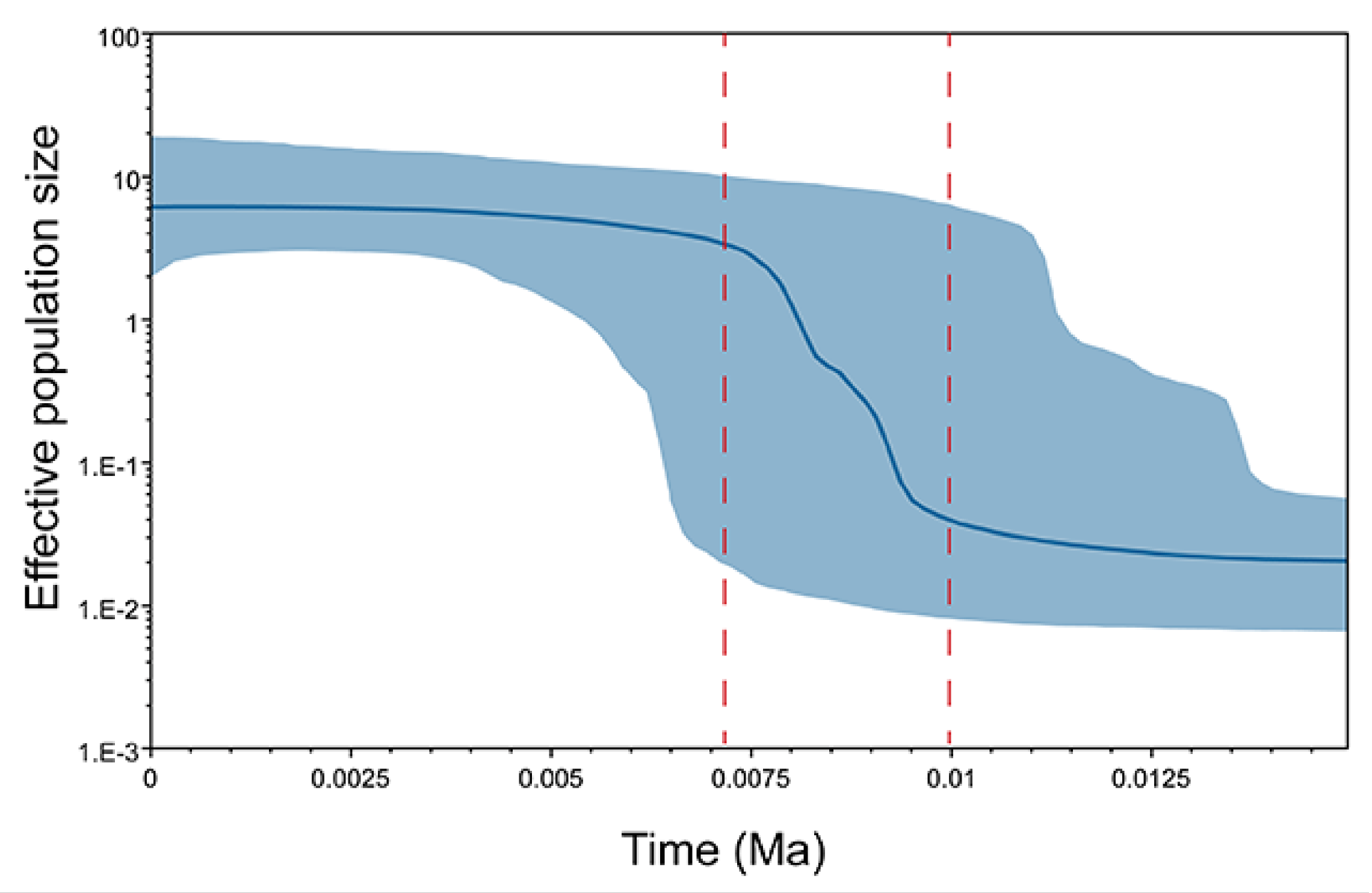
3.4. Demographic History Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rubia-Sanchez, E.; Suzuki, Y.; Arimura, K.; Miyamoto, K.; Matsumura, M.; Watanabe, T. Comparing Nilaparvata lugens (Stal) and Sogatella furcifera (Horvath) (Homoptera: Delphacidae) feeding effects on rice plant growth processes at the vegetative stage. Crop Prot. 2003, 22, 967–974. [Google Scholar] [CrossRef]
- Xiao, T.-G.; Tang, J.-X. Effects of the susceptibility of rice varieties to Sogatella furcifera on nymphal development and reproduction of Microvelia horvathi through a food chain. Insect Sci. 2007, 14, 317–321. [Google Scholar] [CrossRef]
- Dyck, V.A.; Thomas, B. The brown planthopper problem. In Brown Planthopper: Threat to Rice Production in Asia; International Rice Research Institute: Los Baños, Philippines, 1979; pp. 3–17. [Google Scholar]
- Dingle, H.; Drake, V.A. What Is Migration? BioScience 2007, 57, 113–121. [Google Scholar] [CrossRef]
- Alerstam, T.; Chapman, J.W.; Bäckman, J.; Smith, A.D.; Karlsson, H.; Nilsson, C.; Reynolds, D.R.; Klaassen, R.H.G.; Hill, J.K. Convergent patterns of long-distance nocturnal migration in noctuid moths and passerine birds. Proc. R. Soc. B Biol. Sci. 2011, 278, 3074–3080. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Lu, F.; Zhai, B.; Lu, M.; Liu, W.; Zhu, F.; Wu, X.-W.; Chen, G.; Zhang, X. Outbreaks of the Brown Planthopper Nilaparvata lugens (Stål) in the Yangtze River Delta: Immigration or Local Reproduction? PLoS ONE 2014, 9, e88973. [Google Scholar] [CrossRef] [PubMed]
- Otuka, A. Migration of rice planthoppers and their vectored re-emerging and novel rice viruses in East Asia. Front. Microbiol. 2013, 4, 309. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wen, J.; Cai, D.; Li, P.; Xu, D.; Zhang, S. Southern rice black-streaked dwarf virus: A new proposed Fijivirus species in the family Reoviridae. Chin. Sci. Bull. 2008, 53, 3677–3685. [Google Scholar] [CrossRef]
- Zhou, G.; Xu, D.; Xu, D.; Zhang, M. Southern rice black-streaked dwarf virus: A white-backed planthopper-transmitted fijivirus threatening rice production in Asia. Front. Microbiol. 2013, 4, 270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Shen, H.; Yin, Y.; Li, X.; Lyu, J.; Chen, A. The occurrence characteristics and population dynamics of Sogatella furcifera (Horváth) in Yunnan Province. Chin. J. Appl. Entomol. 2013, 51, 516–524. [Google Scholar]
- Win, S.S.; Muhamad, R.; Ahmad, Z.A.M.; Adam, N.A. Population fluctuations of Brown Plant Hopper Nilaparvata lugens Stal. and White Backed Plant Hopper Sogatella furcifera Horvath on rice. J. Entomol. 2011, 8, 183–190. [Google Scholar] [CrossRef]
- Hoang, A.T.; Zhang, H.-M.; Yang, J.; Chen, J.-P.; Hébrard, E.; Zhou, G.; Vinh, V.N.; Cheng, J. Identification, Characterization, and Distribution of Southern rice black-streaked dwarf virus in Vietnam. Plant Dis. 2011, 95, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, X.; Chu, D.; Zhao, X.; Sathya, K.; Douangboupha, B.; Kyaw, M.M.; Kongchuensin, M.; Somrith, A.; Ngo, V.V.; et al. Extensive gene flow of white-backed planthopper in the Greater Mekong Subregion as revealed by microsatellite markers. Sci. Rep. 2017, 7, 15905. [Google Scholar] [CrossRef]
- Huang, S.-H.; Cheng, C.-H.; Chen, C.-N.; Wu, W.-J.; Otuka, A. Estimating the immigration source of rice planthoppers, Nilaparvata lugens (Stål) and Sogatella furcifera (Horváth) (Homoptera: Delphacidae), in Taiwan. Appl. Entomol. Zool. 2010, 45, 521–531. [Google Scholar] [CrossRef]
- Hu, G.; Lu, M.H.; Tuan, H.A.; Liu, W.C.; Xie, M.C.; McInerney, C.E.; Zhai, B.P. Population dynamics of rice planthoppers, Nilaparvata lugens and Sogatella furcifera (Hemiptera, Delphacidae) in Central Vietnam and its effects on their spring migration to China. Bull. Entomol. Res. 2016, 107, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Fu, X.; Wu, K. Seasonal migration of white-backed planthopper Sogatella furcifera Horváth (Hemiptera: Delphacidae) over the Bohai Sea in northern China. J. Asia-Pac. Entomol. 2017, 20, 1358–1363. [Google Scholar] [CrossRef]
- Shen, H.M.; Lyu, J.P.; Zhou, J.Y.; Cheng, X.N.; Zhang, X.X.; Zhai, B.P. Source areas and landing mechanism of early immigration of white-backed planthoppers Sogatella furcifera (Horváth) in Yunnan, 2009. Acta Ecol. Sin. 2011, 31, 4350–4364. [Google Scholar]
- Liu, J.N.; Gui, F.R.; Li, Z.Y. Genetic diversity of the planthopper, Sogatella furcifera in the Greater Mekong Subregion detected by inter-simple sequence repeats (ISSR) markers. J. Insect Sci. 2010, 10, 52. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Matsumura, M.; Sanada-Morimura, S.; Hirai, Y.; Sato, Y.; Noda, H. Mitochondrial cox sequences of Nilaparvata lugens and Sogatella furcifera (Hemiptera, Delphacidae): Low specificity among Asian planthopper populations. Bull. Entomol. Res. 2013, 103, 382–392. [Google Scholar] [CrossRef]
- Li, X.Y.; Chu, D.; Yin, Y.Q.; Zhao, X.Q.; Chen, A.D.; Khay, S.; Douangboupha, B.; Kyaw, M.M.; Kongchuensin, M.; Ngo, V.V.; et al. Possible Source Populations of the White-backed Planthopper in the Greater Mekong Subregion Revealed by Mitochondrial DNA Analysis. Sci. Rep. 2016, 6, 39167. [Google Scholar] [CrossRef]
- Sun, S.; Bao, Y.; Wu, Y.; Lu, M.; Tuan, H. Analysis of the Huge Immigration of Sogatella furcifera (Hemiptera: Delphacidae) to Southern China in the Spring of 2012. Environ. Entomol. 2018, 47, 8–18. [Google Scholar] [CrossRef]
- Hu, S.; Fu, D.; Han, Z.; Ye, H. Density, Demography, and Influential Environmental Factors on Overwintering Populations of Sogatella furcifera (Hemiptera: Delphacidae) in Southern Yunnan, China. J. Insect Sci. 2015, 15, 58. [Google Scholar] [CrossRef]
- Kranthi, S.; Ghodke, A.B.; Puttuswamy, R.K.; Mandle, M.; Nandanwar, R.; Satija, U.; Pareek, R.K.; Desai, H.; Udikeri, S.S.; Balakrishna, D.J.; et al. Mitochondria COI-based genetic diversity of the cotton leafhopper Amrasca biguttula biguttula (Ishida) populations from India. Mitochondrial DNA Part A 2018, 29, 228–235. [Google Scholar] [CrossRef]
- Dumidae, A.; Janthu, P.; Subkrasae, C.; Pumidonming, W.; Dekumyoy, P.; Thanwisai, A.; Vitta, A. Genetic analysis of Cryptozona siamensis (Stylommatophora, Ariophantidae) populations in Thailand using the mitochondrial 16S rRNA and COI sequences. PLoS ONE 2020, 15, e0239264. [Google Scholar] [CrossRef] [PubMed]
- An, H.S.; Jee, Y.J.; Min, K.S.; Kim, B.L.; Han, S.J. Phylogenetic analysis of six species of pacific abalone (Haliotidae) based on DNA sequences of 16s rRNA and cytochrome c oxidase subunit I mitochondrial genes. Mar. Biotechnol. 2005, 7, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-L.; Wang, T.-Z.; Zhu, H.-F.; Pan, H.-B.; Yu, X.-P. Diversity and dynamics of microbial communities in brown planthopper at different developmental stages revealed by high-throughput amplicon sequencing. Insect Sci. 2020, 27, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Othman, S.N.; Putri, E.T.; Messenger, K.R.; Bae, Y.; Yang, Y.; Bova, T.; Reed, T.; Amin, H.; Chuang, M.-F.; Jang, Y.; et al. Impact of the Miocene orogenesis on Kaloula spp. radiation and implication of local refugia on genetic diversification. Integr. Zool. 2022, 17, 261–284. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Watterson, G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genetics 1983, 105, 437–460. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Lynch, M.; Crease, T.J. The analysis of population survey data on DNA sequence variation. Mol. Biol. Evol. 1990, 7, 377–394. [Google Scholar] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y. Statistical Tests of Neutrality of Mutations Against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Beerli, P.; Felsenstein, J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 2001, 98, 4563–4568. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Luo, T.; Wang, Y.; Wang, S.; Liu, T.; Xiao, N.; Zhou, J. Molecular phylogeny and historical biogeography of the cave fish genus Sinocyclocheilus (Cypriniformes: Cyprinidae) in southwest China. Integr. Zool. 2022, 17, 311–325. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Connor, T.R.; Sirén, J.; Aanensen, D.M.; Corander, J. Hierarchical and Spatially Explicit Clustering of DNA Sequences with BAPS Software. Mol. Biol. Evol. 2013, 30, 1224–1228. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Macaulay, V.; Richards, M. Median Networks: Speedy Construction and Greedy Reduction, One Simulation, and Two Case Studies from Human mtDNA. Mol. Phylogenet. Evol. 2000, 16, 8–28. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Anastasiou, I.; Vogler, A.P. Revisiting the Insect Mitochondrial Molecular Clock: The Mid-Aegean Trench Calibration. Mol. Biol. Evol. 2010, 27, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Polzin, T.; Daneshmand, S.V. On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 2003, 31, 12–20. [Google Scholar] [CrossRef]
- Qi, J.; Shi, F.; Zhang, B.; Chen, X.; Xing, J.; Furumitsu, K.; Corush, J.B.; Yamaguchi, A.; Zhang, J. Insights into genetic variation and demographic history of sharpnose rays: Examinations of three species of Telatrygon (Elasmobranchii, Dasyatidae) from the Indo-West Pacific. Integr. Zool. 2022, 17, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.S. Inferring phylogenies from mtDNA variation: Mitochondrial-gene trees versus nuclear-gene trees. Evolution 1995, 49, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, J. Allozyme diversity in plant species. In Plant Population Genetics Breeding & Genetic Resources; Sinauer Associates Inc.: Sunderland, MA, USA, 1989. [Google Scholar]
- Ma, M.; Peng, Z.; He, Y. Effects of Temperature on Functional Response of Anagrus nilaparvatae Pang et Wang (Hymenoptera: Mymaridae) on the Eggs of Whitebacked Planthopper, Sogatella furcifera Horváth and Brown Planthopper, Nilaparvata lugens Stål. J. Integr. Agric. 2012, 11, 1313–1320. [Google Scholar] [CrossRef]
- Xu, D.; Zhong, T.; Feng, W.; Zhou, G. Tolerance and responsive gene expression of Sogatella furcifera under extreme temperature stresses are altered by its vectored plant virus. Sci. Rep. 2016, 6, 31521. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, S.; Jiang, Z.; Chen, A.; Ma, M.; Zhang, G.; Zhai, B.; Li, C. Immigration Pathways of White-Backed Planthopper in the Confluence Area of the Two Monsoon Systems. J. Econ. Entomol. 2022, 115, 1480–1489. [Google Scholar] [CrossRef]
- Otuka, A.; Watanabe, T.; Suzuki, Y.; Matsumura, M.; Furuno, A.; Chino, M. Real-time prediction system for migration of rice planthoppers Sogatella furcifera (Horváth) and Nilaparvata lugens (Stål) (Homoptera: Delphacidae). Appl. Entomol. Zool. 2005, 40, 221–229. [Google Scholar] [CrossRef]
- Dsouli Aymes, N.; Michaux, J.; De Stordeur, E.; Couloux, A.; Veuille, M.; Duvallet, G. Global population structure of the stable fly (Stomoxys calcitrans) inferred by mitochondrial and nuclear sequence data. Infect. Genet. Evol. 2011, 11, 334–342. [Google Scholar] [CrossRef]
- Ma, C.; Yang, P.; Jiang, F.; Chapuis, M.-P.; Shali, Y.; Sword, G.A.; Kang, L.E. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 2012, 21, 4344–4358. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Lin, C.; Liang, A. Comparative phylogeography of two hemipteran species (Geisha distinctissima and Megacopta cribraria) in the Zhoushan Archipelago of China reveals contrasting genetic structures despite concordant historical demographies. Heredity 2020, 124, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Xun, H.; Li, H.; Li, S.; Wei, S.; Zhang, L.; Song, F.; Jiang, P.; Yang, H.; Han, F.; Cai, W. Population genetic structure and post-LGM expansion of the plant bug Nesidiocoris tenuis (Hemiptera: Miridae) in China. Sci. Rep. 2016, 6, 26755. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.W.; Bell, J.R.; Burgin, L.E.; Reynolds, D.R.; Pettersson, L.B.; Hill, J.K.; Bonsall, M.B.; Thomas, J.A. Seasonal migration to high latitudes results in major reproductive benefits in an insect. Proc. Natl. Acad. Sci. USA 2012, 109, 14924–14929. [Google Scholar] [CrossRef]
- Chapman, J.W.; Reynolds, D.R.; Wilson, K. Long-range seasonal migration in insects: Mechanisms, evolutionary drivers and ecological consequences. Ecol. Lett. 2015, 18, 287–302. [Google Scholar] [CrossRef]
- Bebber, D.P.; Holmes, T.; Gurr, S.J. The global spread of crop pests and pathogens. Glob. Ecol. Biogeogr. 2014, 23, 1398–1407. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
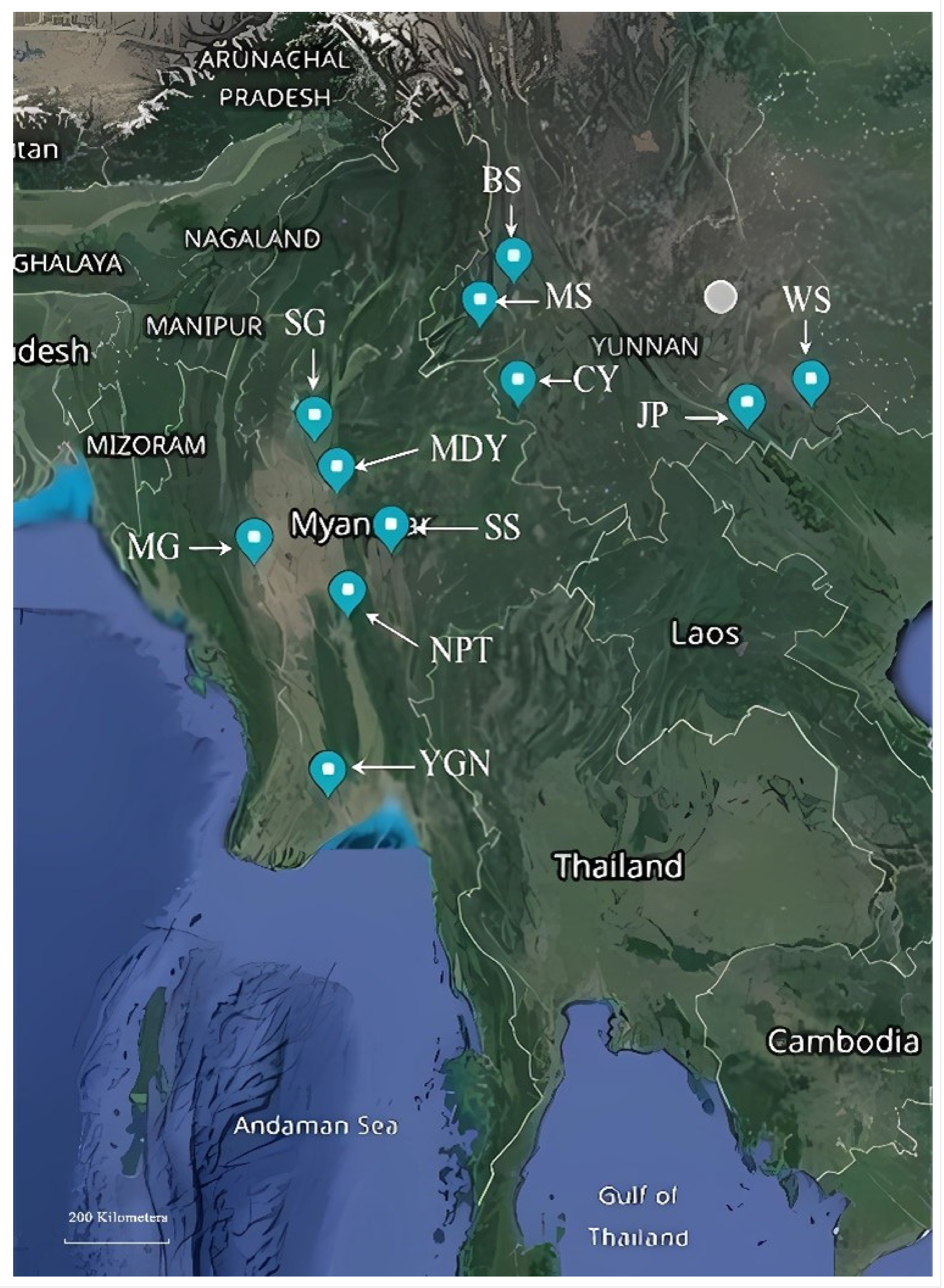
| Code | Location | Longitude | Latitude | Date |
|---|---|---|---|---|
| YGN | Tontay, Yangon Division, Myanmar | 16.71 | 95.94 | 5 September 2018 |
| NPT | Pyinmana, Naypyitaw, Myanmar | 19.71 | 96.28 | 10 May 2018 |
| MG | Salin, Magway Division, Myanmar | 20.58 | 94.65 | 8 June 2018 |
| SG | Shwebo, Sagaing Division, Myanmar | 22.58 | 95.70 | 15 May 2018 |
| MDY | Sintkaing, Mandalay Division, Myanmar | 21.74 | 96.09 | 11 April 2018 |
| SS | Taunggyi, Shan State, Myanmar | 20.91 | 96.94 | 10 June 2019 |
| BS | Baoshan, Yunnan, China | 25.13 | 99.17 | 10 May 2019 |
| CY | Cangyuan, Yunnan, China | 23.15 | 99.25 | 17 April 2019 |
| JP | Jinping, Yunnan, China | 22.78 | 103.23 | 15 May 2019 |
| MS | Mangshi, Yunnan, China | 24.44 | 98.58 | 10 May 2019 |
| WS | Wenshan, Yunnan, China | 23.16 | 104.35 | 5 May 2019 |
| Population Code (No. of Individuals Tested) | S | η | H | D (p) | Fs (p) | Hd (SD) | π (SD) | K | π (JC) |
|---|---|---|---|---|---|---|---|---|---|
| YGN (43) | 2 | 2 | 3 | −0.10389 (p > 0.10) | 0.011 (0.280) | 0.424 (0.068) | 0.00055 (0.00010) | 0.436 | 0.00055 |
| NPT(15) | 2 | 2 | 3 | −0.59419 (p > 0.10) | 0.518 (0.251) | 0.362 (0.145) | 0.00060 (0.00027) | 0.476 | 0.00060 |
| MDY (48) | 5 | 5 | 6 | −1.20378 (p > 0.10) | −2.582 (0.039) | 0.427 (0.085) | 0.00072 (0.00017) | 0.572 | 0.00073 |
| MG (47) | 6 | 6 | 6 | −1.24044 (p > 0.10) | −2.187 (0.067) | 0.574 (0.047) | 0.00089 (0.00014) | 0.705 | 0.00090 |
| SG (47) | 10 | 10 | 10 | −1.88055 (p < 0.05) | −7.005 (0.001) | 0.599 (0.068) | 0.00100 (0.00020) | 0.792 | 0.00100 |
| SS (38) | 10 | 10 | 8 | −1.72907 (p > 0.10) | −3.424 (0.022) | 0.662 (0.060) | 0.00104 (0.00017) | 0.825 | 0.00129 |
| BS (26) | 5 | 5 | 6 | −1.05038 (p > 0.10) | −2.447 (0.056) | 0.662 (0.060) | 0.00104 (0.00017) | 0.825 | 0.00105 |
| CY (36) | 5 | 5 | 5 | −1.29300 (p > 0.05) | −1.767 (0.098) | 0.505 (0.078) | 0.00077 (0.00017) | 0.608 | 0.00077 |
| JP (39) | 5 | 5 | 5 | −1.21593 (p > 0.10) | −1.632 (0.107) | 0.521 (0.068) | 0.00079 (0.00015) | 0.621 | 0.00079 |
| MS (38) | 7 | 7 | 8 | −1.52318 (p > 0.05) | −4.725 (0.007) | 0.623 (0.065) | 0.00096 (0.00015) | 0.760 | 0.00096 |
| WS (40) | 6 | 6 | 7 | −1.51888 (p > 0.10) | −4.204 (0.011) | 0.488 (0.087) | 0.00077 (0.00017) | 0.608 | 0.00077 |
| YGN | NPT | MDY | MG | SG | SS | BS | CY | JP | MS | WS | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| YGN | --- | ||||||||||
| NPT | 0.33603 | --- | |||||||||
| MDY | 0.03923 * | 0.62196 | --- | ||||||||
| MG | 0.27267 | 0.28552 | 0.00080 * | --- | |||||||
| SG | 0.98360 | 0.83533 | 0.02784 * | 0.25266 | --- | ||||||
| SS | 0.29515 | 0.44171 | 0.00056 * | 0.23759 | 0.30503 | --- | |||||
| BS | 0.02709 * | 0.09100 | 0.00051 * | 0.25605 | 0.23489 | 0.50366 | --- | ||||
| CY | 0.55549 | 0.56302 | 0.06487 | 0.41749 | 0.98038 | 0.27200 | 0.06905 | --- | |||
| JP | 0.60843 | 0.45765 | 0.01922 * | 0.54649 | 0.99198 | 0.30037 | 0.12561 | 0.85094 | --- | ||
| MS | 0.31108 | 0.73535 | 0.00908 * | 0.43340 | 0.87622 | 0.58144 | 0.39849 | 0.68066 | 0.64490 | --- | |
| WS | 0.38822 | 0.88951 | 0.11603 | 0.08123 | 0.83152 | 0.06724 * | 0.04464 | 0.68255 | 0.38785 | 0.26380 | --- |
| Source of Variation | Sum of Squares | Variance Components | Percentage Variation (p Value) |
|---|---|---|---|
| Among areas (ΦCT) | 0.203 | −0.00189 | −0.55% (0.90029) |
| Among population within areas (ΦSC) | 5.208 | 0.00638 | 1.85% (0.02639) |
| Within population (ΦST) | 137.766 | 0.33933 | 98.69% (0.03519) |
| Population Code (ἰ) | ϴἰ | YGN→ἰ | NPT→ἰ | MDY→ἰ | MG→ἰ | SG→ἰ | SS→ἰ | BS→ἰ | CY→ἰ | JP→ἰ | MS→ἰ | WS→ἰ | ∑(immigration) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| YGN | 0.0769 | --- | 4.8837 | 2.0503 | 50.8836 | 35.6291 | 49.9904 | 1.4941 | 14.9216 | 14.2943 | 4.3621 | 22.7011 | 201.2103 |
| NPT | 0.0070 | 19.7556 | --- | 3.1556 | 0.0284 | 25.2077 | 5.8986 | 8.5400 | 16.3450 | 5.3087 | 4.0499 | 17.8145 | 106.104 |
| MDY | 0.0033 | 2.0240 | 1.7057 | --- | 8.6284 | 35.7985 | 13.5892 | 6.5054 | 29.7385 | 5.7505 | 7.1291 | 32.4422 | 143.3115 |
| MG | 0.0860 | 4.0226 | 4.2957 | 1.9391 | --- | 6.5406 | 23.6466 | 7.6845 | 21.8516 | 1.5264 | 26.3409 | 6.1254 | 103.9734 |
| SG | 0.0363 | 0.0254 | 0.3617 | 1.4617 | 73.4156 | --- | 28.8666 | 1.1993 | 3.1616 | 25.4497 | 5.1209 | 3.6489 | 142.7114 |
| SS | 0.0522 | 2.9464 | 2.3777 | 0.0534 | 23.5356 | 11.2074 | --- | 3.2281 | 1.0616 | 8.8223 | 12.0602 | 21.2152 | 86.5079 |
| BS | 0.0087 | 0.0254 | 0.4643 | 2.6978 | 3.8124 | 14.1333 | 23.7510 | --- | 22.1785 | 12.7008 | 25.2923 | 7.5781 | 112.6339 |
| CY | 0.0350 | 1.4090 | 5.5603 | 3.1468 | 47.9596 | 35.7742 | 2.6972 | 8.2047 | --- | 5.8271 | 7.1291 | 19.4985 | 137.2065 |
| JP | 0.0288 | 0.8456 | 4.0950 | 1.1783 | 16.4836 | 18.1713 | 51.3476 | 1.1011 | 33.6350 | --- | 13.2876 | 19.9606 | 160.1057 |
| MS | 0.0335 | 2.5882 | 6.8810 | 3.2079 | 10.7500 | 16.7448 | 18.2528 | 8.5573 | 33.0050 | 0.0095 | --- | 18.8051 | 118.8016 |
| WS | 0.0495 | 2.4852 | 0.9777 | 2.9136 | 0.0284 | 3.2523 | 37.1142 | 3.7773 | 2.8116 | 28.3487 | 24.5335 | --- | 106.2425 |
| ∑(emigration) | --- | 36.1274 | 31.6028 | 21.8045 | 235.5256 | 202.4592 | 255.1542 | 50.2918 | 178.7100 | 108.0380 | 129.3056 | 169.7896 | --- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Chan, K.N.; Li, X.; Zhao, X.; Chu, D.; Yin, Y.; Liu, Y.; Chen, A. The Genetic Diversity of White-Backed Planthoppers (Sogatella furcifera) between Myanmar and Yunnan Province of China. Genes 2023, 14, 2164. https://doi.org/10.3390/genes14122164
Liu Y, Chan KN, Li X, Zhao X, Chu D, Yin Y, Liu Y, Chen A. The Genetic Diversity of White-Backed Planthoppers (Sogatella furcifera) between Myanmar and Yunnan Province of China. Genes. 2023; 14(12):2164. https://doi.org/10.3390/genes14122164
Chicago/Turabian StyleLiu, Yue, Khin Nyein Chan, Xiangyong Li, Xueqing Zhao, Dong Chu, Yanqiong Yin, Ying Liu, and Aidong Chen. 2023. "The Genetic Diversity of White-Backed Planthoppers (Sogatella furcifera) between Myanmar and Yunnan Province of China" Genes 14, no. 12: 2164. https://doi.org/10.3390/genes14122164