
Transcriptome Analysis and QTL Mapping Identify Candidate Genes and Regulatory Mechanisms Related to Low-Temperature Germination Ability in Maize
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotypic Evaluation of Low-Temperature Tolerance during Germination
2.3. Measurement of Enzyme Activity
2.4. RNA-Sequencing and Data Analysis
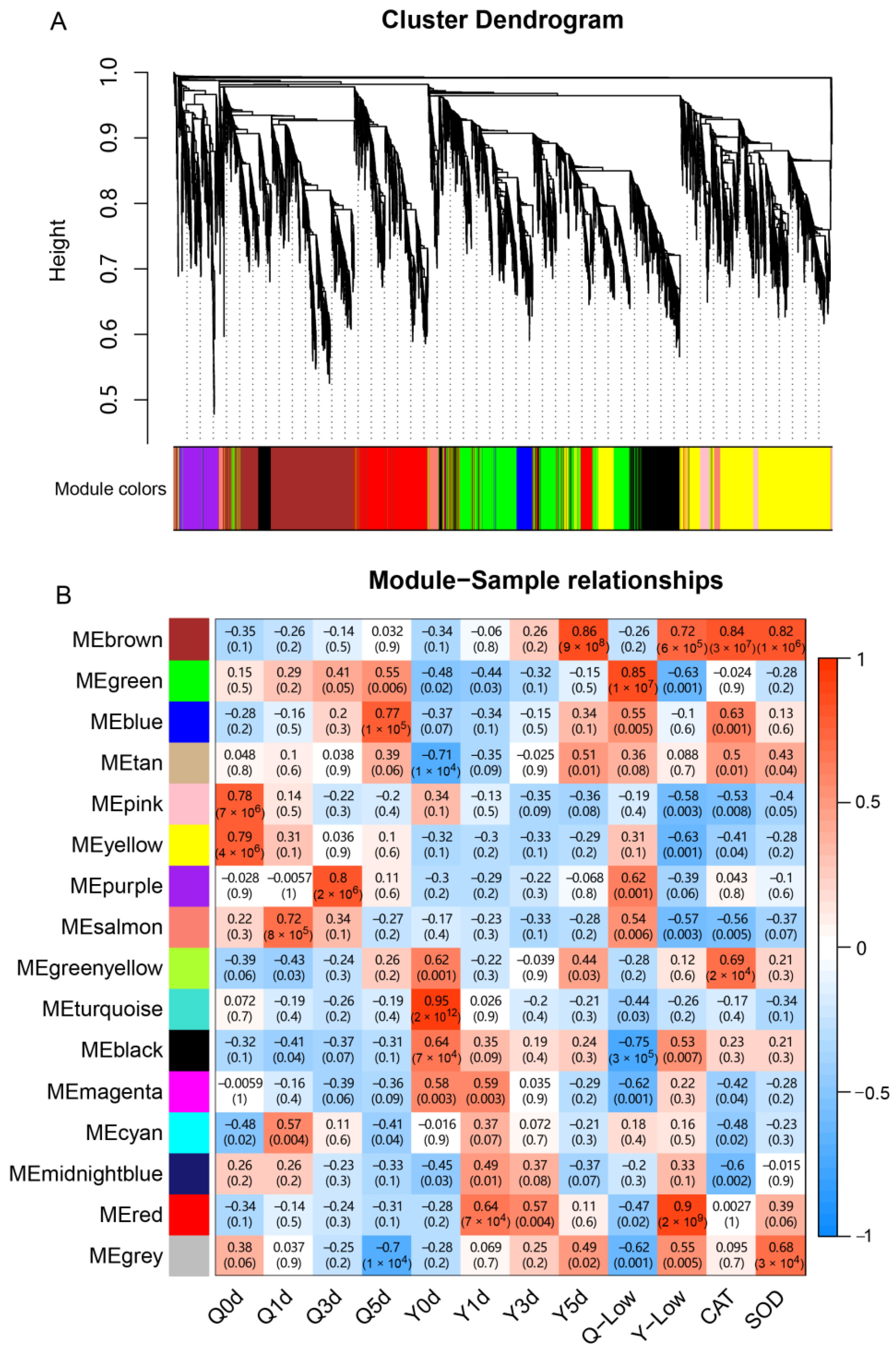
2.5. WGCNA Co-Expression Network Construction Module Identification
2.6. QTL Analysis and Investigate Effects of the Target Genes
3. Results
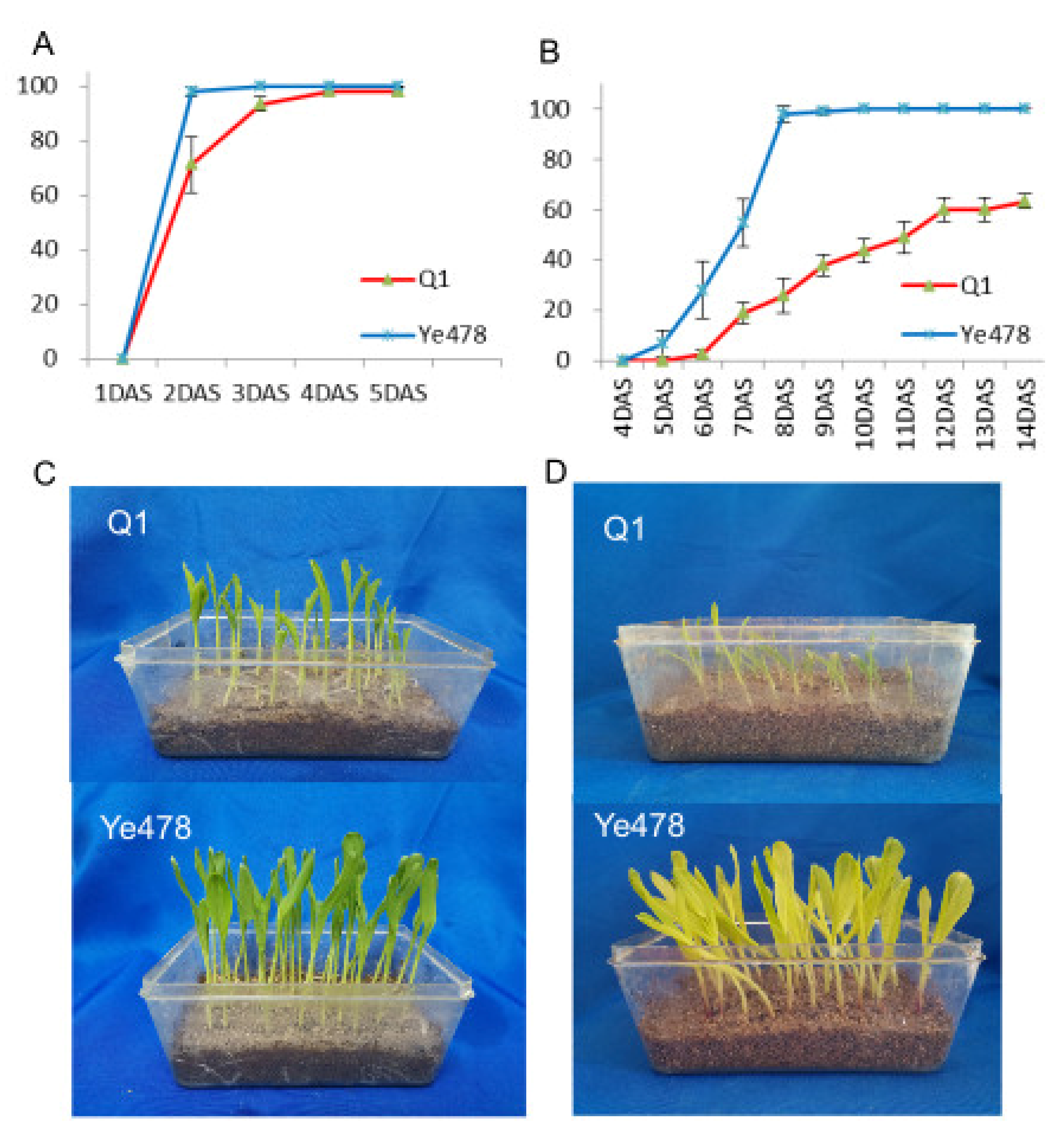
3.1. Germination Rate Analysis of the Two Genotypes
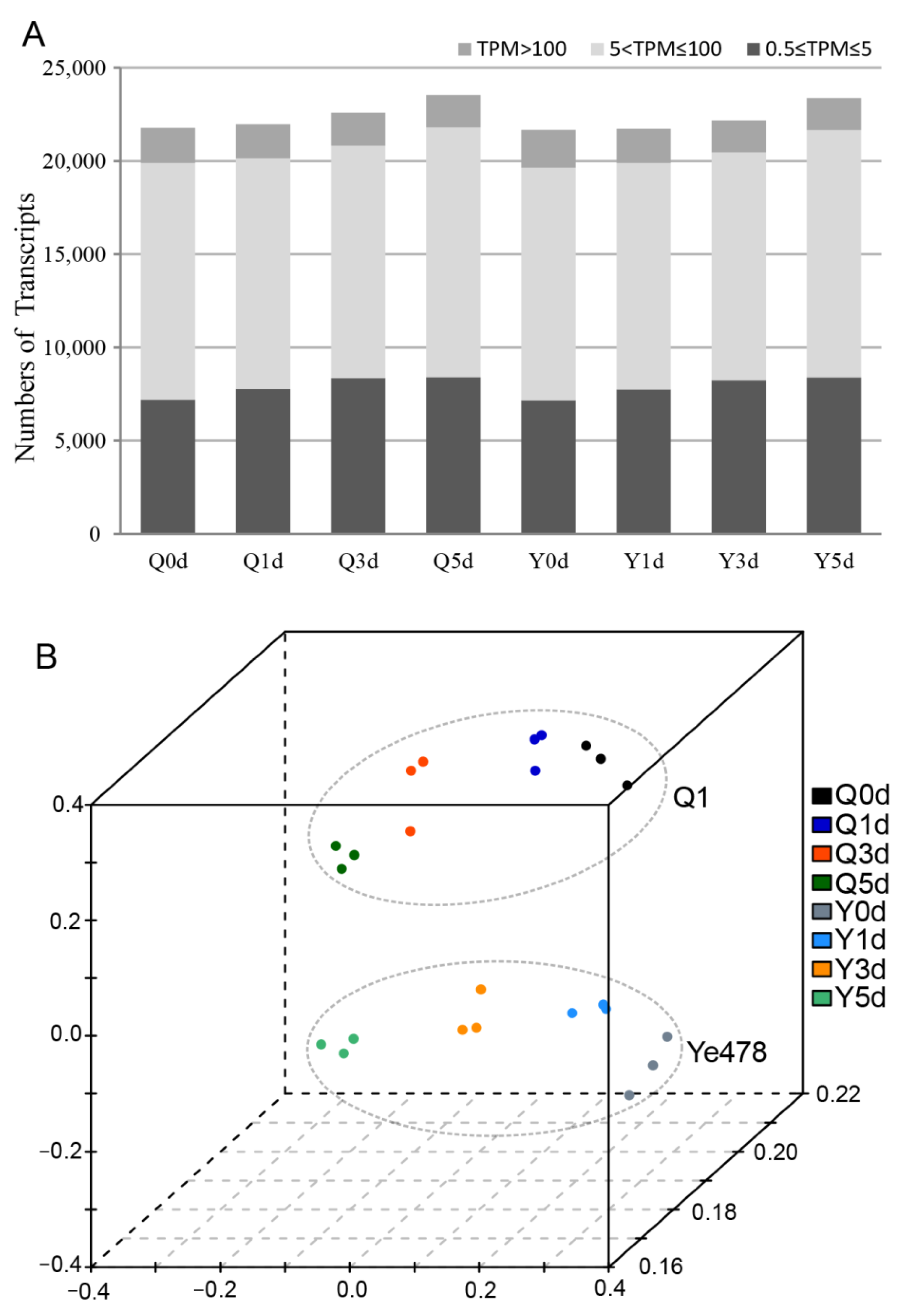
3.2. RNA-Sequencing Analysis for the Two Genotypes during Seed Germination
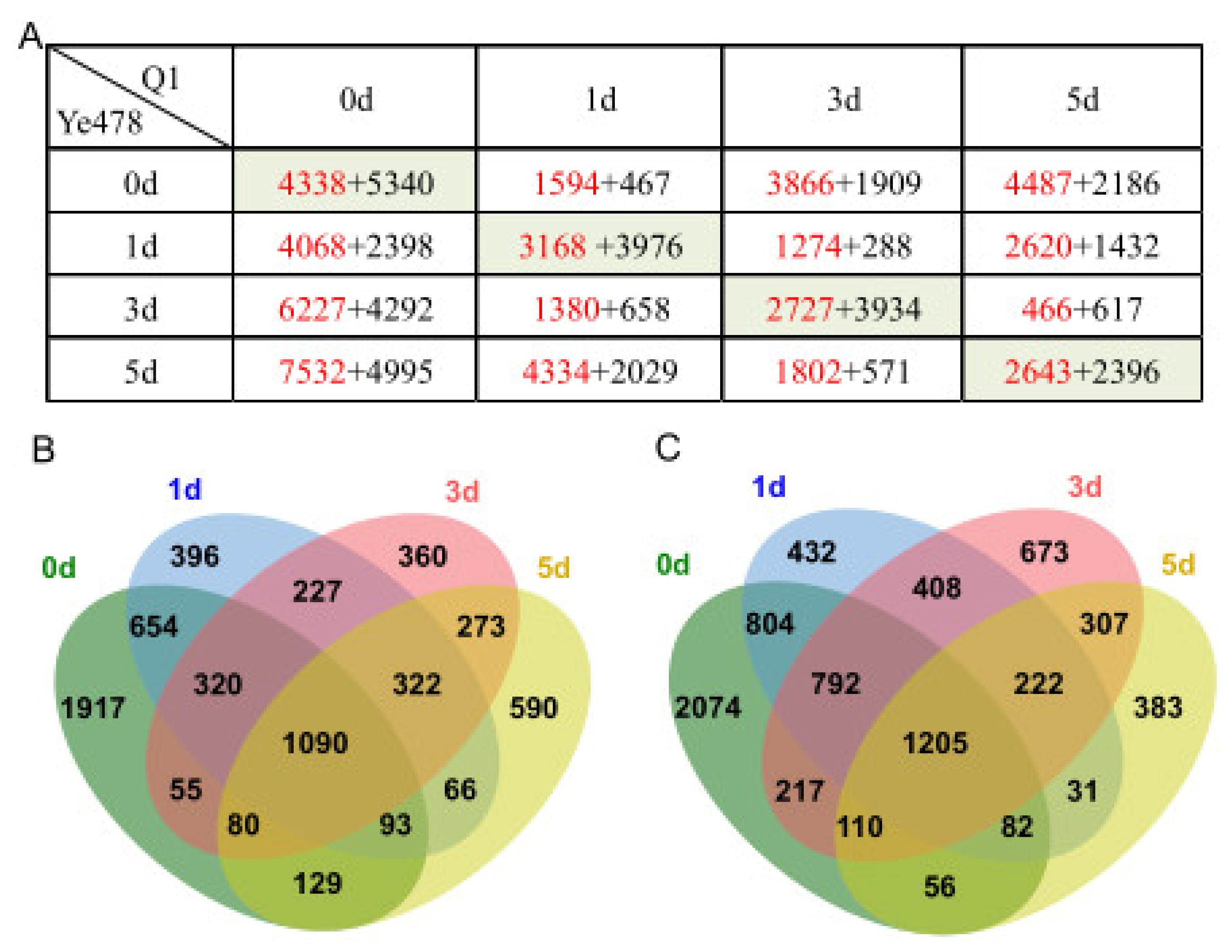
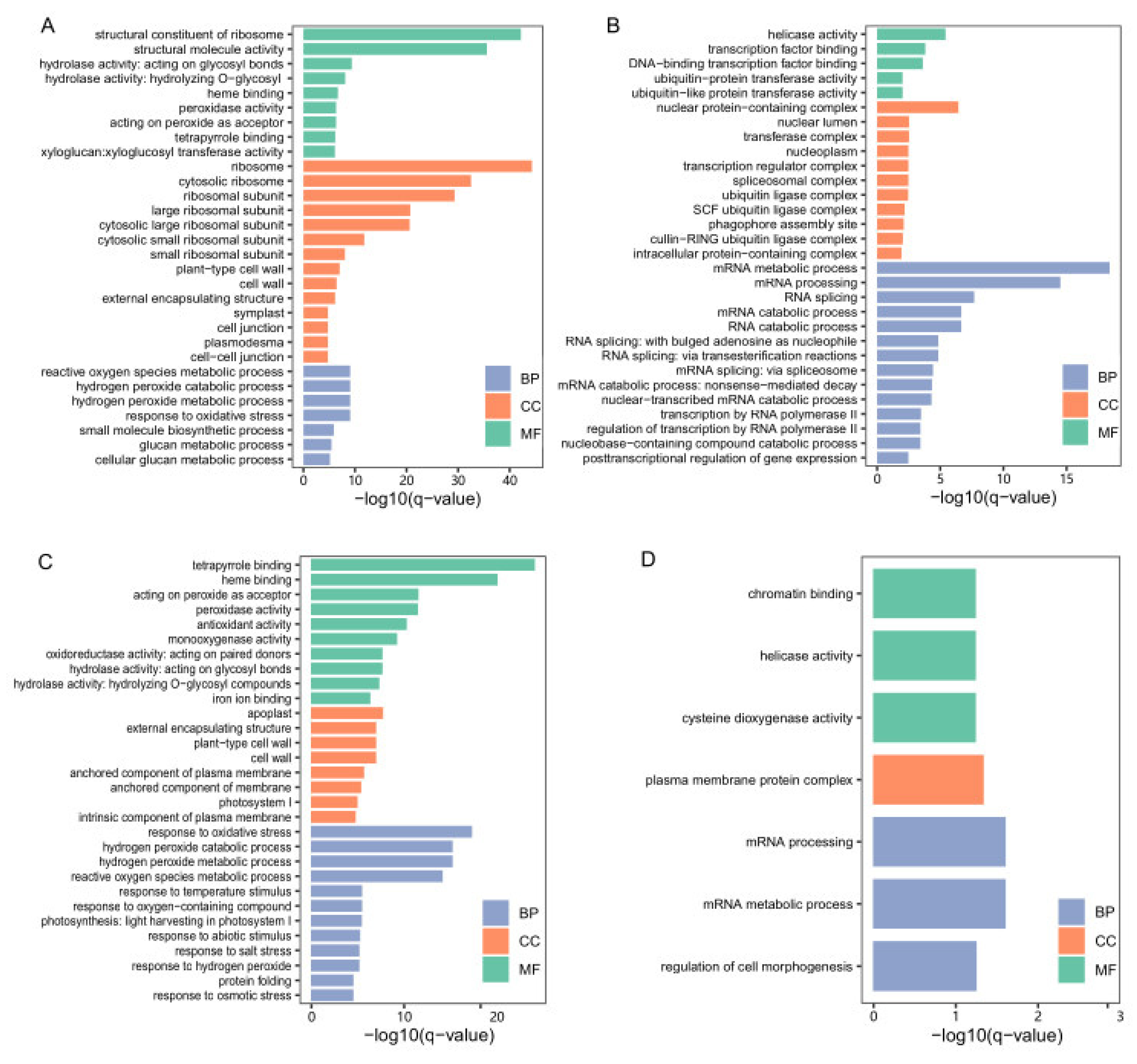
3.3. Identification of DEGs at the Low-Temperature Germination Stage for Ye478
3.4. Identification of DEGs at the Low-Temperature Germination Stage for the Q1
3.5. Identification of DEGs between the Two Genotypes
3.6. WGCNA Analysis
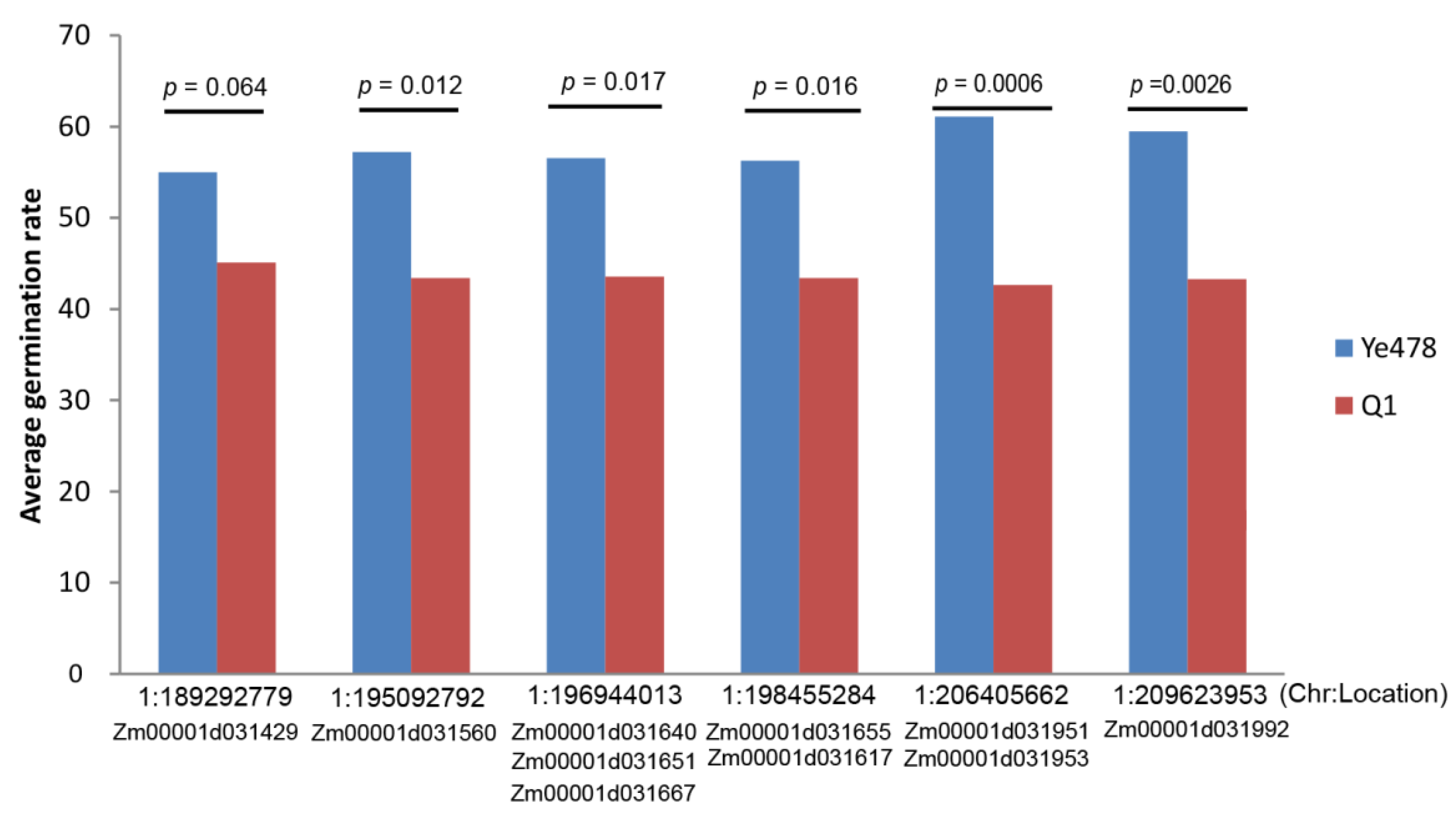
3.7. QTL Analysis and Investigate Effects of the Target Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, F.; Shen, H.; Lin, C.; Fu, H.; Sheteiwy, M.S.; Guan, Y.; Huang, Y.; Hu, J. Transcriptome analysis of chilling-imbibed embryo revealed membrane recovery related genes in maize. Front. Plant Sci. 2016, 7, 1978. [Google Scholar] [CrossRef] [PubMed]
- Kołodziejczyk, I.; Kazmierczak, A.; Posmyk, M.M. Melatonin application modifies antioxidant defense and induces endoreplication in maize seeds exposed to chilling stress. Int. J. Mol. Sci. 2021, 22, 8628. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, J.; Li, W.; Hu, W.; Duan, L.; Feng, Y.; Qiu, F.; Yue, B. Genome-wide association analysis of ten chilling tolerance indices at the germination and seedling stages in maize. J. Integr. Plant. Biol. 2013, 55, 735–744. [Google Scholar] [CrossRef]
- China Statistics Press. China Statistical Yearbook; China Statistics Press: Beijing, China, 2022. [Google Scholar]
- Qin, F.; Sakuma, Y.; Li, J.; Liu, Q.; Li, Y.Q.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Cloning and functional analysis of a novel DREB1/CBF transcription factor involved in cold-responsive gene expression in Zea mays L. Plant Cell Physiol. 2004, 45, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Li, Z.; Shi, Y.; Fu, D.; Yin, P.; Cheng, J.K.; Jiang, C.F.; Yang, S.H. Natural variation in a type-A response regulator confers maize chilling tolerance. Nat. Commun. 2021, 12, 4713. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Fu, D.Y.; Wang, X.; Zeng, R.; Zhang, X.; Tian, J.G.; Zhang, S.S.; Yang, X.H.; Tian, F.; Lai, J.S.; et al. The transcription factor bZIP68 negatively regulates cold tolerance in maize. Plant Cell 2022, 34, 2833–2851. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shi, Y.; Liu, J.; Li, Z.; Fu, D.Y.; Wu, S.F.; Li, M.Z.; Yang, Z.J.; Shi, Y.L.; Lai, J.S.; et al. Natural polymorphism of ZmICE1 contributes to amino acid metabolism that impacts cold tolerance in maize. Nat. Plants 2022, 8, 1176–1190. [Google Scholar] [CrossRef]
- He, R.Y.; Yang, T.; Zheng, J.J.; Pan, Z.Y.; Chen, Y.; Zhou, Y.; Li, X.F.; Li, Y.Z.; Iqbal, M.Z.; Yang, C.Y.; et al. QTL mapping and a transcriptome integrative analysis uncover the candidate genes that control the cold tolerance of maize introgression lines at the seedling stage. Int. J. Mol. Sci. 2023, 24, 2629. [Google Scholar] [CrossRef] [PubMed]
- He, R.Y.; Zheng, J.J.; Chen, Y.; Pan, Z.Y.; Yang, T.; Zhou, Y.; Li, X.F.; Li, Y.Z.; Cheng, M.J.; Li, Y.; et al. QTL-seq and transcriptomic integrative analyses reveal two positively regulated genes that control the low-temperature germination ability of MTP–maize introgression lines. Theor. Appl. Genet. 2023, 136, 116. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Li, B.; Nakayama, T.; Kawamura, Y.; Uemura, M. Plant plasma membrane proteomics for improving cold tolerance. Front. Plant Sci. 2013, 4, 90. [Google Scholar] [CrossRef]
- Martiniere, A.; Shvedunova, M.; Thomson, A.J.; Evans, N.H.; Penfield, S.; Runions, J.; McWatters, H.G. Homeostasis of plasma membrane viscosity in fluctuating temperatures. New Phytol. 2011, 192, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Cao, Y.; Fan, X.R.; Li, M.; Wang, Y.F.; Ming, F. A rice microsomal delta-12 fatty acid desaturase can enhance resistance to cold stress in yeast and Oryza sativa. Mol. Breed. 2012, 29, 743–757. [Google Scholar] [CrossRef]
- Wang, X.; Yu, C.; Liu, Y.; Yang, L.; Li, Y.; Yao, W.; Cai, Y.; Yan, X.; Li, S.; Cai, Y.; et al. GmFAD3A, A ω-3 fatty acid desaturase gene, enhances cold tolerance and seed germination rate under low temperature in rice. Int. J. Mol. Sci. 2019, 20, 3796. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Dai, X.; Xu, Y.; Luo, W.; Zheng, X.; Zeng, D.; Pan, Y.; Lin, X.; Liu, H.; Zhang, D.; et al. COLD1 confers chilling tolerance in Rice. Cell 2015, 160, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Renhu, N.; Naito, M.; Nakamura, A.; Shiba, H.; Yamamoto, T.; Suzaki, T.; Iida, H.; Miura, K. Ca2+-permeable mechanosensitive channels MCA1 and MCA2 mediate cold-induced cytosolic Ca2+ increase and cold tolerance in Arabidopsis. Sci. Rep. 2018, 8, 550. [Google Scholar] [CrossRef]
- Liu, Q.; Ding, Y.; Shi, Y.; Ma, L.; Wang, Y.; Song, C.; Wilkins, K.A.; Davies, J.M.; Knight, H.; Knight, M.R.; et al. The calcium transporter ANNEXIN1 mediates cold-induced calcium signaling and freezing tolerance in plants. EMBO J. 2021, 40, e104559. [Google Scholar] [CrossRef]
- Mega, R.; Meguro-Maoka, A.; Endo, A.; Shimosaka, E.; Murayama, S.; Nambara, E.; Seo, M.; Kanno, Y.; Abrams, S.R.; Sato, Y. Sustained low abscisic acid levels increase seedling vigor under cold stress in rice (Oryza sativa L.). Sci. Rep. 2015, 5, 13819. [Google Scholar] [CrossRef]
- Du, H.; Wu, N.; Fu, J.; Wang, S.; Li, X.; Xiao, J.; Xiong, L. A GH3 family member, OsGH3-2, modulates auxin and abscisic acid levels and differentially affects drought and cold tolerance in rice. J. Exp. Bot. 2012, 63, 6467–6480. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, K.X.; Wang, W.S.; Gong, W.; Liu, W.C.; Chen, H.G.; Xu, H.H.; Lu, Y.T. Low temperature inhibits root growth by reducing auxin accumulation via ARR1/12. Plant Cell Physiol. 2015, 56, 727–736. [Google Scholar] [CrossRef]
- Zwack, P.J.; Compton, M.A.; Adams, C.I.; Rashotte, A.M. Cytokinin response factor 4 (CRF4) is induced by cold and involved in freezing tolerance. Plant Cell Rep. 2016, 35, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Bolt, S.; Zuther, E.; Zintl, S.; Hincha, D.K.; Schmulling, T. ERF105 is a transcription factor gene of Arabidopsis thaliana required for freezing tolerance and cold acclimation. Plant Cell Environ. 2017, 40, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Catala, R.; Salinas, J. The Arabidopsis ethylene overproducer mutant eto1-3 displays enhanced freezing tolerance. Plant Signal. Behav. 2015, 10, e989768. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ding, Y.; Shi, Y.; Zhang, X.; Zhang, S.; Gong, Z.; Yang, S. MPK3- and MPK6-Mediated ICE1 Phosphorylation Negatively Regulates ICE1 Stability and Freezing Tolerance in Arabidopsis. Dev. Cell 2017, 43, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, P.; Si, T.; Hsu, C.C.; Wang, L.; Zayed, O.; Yu, Z.; Zhu, Y.; Dong, J.; Tao, W.A.; et al. MAP Kinase Cascades Regulate the Cold Response by Modulating ICE1 Protein Stability. Dev. Cell 2017, 43, 618–629. [Google Scholar] [PubMed]
- Zhang, Z.; Li, J.H.; Li, F.; Liu, H.H.; Yang, W.S.; Chong, K.; Cu, Y.Y. OsMAPK3 phosphorylates OsbHLH002/OsICE1 and inhibits its ubiquitination to activate OsTPP1 and enhances rice chilling tolerance. Dev. Cell 2017, 43, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.R.; Meng, X.B.; Song, X.G.; Zhang, D.H.; Kou, L.Q.; Zhang, J.H.; Jing, Y.H.; Liu, G.F.; Liu, H.H.; Huang, X.H.; et al. Chilling-induced phosphorylation of IPA1 by OsSAPK6 activates chilling tolerance responses in rice. Cell Discov. 2022, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- An, J.P.; Li, R.; Qu, F.J.; You, C.X.; Wang, X.F.; Hao, Y.J. An apple NAC transcription factor negatively regulates cold tolerance via CBF-dependent pathway. J. Plant Physiol. 2018, 221, 74–80. [Google Scholar] [CrossRef]
- Liu, C.T.; Schläppi, M.R.; Mao, B.G.; Wang, W.; Wang, A.J.; Chu, C.C. The bZIP73 transcription factor controls rice cold tolerance at the reproductive stage. Plant Biotechnol. J. 2019, 17, 1834–1849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Wu, T.; Li, Z.; Huang, K.; Kim, N.E.; Ma, Z.M.; Kwon, S.W.; Jiang, W.Z.; Du, X.L. OsGATA16, a GATA Transcription Factor, Confers Cold Tolerance by Repressing OsWRKY45–1 at the Seedling Stage in Rice. Rice 2021, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Chan, Z. ROS regulation during abiotic stress responses in crop plants. Front. Plant Sci. 2015, 6, 1092. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Masuta, Y.; Saito, K.; Ozawa, K. Enhanced chilling tolerance at the booting stage in rice by transgenic overexpression of the ascorbate peroxidase gene, OsAPXa. Plant Cell Rep. 2011, 30, 399–406. [Google Scholar] [PubMed]
- Xu, J.; Duan, X.G.; Yang, J.; Beeching, J.R.; Zhang, P. Coupled expression of Cu/Zn-superoxide dismutase and catalase in cassava improves tolerance against cold and drought stresses. Plant Signal. Behav. 2013, 8, e24525. [Google Scholar] [CrossRef] [PubMed]
- Kocsy, C.; Galiba, C.; Brunold, C. Role of glutathione in adaptation and signalling during chilling and cold acclimation in plants. Physiol. Plant. 2008, 113, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ming, R.H.; Khan, M.; Wang, Y.; Dahro, B.; Xiao, W.; Li, C.L.; Liu, J.H. ERF9 of Poncirus trifoliata (L.) Raf. undergoes feedback regulation by ethylene and modulates cold tolerance via regulating a glutathione S-transferase U17 gene. Plant Biotechnol. J. 2022, 20, 183–200. [Google Scholar] [CrossRef]
- Ma, Q.B.; Dai, X.Y.; Xu, Y.Y.; Guo, J.; Liu, Y.J.; Chen, N.; Xiao, J.; Zhang, D.J.; Xu, Z.H.; Zhang, X.S.; et al. Enhanced tolerance to chilling stress in OsMYB3R-2 transgenic rice is mediated by alteration in cell cycle and ectopic expression of stress genes. Plant Physiol. 2009, 150, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Rymen, B.; Fiorani, F.; Kartal, F.; Vandepoele, K.; Inzé, D.; Beemster, G.T. Cold nights impair leaf growth and cell cycle progression in maize through transcriptional changes of cell cycle genes. Plant Physiol. 2007, 143, 1429–1438. [Google Scholar] [CrossRef]
- Zhang, J.X.; Yuan, H.; Yang, Y.; Fish, T.; Lyi, S.M.; Thannhauser, T.M.; Zhang, L.; Li, L. Plastid ribosomal protein S5 is involved in photosynthesis, plant development, and cold stress tolerance in Arabidopsis. J. Exp. Bot. 2016, 67, 2731–2744. [Google Scholar] [CrossRef] [PubMed]
- Pulido, P.; Zagari, N.; Manavski, N.; Gawronski, P.; Matthes, A.; Scharff, L.B.; Meurer, J.; Leister, D. CHLOROPLAST RIBOSOME ASSOCIATED Supports Translation under Stress and Interacts with the Ribosomal 30S Subunit. Plant Physiol. 2018, 177, 1539–1554. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Kong, X.F.; Huang, H.; Wu, W.W.; Park, J.; Yun, D.J.; Lee, B.H.; Shi, H.Z.; Zhu, J.K. STCH4/REIL2 Confers Cold Stress Tolerance in Arabidopsis by Promoting rRNA Processing and CBF Protein Translation. Cell Rep. 2020, 30, 229–242.e5. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Xie, H.; Wei, X.; Dossa, K.; Yu, Y.; Hui, S.; Tang, G.; Zeng, X.; Yu, Y.; Hu, P.; et al. WGCNA analysis of salt-responsive core transcriptome identifies novel hub genes in rice. Genes 2019, 10, 719. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zhang, J.; Cao, J.; Ma, X.; Li, W.; Yang, G. Hub gene mining and co-expression network construction of low-temperature response in maize of seedling by WGCNA. Genes 2023, 14, 1598. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Yu, F.; Zhang, H.; Wang, B.; Ma, K.; Yu, C.; Xin, W.; Huang, X.; Liu, Y.; Liu, K. Genetic mapping of quantitative trait loci and a major locus for resistance to grey leaf spot in maize. Theor. Appl. Genet. 2020, 133, 2521–2533. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Vienna, Austria. 2014. Available online: http://www.R-project.org/ (accessed on 16 June 2023).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wang, S.; Basten, C.; Zeng, Z. Windows QTL Cartographer 2.5; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 2012. [Google Scholar]
- Liu, Z.J.; Hubbard, K.G.; Lin, X.M.; Yang, X.G. Negative effects of climate warming on maize yield are reversed by the changing of sowing date and cultivar selection in Northeast China. Glob. Chang. Biol. 2013, 19, 3481–3492. [Google Scholar] [CrossRef]
- Martinez-Seidel, F.; Suwanchaikasem, P.; Gentry-Torfer, D.; Rajarathinam, R.; Ebert, A.; Erban, A.; Firmino, A.A.P.; Nie, S.; Leeming, M.G.; Williamson, N.A.; et al. Remodelled ribosomes synthesise a specific proteome in proliferating plant tissue during cold. bioRxiv 2022. [Google Scholar] [CrossRef]
- Martinez-Seidel, F.; Beine-Golovchuk, O.; Hsieh, Y.-C.; Eshraky, K.E.; Gorka, M.; Cheong, B.-E.; Jimenez-Posada, E.V.; Walther, D.; Skirycz, A.; Roessner, U.; et al. Spatially enriched paralog rearrangements argue functionally diverse ribosomes arise during cold acclimation in Arabidopsis. Int. J. Mol. Sci. 2021, 22, 6160. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, J.; Zu, X.F.; Gong, J.; Deng, H.J.; Hang, R.; Zhang, X.F.; Liu, C.Y.; Deng, X.; Luo, L.L.; et al. Pseudouridylation of chloroplast ribosomal RNA contributes to low temperature acclimation in rice. New Phytol. 2022, 236, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Wang, C.M.; Zheng, M.; Lyu, J.; Xu, Y.; Li, X.H.; Niu, M.; Long, W.H.; Wang, D.; Wang, H.Y.; et al. WHITE PANICLE1, a Val-tRNA synthetase regulating chloroplast ribosome biogenesis in rice, is essential for early chloroplast development. Plant Physiol. 2016, 170, 2110–2123. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.P.; Cai, W.M. RAN1 is involved in plant cold resistance and development in rice (Oryza sativa). J. Exp. Bot. 2014, 65, 3277–3287. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ontology | ID | Description | q-Value |
|---|---|---|---|
| MEbrown: | |||
| BP | GO:0043549 | regulation of kinase activity | 1.5 × 105 |
| BP | GO:0045859 | regulation of protein kinase activity | 1.5 × 105 |
| BP | GO:0000079 | regulation of cyclin-dependent protein serine/threonine kinase activity | 1.5 × 105 |
| BP | GO:1904029 | regulation of cyclin-dependent protein kinase activity | 1.5 × 105 |
| BP | GO:0001932 | regulation of protein phosphorylation | 4.1 × 105 |
| BP | GO:0042325 | regulation of phosphorylation | 4.1 × 105 |
| BP | GO:0071900 | regulation of protein serine/threonine kinase activity | 4.1 × 105 |
| MF | GO:0046983 | protein dimerization activity | 9.2 × 105 |
| BP | GO:0007049 | cell cycle | 1.8 × 104 |
| BP | GO:0051338 | regulation of transferase activity | 2.3 × 104 |
| BP | GO:0051726 | regulation of cell cycle | 2.3 × 104 |
| BP | GO:0040008 | regulation of growth | 1.0 × 103 |
| BP | GO:0000278 | mitotic cell cycle | 1.2 × 103 |
| BP | GO:0048638 | regulation of developmental growth | 1.3 × 103 |
| BP | GO:0044770 | cell cycle phase transition | 1.3 × 103 |
| MEred: | |||
| CC | GO:0005840 | ribosome | 2.8 × 10140 |
| MF | GO:0003735 | structural constituent of ribosome | 8.4 × 10137 |
| MF | GO:0005198 | structural molecule activity | 4.8 × 10124 |
| CC | GO:0022626 | cytosolic ribosome | 9.8 × 1089 |
| CC | GO:0044391 | ribosomal subunit | 2.5 × 1085 |
| CC | GO:1990904 | ribonucleoprotein complex | 3.2 × 1072 |
| CC | GO:0022625 | cytosolic large ribosomal subunit | 6.5 × 1056 |
| CC | GO:0015934 | large ribosomal subunit | 6.1 × 1054 |
| CC | GO:0022627 | cytosolic small ribosomal subunit | 1.3 × 1032 |
| CC | GO:0015935 | small ribosomal subunit | 7.0 × 1030 |
| BP | GO:0042254 | ribosome biogenesis | 1.5 × 1025 |
| BP | GO:0022613 | ribonucleoprotein complex biogenesis | 3.0 × 1023 |
| BP | GO:0042273 | ribosomal large subunit biogenesis | 1.5 × 1017 |
| BP | GO:0042255 | ribosome assembly | 7.8 × 1017 |
| BP | GO:0002181 | cytoplasmic translation | 6.7 × 1016 |
| MEgreen: | |||
| CC | GO:0016469 | proton-transporting two-sector ATPase complex | 8.6 × 108 |
| CC | GO:0033178 | proton-transporting two-sector ATPase complex | 7.4 × 107 |
| CC | GO:0033176 | proton-transporting V-type ATPase complex | 9.7 × 107 |
| CC | GO:0033180 | proton-transporting V-type ATPase, V1 domain | 1.3 × 105 |
| CC | GO:0098796 | membrane protein complex | 1.6 × 103 |
| CC | GO:1990204 | oxidoreductase complex | 4.4 × 102 |
| MF | GO:0044769 | ATPase activity, coupled to transmembrane movement of ions | 1.7 × 107 |
| MF | GO:0046961 | proton-transporting ATPase activity, rotational mechanism | 1.7 × 107 |
| MF | GO:0019829 | ATPase-coupled cation transmembrane transporter activity | 1.3 × 104 |
| MF | GO:0042625 | ATPase-coupled ion transmembrane transporter activity | 1.3 × 104 |
| MF | GO:0015078 | proton transmembrane transporter activity | 8.3 × 104 |
| MF | GO:0016627 | oxidoreductase activity | 3.9 × 102 |
| Chr. | Peak (cM) | LOD | Add | R2 (%) | 2 LOD (cM) | Location (Mb) |
|---|---|---|---|---|---|---|
| 1 | 105.0 | 5.0 | −23.7 | 13.8 | 93.2–106.1 | 118.5–174.8 |
| 1 | 128.8 | 7.2 | 19.0 | 20.6 | 117.8–139.7 | 191.3–209.5 |
| 2 | 114.2 | 3.4 | 9.4 | 8.9 | 109.3–119.1 | 223.6–229.7 |
| GeneID | KME | GS | NR Annotation | GO Term | Location (Chr. 1) |
|---|---|---|---|---|---|
| MEbrown: | |||||
| Zm00001d031655 | 0.90 | 0.41 | uncharacterized protein LOC100281072 | regulation of transcription | 197,545,631–197,549,052 |
| Zm00001d031951 | 0.98 | 0.63 | uncharacterized protein LOC100272476 | glutathione metabolic process | 207,697,908–207,854,723 |
| Zm00001d031953 | 0.92 | 0.69 | uncharacterized protein LOC100272476 | glutathione metabolic process | 207,932,227–207,935,640 |
| MEred: | |||||
| Zm00001d031429 | 0.96 | 0.84 | pfkB-like carbohydrate kinase family protein | structural constituent of ribosome | 189,856,343–189,862,099 |
| Zm00001d031560 | 0.95 | 0.81 | 60S ribosomal protein L32 | structural constituent of ribosome | 194,266,307–194,270,465 |
| Zm00001d031640 | 0.90 | 0.66 | hypothetical protein SETIT_2G267400v2 | * | 197,108,702–197,111,394 |
| Zm00001d031651 | 0.92 | 0.88 | Mitochondrial import inner membrane translocase subunit Tim8 | mitochondrial intermembrane space | 197,393,450–197,395,554 |
| MEgreen: | |||||
| Zm00001d031667 | 0.92 | −0.42 | hypothetical protein Zm00014a_031611 | * | 196,224,574–196,228,047 |
| Zm00001d031617 | 0.92 | −0.50 | Discolored-paralog2 | oxidoreductase activity | 198,119,074–198,130,785 |
| Zm00001d031992 | 0.93 | −0.49 | indeterminate domain p1 | * | 208,942,917–208,950,782 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, L.; Peng, X.; Zhang, H.; Xin, W.; Ma, K.; Liu, Y.; Hu, G. Transcriptome Analysis and QTL Mapping Identify Candidate Genes and Regulatory Mechanisms Related to Low-Temperature Germination Ability in Maize. Genes 2023, 14, 1917. https://doi.org/10.3390/genes14101917
Du L, Peng X, Zhang H, Xin W, Ma K, Liu Y, Hu G. Transcriptome Analysis and QTL Mapping Identify Candidate Genes and Regulatory Mechanisms Related to Low-Temperature Germination Ability in Maize. Genes. 2023; 14(10):1917. https://doi.org/10.3390/genes14101917
Chicago/Turabian StyleDu, Lei, Xin Peng, Hao Zhang, Wangsen Xin, Kejun Ma, Yongzhong Liu, and Guangcan Hu. 2023. "Transcriptome Analysis and QTL Mapping Identify Candidate Genes and Regulatory Mechanisms Related to Low-Temperature Germination Ability in Maize" Genes 14, no. 10: 1917. https://doi.org/10.3390/genes14101917