
Comparative Mitogenomic Analyses of Darkling Beetles (Coleoptera: Tenebrionidae) Provide Evolutionary Insights into tRNA-like Sequences

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. Mitogenome Sequencing, Assembly, and Annotation
2.3. Comparative Mitogenomic Analysis of Tenebrionidae
2.4. Phylogenetic Analysis
3. Results
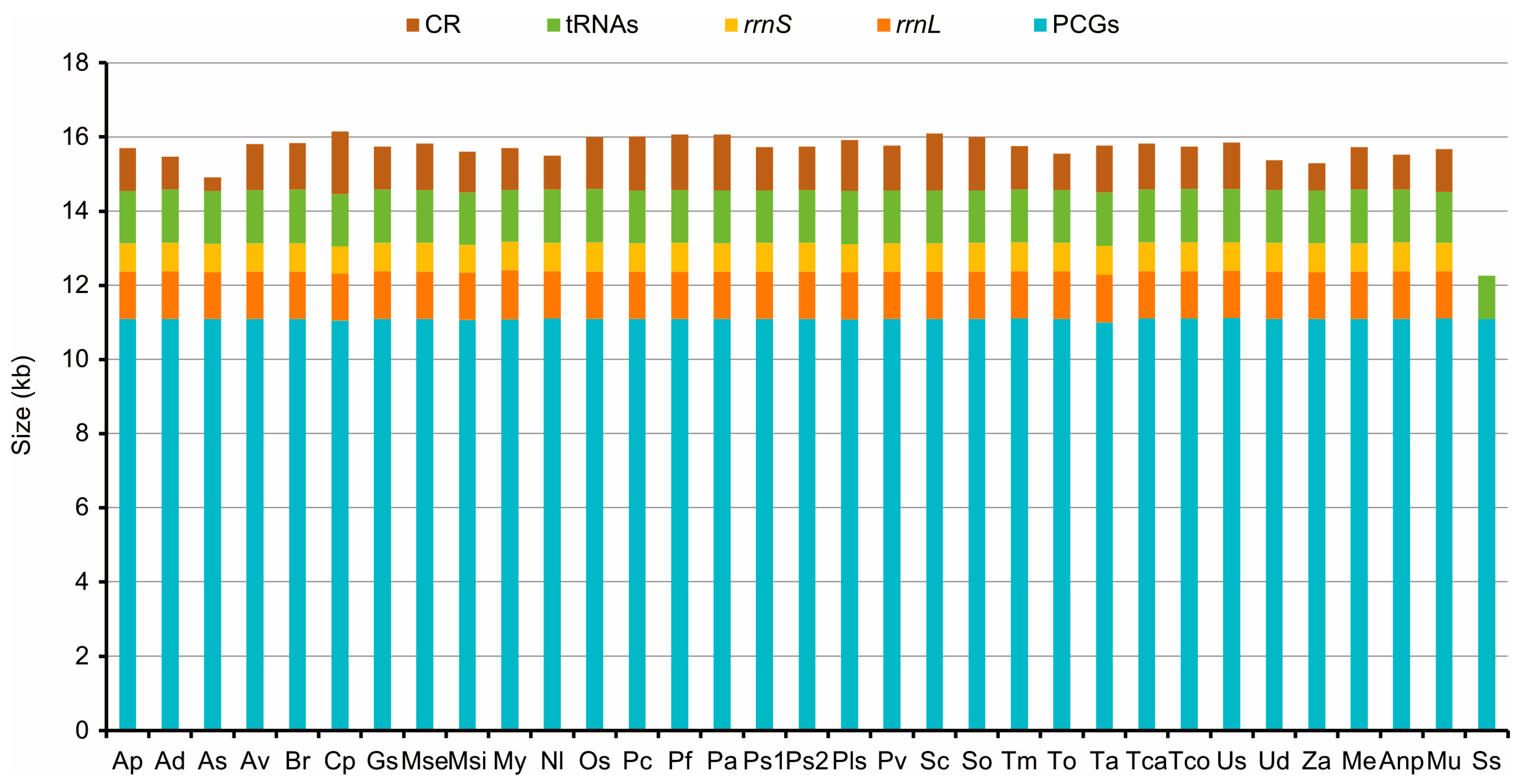
3.1. General Characteristics of Tenebrionidae Mitogenomes
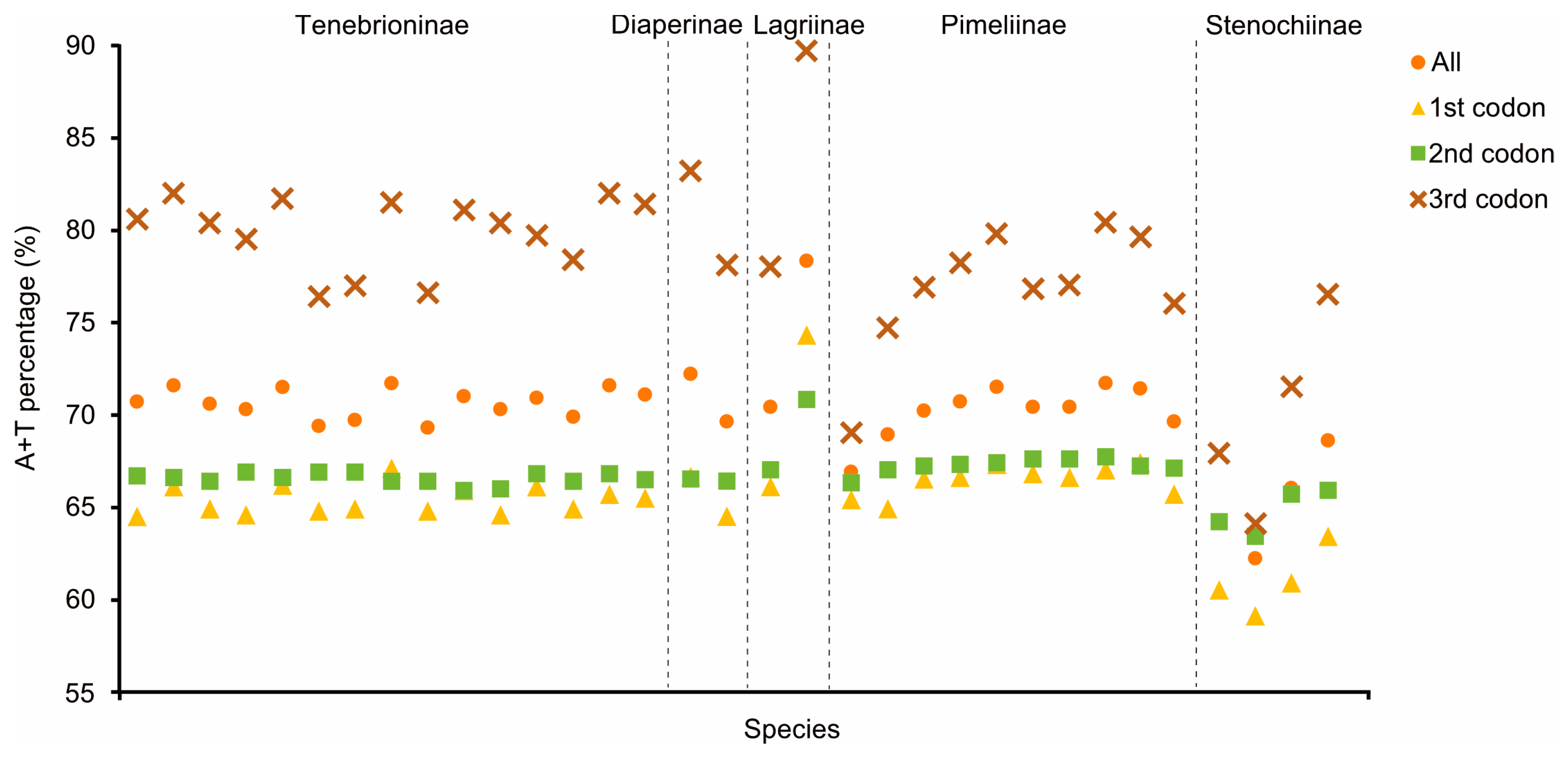
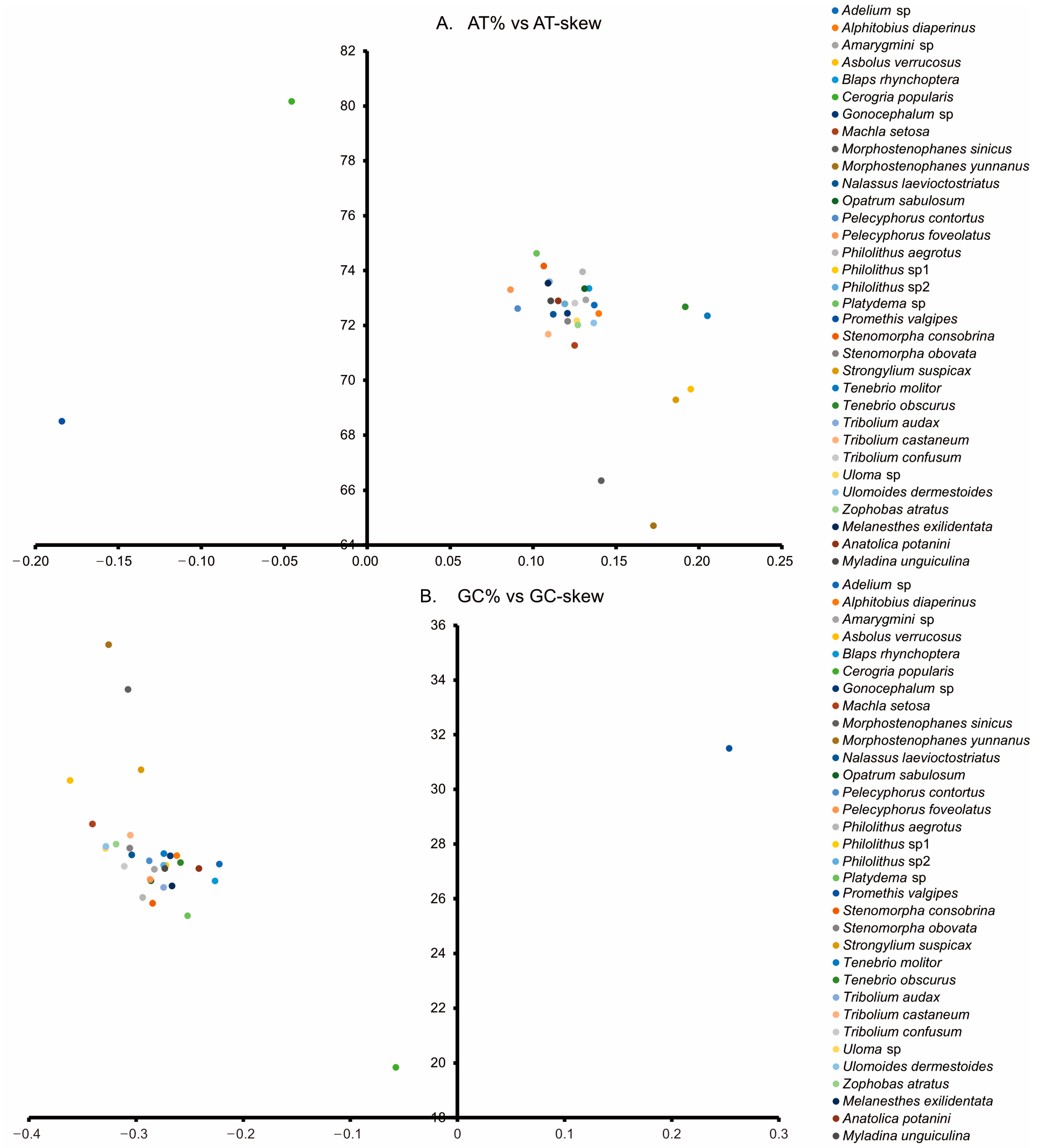
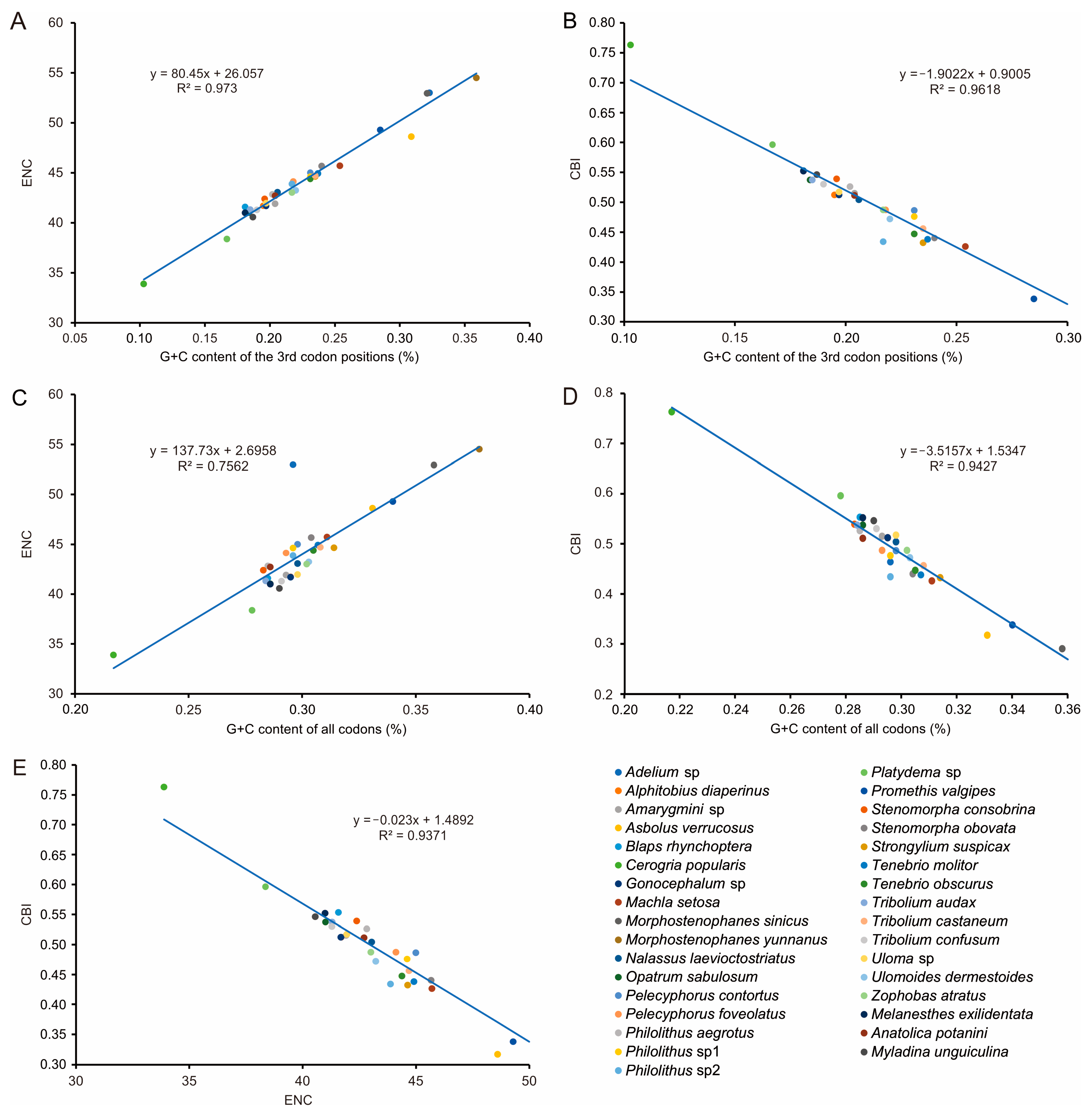
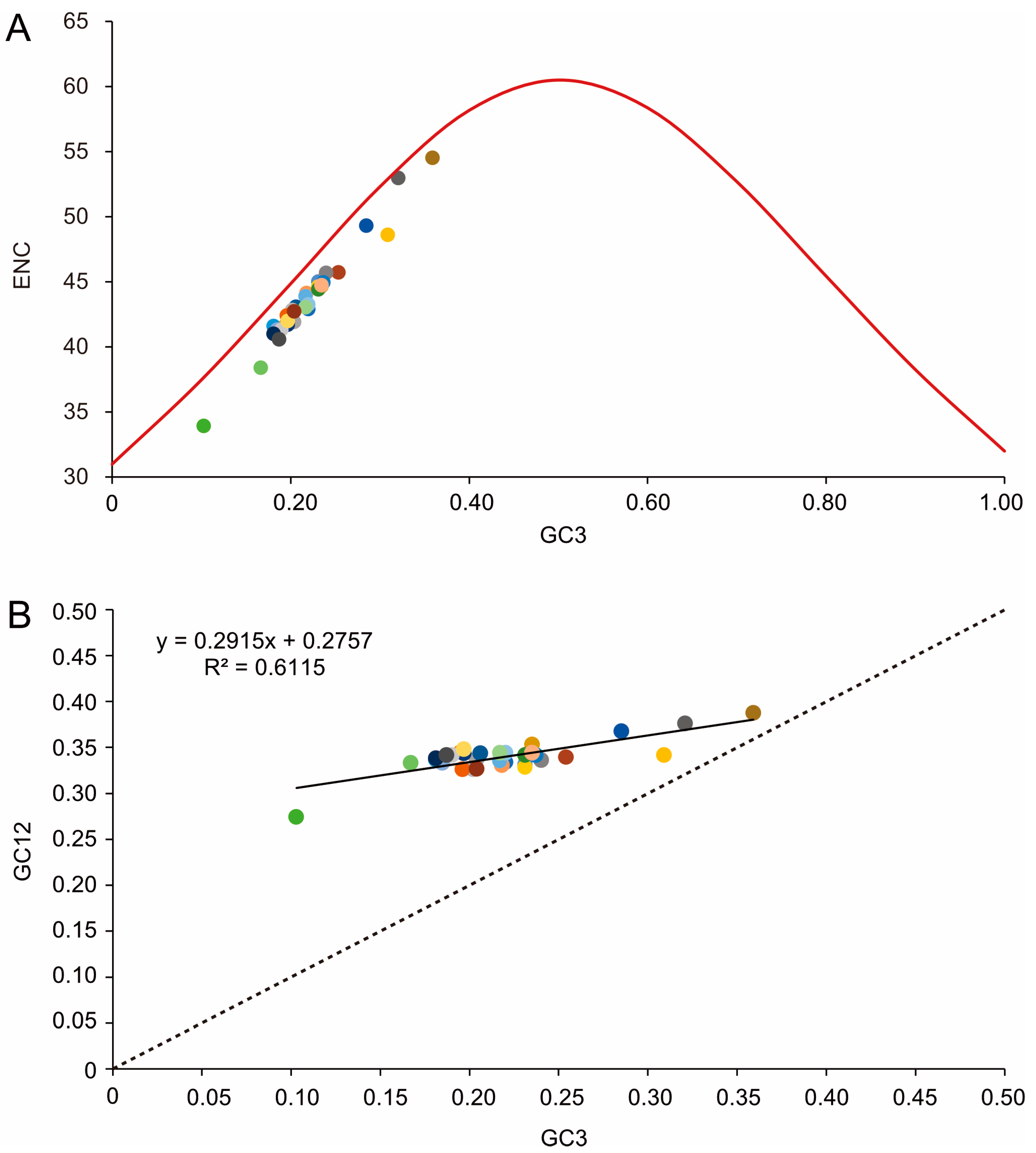
3.2. Nucleotide Composition and Codon Usage
3.3. Non-Coding Region
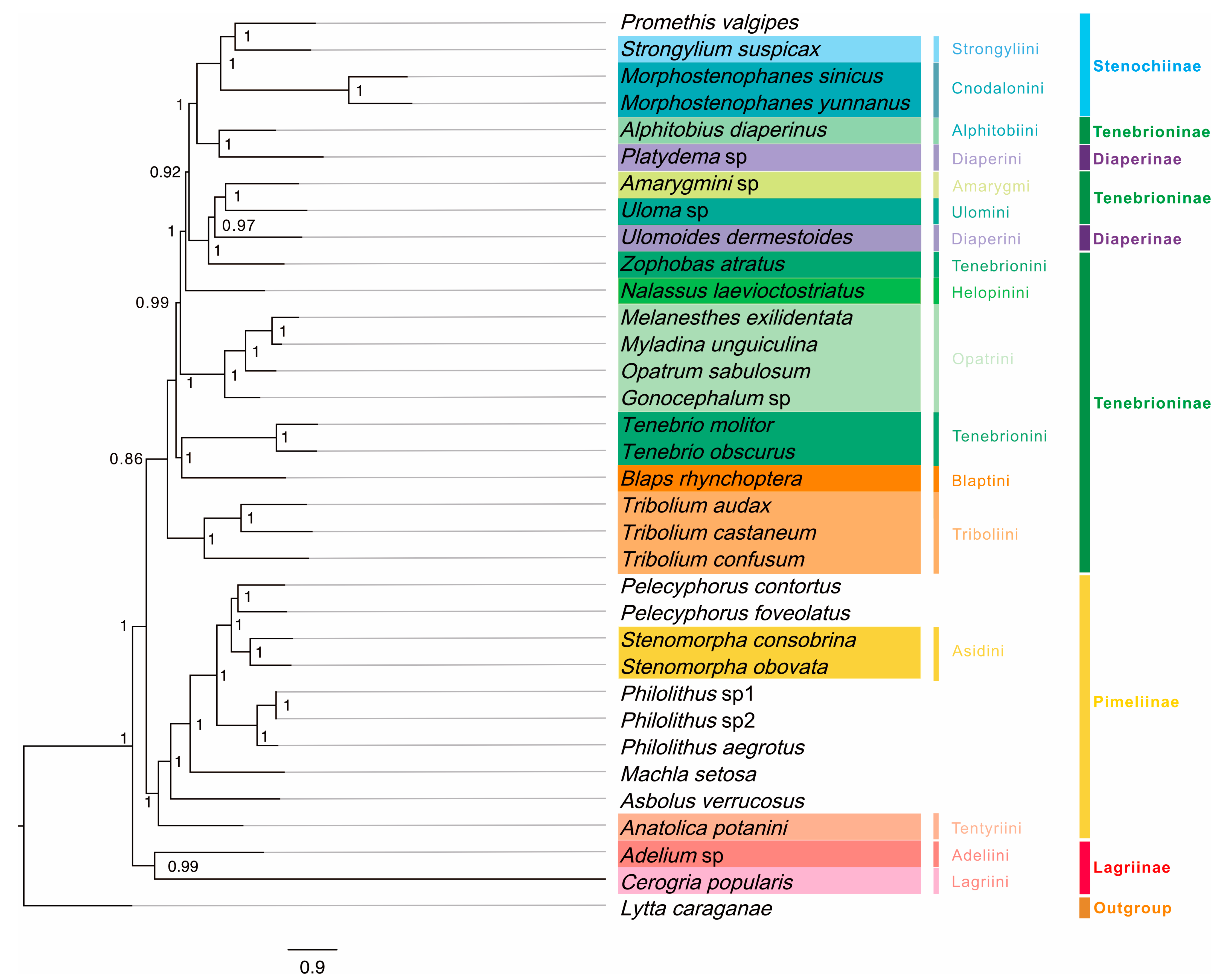
3.4. Mitochondrial Phylogeny of Tenebrionidae
4. Discussion
4.1. Mitochondrial Genome Organization and Composition
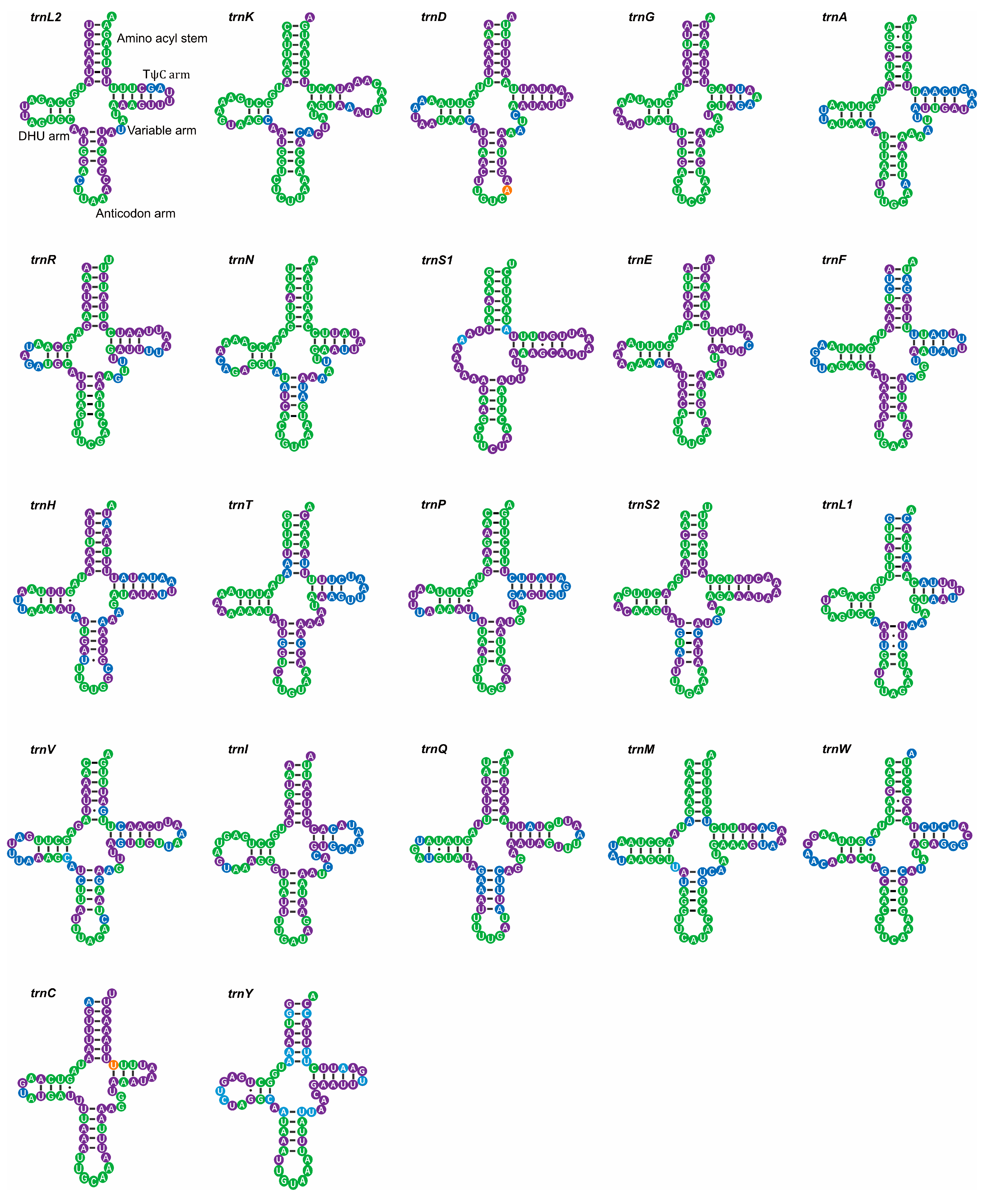
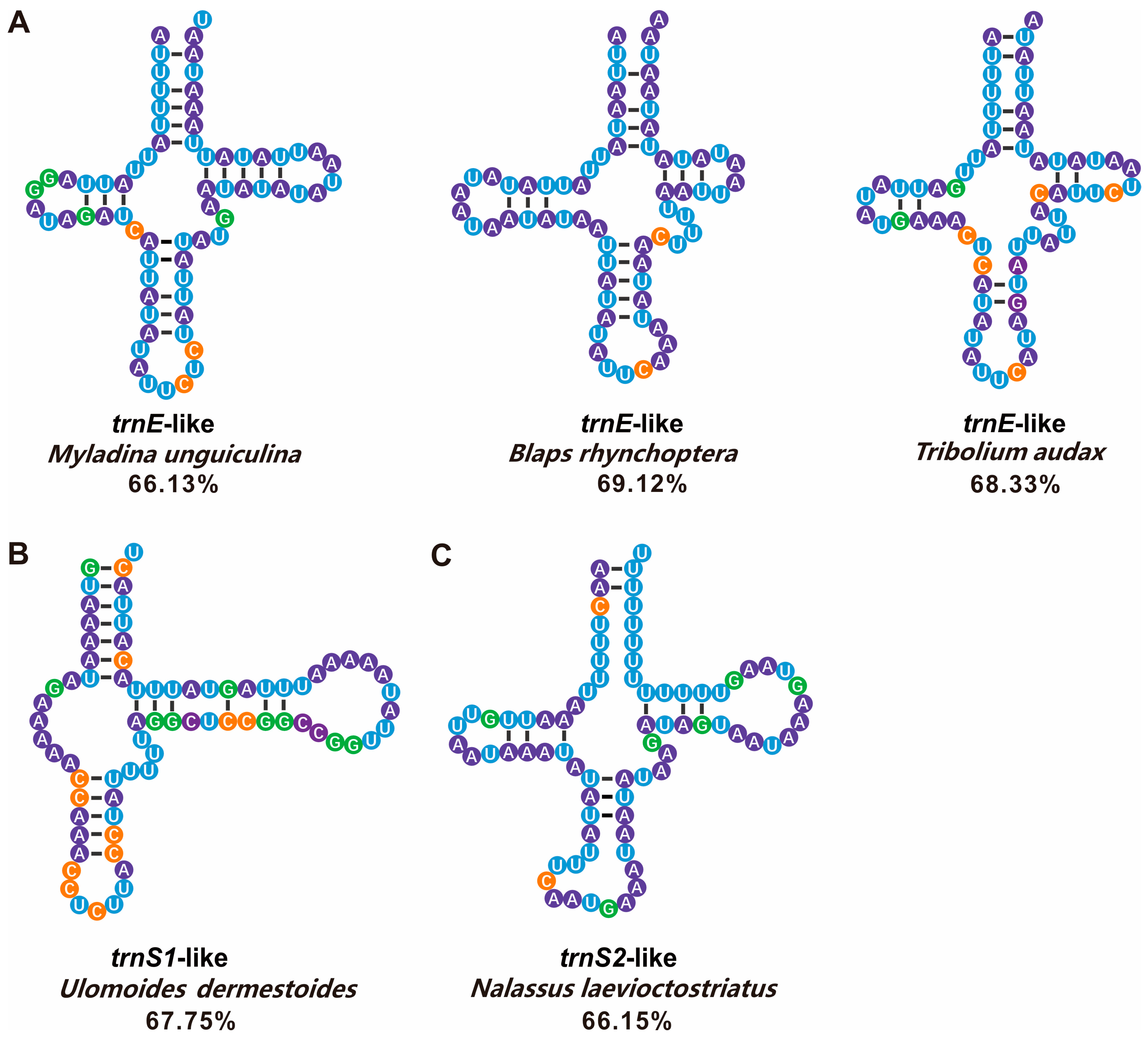
4.2. tRNA-like Structures
4.3. Phylogeny of Tenebrionidae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gunter, N.L.; Levkaničová, Z.; Weir, T.H.; Ślipiński, A.; Cameron, S.L.; Bocak, L. Towards a phylogeny of the Tenebrionoidea (Coleoptera). Mol. Phylogenet. Evol. 2014, 79, 305–312. [Google Scholar] [CrossRef]
- Aalbu, R.L.; Triplehorn, C.A.; Campbell, J.M.; Brown, K.W.; Somerby, R.E.; Thomas, D.B. Tenebrionidae Latreille 1802. In Polyphaga: Scarabaeoidea through Curculionoidea; Arnett, R.H., Jr., Thomas, M.C., Skelley, P.E., Frank, J.H., Eds.; CRC Press: Boca Raton, FL, USA, 2022; Volume 3, pp. 463–509. [Google Scholar]
- Bouchard, P.; Lawrence, J.F.; Davies, A.E.; Newton, A.F. Synoptic classification of the world Tenebrionidae (Insecta: Coleoptera) with a review of family-group names. In Annales Zoologici. Museum and Institute of Zoology; Polish Academy of Sciences: Warsaw, Poland, 2005; Volume 55, pp. 499–530. [Google Scholar]
- Levkaničová, Z. Molecular Phylogeny of the Superfamily Tenebrionoidea (Coleoptera: Cucujiformia). Ph.D. Thesis, Palaký University, Olomouc, Czech Republic, 2009. [Google Scholar]
- Throne, J.E.; Hallman, G.J.; Johnson, J.A.; Follett, P.A. Post-harvest entomology research in the United States Department of Agriculture-Agricultural Research Service. Pest Manag. Sci. 2003, 59, 619–628. [Google Scholar] [CrossRef]
- Selaledi, L.; Mbajiorgu, C.A.; Mabelebele, M. The use of yellow mealworm (T. molitor) as alternative source of protein in poultry diets: A review. Trop. Anim. Health Prod. 2020, 52, 7–16. [Google Scholar] [CrossRef]
- Kundungal, H.; Synshiang, K.; Devipriya, S.P. Biodegradation of polystyrene wastes by a newly reported honey bee pest Uloma sp. larvae: An insight to the ability of polystyrene-fed larvae to complete its life cycle. Environ. Chall. 2021, 4, 100083. [Google Scholar] [CrossRef]
- Pivato, A.F.; Miranda, G.M.; Prichula, J.; Lima, J.E.A.; Ligabue, R.A.; Seixas, A.; Trentin, D.S. Hydrocarbon-based plastics: Progress and perspectives on consumption and biodegradation by insect larvae. Chemosphere 2022, 293, 133600. [Google Scholar] [CrossRef]
- Doyen, J.T.; Matthews, E.; Lawrence, J.F. Classification and annotated checklist of the Australian genera of Tenebrionidae (Coleoptera). Invertebr. Syst. 1989, 3, 229–260. [Google Scholar] [CrossRef]
- Maeno, K.O.; Nakamura, S.; Babah, M.A.O. Sexing Live Adults of the Three Species of Darkling Beetle (Coleoptera: Tenebrionidae) and Morphological Characteristics. Ann. Entomol. Soc. Am. 2012, 105, 726–730. [Google Scholar] [CrossRef]
- Song, N.; Zhang, H.; Li, H.; Cai, W. All 37 Mitochondrial Genes of Aphid Aphis craccivora Obtained from Transcriptome Sequencing: Implications for the Evolution of Aphids. PLoS ONE 2016, 11, e0157857. [Google Scholar] [CrossRef]
- Angelini, D.R.; Jockusch, E.L. Relationships among pest flour beetles of the genus Tribolium (Tenebrionidae) inferred from multiple molecular markers. Mol. Phylogenet. Evol. 2008, 46, 127–141. [Google Scholar] [CrossRef]
- Ge, X.; Yuan, L.; Kang, Y.; Liu, T.; Liu, H.; Yang, Y. Characterization of the First Complete Mitochondrial Genome of Cyphonocerinae (Coleoptera: Lampyridae) with Implications for Phylogeny and Evolution of Fireflies. Insects 2021, 12, 570. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, Y.; Tian, T.; Li, T.J.; Chen, B. First report of complete mitochondrial genome in the subfamily Alleculinae and mitochondrial genome-based phylogenetics in Tenebrionidae (Coleoptera: Tenebrionoidea). Insect Sci. 2022, 29, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.; Bergsten, J.; Levkanicova, Z.; Papadopoulou, A.; John, O.S.; Wild, R.; Hammond, P.M.; Ahrens, D.; Balke, M.; Caterino, M.S.; et al. A Comprehensive Phylogeny of Beetles Reveals the Evolutionary Origins of a Superradiation. Science 2007, 318, 1913–1916. [Google Scholar] [CrossRef] [PubMed]
- Kergoat, G.J.; Soldati, L.; Clamens, A.L.; Jourdan, H.; Jabbour-Zahab, R.; Genson, G.; Bouchard, P.; Condamine, F.L. Higher level molecular phylogeny of darkling beetles (Coleoptera: Tenebrionidae). Syst. Entomol. 2014, 39, 486–499. [Google Scholar] [CrossRef]
- Song, N.; Liu, H.-Y.; Yang, X.-J.; Zhao, X.-C.; Lin, A.-L. Complete mitochondrial genome of the darkling beetle Gonocephalum outreyi (Coleoptera: Tenebrionidae) with phylogenetic implications. J. Asia-Pac. Entomol. 2018, 21, 721–730. [Google Scholar] [CrossRef]
- Yot, P.; Pinck, M.; Haenni, A.-L.; Duranton, H.M.; Chapeville, F. Valine-Specific tRNA-like Structure in Turnip Yellow Mosaic Virus RNA. Proc. Natl. Acad. Sci. USA 1970, 67, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kim, C.H.; Sivakumaran, K.; Kao, C. Stable RNA structures can repress RNA synthesis in vitro by the brome mosaic virus replicase. RNA 2003, 9, 555–565. [Google Scholar] [CrossRef]
- Vieweger, M.; Holmstrom, E.D.; Nesbitt, D.J. Single-Molecule FRET Reveals Three Conformations for the TLS Domain of Brome Mosaic Virus Genome. Biophys. J. 2015, 109, 2625–2636. [Google Scholar] [CrossRef]
- Ni, P.; Vaughan, R.C.; Tragesser, B.; Hoover, H.; Kao, C.C. The Plant Host Can Affect the Encapsidation of Brome Mosaic Virus (BMV) RNA: BMV Virions Are Surprisingly Heterogeneous. J. Mol. Biol. 2014, 426, 1061–1076. [Google Scholar] [CrossRef]
- Comandur, R.; Olson, E.D.; Musier-Forsyth, K. Conservation of tRNA mimicry in the 5′-untranslated region of distinct HIV-1 subtypes. RNA 2017, 23, 1850–1859. [Google Scholar] [CrossRef]
- Johanson, K.; Hoang, T.; Sheth, M.; Hyman, L.E. GRS1, a Yeast tRNA Synthetase with a Role in mRNA 3′ End Formation. J. Biol. Chem. 2003, 278, 35923–35930. [Google Scholar] [CrossRef]
- Plewka, P.; Thompson, A.; Szymanski, M.; Nuc, P.; Knop, K.; Rasinska, A.; Bialkowska, A.; Szweykowska-Kulinska, Z.; Karlowski, W.M.; Jarmolowski, A. A stable tRNA-like molecule is generated from the long noncoding RNA GUT15 in Arabidopsis. RNA Biol. 2018, 15, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Liao, J.Y.; Zhou, H.; Qu, L.H. The rat mitochondrial Ori L encodes a novel small RNA resembling an ancestral tRNA. Biochem. Biophys. Res. Commun. 2008, 372, 634–638. [Google Scholar] [CrossRef]
- Cha, S.Y.; Yoon, H.J.; Lee, E.M.; Yoon, M.H.; Hwang, J.S.; Jin, B.R.; Han, Y.S.; Kim, I. The complete nucleotide sequence and gene organization of the mitochondrial genome of the bumblebee, Bombus ignitus (Hymenoptera: Apidae). Gene 2007, 392, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.I.; Baek, J.Y.; Kim, M.J.; Jeong, H.C.; Kim, K.G.; Bae, C.H.; Han, Y.S.; Jin, B.R.; Kim, I. Complete Nucleotide Sequence and Organization of the Mitogenome of the Red-Spotted Apollo Butterfly, Parnassius bremeri (Lepidoptera: Papilionidae) and Comparison with Other Lepidopteran Insects. Mol. Cells 2009, 28, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.Y.; Cha, S.Y.; Kim, D.Y.; Yoon, H.J.; Kim, S.R.; Hwang, J.S.; Kim, K.G.; Han, Y.S.; Kim, I.S. Presence of several tRNA-like sequences in the mitochondrial genome of the bumblebee, Bombus hypocrita sapporoensis (Hymenoptera: Apidae). Genes Genom. 2008, 30, 307–318. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of Animal Mitochondrial DNA: Relevance for Population Biology and Systematics. Annu. Rev. Ecol. Evol. S 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [PubMed]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of Nueleotide Composition at Fourfold Degenerate Sites of Animal Mitochondrial Genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Zang, M.; He, W.; Du, F.; Wu, G.; Wu, B.; Zhou, Z. Analysis of the codon usage of the ORF2 gene of feline calicivirus. Infect. Genet. Evol. 2017, 54, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Wei, S.J.; Shi, M.; Chen, X.X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.Y.; He, J.H. New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef]
- Wei, S.J.; Shi, M.; Sharkey, M.J.; van Achterberg, C.; Chen, X.X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics 2010, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhou, L.; Zhou, X.Y.; Yang, W.T.; Zhang, J.; Zhang, X.J.; Wang, Y.; Gui, J.F. Unusual AT-skew of Sinorhodeus microlepis mitogenome provides new insights into mitogenome features and phylogenetic implications of bitterling fishes. Int. J. Biol. Macromol. 2019, 129, 339–350. [Google Scholar] [CrossRef]
- Ou, J.; Liu, J.B.; Yao, F.J.; Wang, X.G.; Wei, Z.M. The complete mitochondrial genome of the American black flour beetle Tribolium audax (Coleoptera: Tenebrionidae). Mitochondrial. DNA A 2016, 27, 958–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.N.; Wang, C.Y. Complete mitochondrial genome of yellow meal worm (Tenebrio molitor). Zool Res. 2014, 35, 537–545. [Google Scholar]
- Knight, R.D.; Freeland, S.J.; Landweber, L.F. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001, 2, RESEARCH0010. [Google Scholar] [PubMed]
- Eyre-Walker, A. Differentiating between selection and mutation bias. Genetics 1997, 147, 1983–1987. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. A Comparative Analysis of Mitochondrial Genomes in Coleoptera (Arthropoda: Insecta) and Genome Descriptions of Six New Beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef]
- Du, C.; He, S.; Song, X.; Liao, Q.; Zhang, X.; Yue, B. The complete mitochondrial genome of Epicauta chinensis (Coleoptera: Meloidae) and phylogenetic analysis among Coleopteran insects. Gene 2016, 578, 274–280. [Google Scholar] [CrossRef]
- Liao, F. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int. J. Biol. Sci. 2010, 6, 172–186. [Google Scholar] [CrossRef]
- Li, X.; Ogoh, K.; Ohba, N.; Liang, X.; Ohmiya, Y. Mitochondrial genomes of two luminous beetles, Rhagophthalmus lufengensis and R. ohbai (Arthropoda, Insecta, Coleoptera). Gene 2007, 392, 196–205. [Google Scholar] [CrossRef]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Roberti, M.; Polosa, P.L.; Bruni, F.; Musicco, C.; Gadaleta, M.N.; Cantatore, P. DmTTF, a novel mitochondrial transcription termination factor that recognises two sequences of Drosophila melanogaster mitochondrial DNA. Nucleic Acids Res. 2003, 31, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Yuan, L.; Kang, Y.; Liu, T.; Liu, H.; Yang, Y. First complete mitochondrial genomes of Ototretinae (Coleoptera, Lampyridae) with evolutionary insights into the gene rearrangement. Genomics 2022, 114, 110305. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Kawai, G.; Watanabe, K. The minimal tRNA: Unique structure of Ascaris suum mitochondrial tRNA(Ser)(UCU) having a short T arm and lacking the entire D arm. FEBS Lett. 2002, 514, 37–43. [Google Scholar] [CrossRef]
- Lorenz, C.; Lunse, C.E.; Morl, M. tRNA Modifications: Impact on Structure and Thermal Adaptation. Biomolecules 2017, 7, 35. [Google Scholar] [CrossRef]
- Hong, M.Y.; Jeong, H.C.; Kim, M.J.; Jeong, H.U.; Lee, S.H.; Kim, I. Complete mitogenome sequence of the jewel beetle, Chrysochroa fulgidissima (Coleoptera: Buprestidae). Mitochondrial. DNA 2009, 20, 46–60. [Google Scholar] [CrossRef]
- Kim, M.J.; Jeong, H.C.; Kim, S.R.; Kim, I. Complete mitochondrial genome of the nerippe fritillary butterfly, Argynnis nerippe (Lepidoptera: Nymphalidae). Mitochondrial. DNA 2011, 22, 86–88. [Google Scholar] [CrossRef]
- Zhang, B.; Ma, C.; Edwards, O.; Fuller, S.; Kang, L. The mitochondrial genome of the Russian wheat aphid Diuraphis noxia: Large repetitive sequences between trnE and trnF in aphids. Gene 2014, 533, 253–260. [Google Scholar] [CrossRef]
- Wu, S.; Li, X.; Wang, G. tRNA-like structures and their functions. FEBS J. 2022, 289, 5089–5099. [Google Scholar] [CrossRef]
- Li, M.; Chen, W.T.; Zhang, Q.L.; Liu, M.; Xing, C.W.; Cao, Y.; Luo, F.Z.; Yuan, M.L. Mitochondrial phylogenomics provides insights into the phylogeny and evolution of spiders (Arthropoda: Araneae). Zool Res. 2022, 43, 566–584. [Google Scholar] [CrossRef]
- Macey, J.R.; Larson, A.; Ananjeva, N.B.; Fang, Z.; Papenfuss, T.J. Two Novel Gene Orders and the Role of Light-Strand Replication in Rearrangement of the Vertebrate Mitochondrial Genome. Mol. Biol. Evol. 1997, 14, 91–104. [Google Scholar] [CrossRef]
- Brown, C.R. Cliff swallow colonies as information centers. Science 1986, 234, 83–85. [Google Scholar] [CrossRef]
- Cantatore, P.; Gadaleta, M.N.; Roberti, M.; Saccone, C.; Wilson, A.C. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 1987, 329, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Goddard, J.M.; Wolstenholme, D.R. Origin and direction of replication in mitochondrial DNA molecules from the genus Drosophila. Nucleic Acids Res. 1980, 8, 741–757. [Google Scholar] [PubMed]
- Timmermans, M.J.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-Level Sampling of Mitochondrial Genomes in Coleoptera: Compositional Heterogeneity and Phylogenetics. Genome Biol. Evol. 2015, 8, 161–175. [Google Scholar] [CrossRef]
- Doyen, J.T.; TSCHINKEL, W.R. Phenetic and cladistic relationships among tenebrionid beetles (Coleoptera). Syst. Entomol. 1982, 7, 127–183. [Google Scholar] [CrossRef]
- Zhang, H.L.; Liu, B.B.; Wang, X.Y.; Han, Z.P.; Zhang, D.X.; Su, C.N. Comparative Mitogenomic Analysis of Species Representing Six Subfamilies in the Family Tenebrionidae. Int. J. Mol. Sci. 2016, 17, 841. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Chen, J.; Li, G.; Luo, J.; Wang, H.; Yang, Y.; Liang, S.; Ouyang, B. Complete mitochondrial genome of Promethis valgipes valgipes (Marseul) (Insecta: Coleoptera: Tenebrionidae). Mitochondrial. DNA B 2021, 6, 538–539. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Gao, X.; Yu, Y.; Long, X.; Zeng, X.; Wei, D.; Ye, L. Complete mitochondrial genome of Morphostenophanes sinicus (Zhou, 2020) (Insecta: Coleoptera: Tenebrionidae). Mitochondrial. DNA B 2021, 6, 2946–2948. [Google Scholar] [CrossRef]
- Hong, K.J.; Ki, W.; Park, D.S.; Yang, B.K.; Lee, H.; Park, J.; Lee, W. The complete mitochondrial genome of Alphitobius diaperinus Panzer, 1797 (Coleoptera: Tenebrionidae). Mitochondrial. DNA B 2020, 5, 2291–2293. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Species | Sampling Site | Voucher Specimen Code | Coordinate |
|---|---|---|---|---|
| Tenebrionidae | M. exilidentata | Lingwu County, Ningxia Province, China | YCLW-BJ1 | 106°34′ E, 38°10′ N |
| Tenebrionidae | A. potanini | Lingwu County, Ningxia Province, China | YCLW-BJ2 | 106°34′ E, 38°10′ N |
| Tenebrionidae | M. unguiculina | Lingwu County, Ningxia Province, China | YCLW-BJ3 | 106°34′ E, 38°10′ N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.-H.; Hu, S.-Y.; Li, M.; Liu, M.; Sun, H.; Zhao, J.-R.; Chen, W.-T.; Yuan, M.-L. Comparative Mitogenomic Analyses of Darkling Beetles (Coleoptera: Tenebrionidae) Provide Evolutionary Insights into tRNA-like Sequences. Genes 2023, 14, 1738. https://doi.org/10.3390/genes14091738
Wang S-H, Hu S-Y, Li M, Liu M, Sun H, Zhao J-R, Chen W-T, Yuan M-L. Comparative Mitogenomic Analyses of Darkling Beetles (Coleoptera: Tenebrionidae) Provide Evolutionary Insights into tRNA-like Sequences. Genes. 2023; 14(9):1738. https://doi.org/10.3390/genes14091738
Chicago/Turabian StyleWang, Su-Hao, Shi-Yun Hu, Min Li, Min Liu, Hao Sun, Jia-Rui Zhao, Wen-Ting Chen, and Ming-Long Yuan. 2023. "Comparative Mitogenomic Analyses of Darkling Beetles (Coleoptera: Tenebrionidae) Provide Evolutionary Insights into tRNA-like Sequences" Genes 14, no. 9: 1738. https://doi.org/10.3390/genes14091738