
Tissue-Specific Tumour Suppressor and Oncogenic Activities of the Polycomb-like Protein MTF2
 ,
,
Abstract
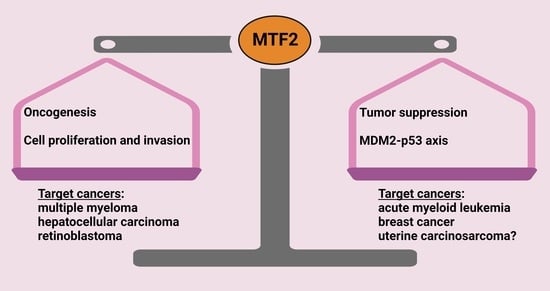
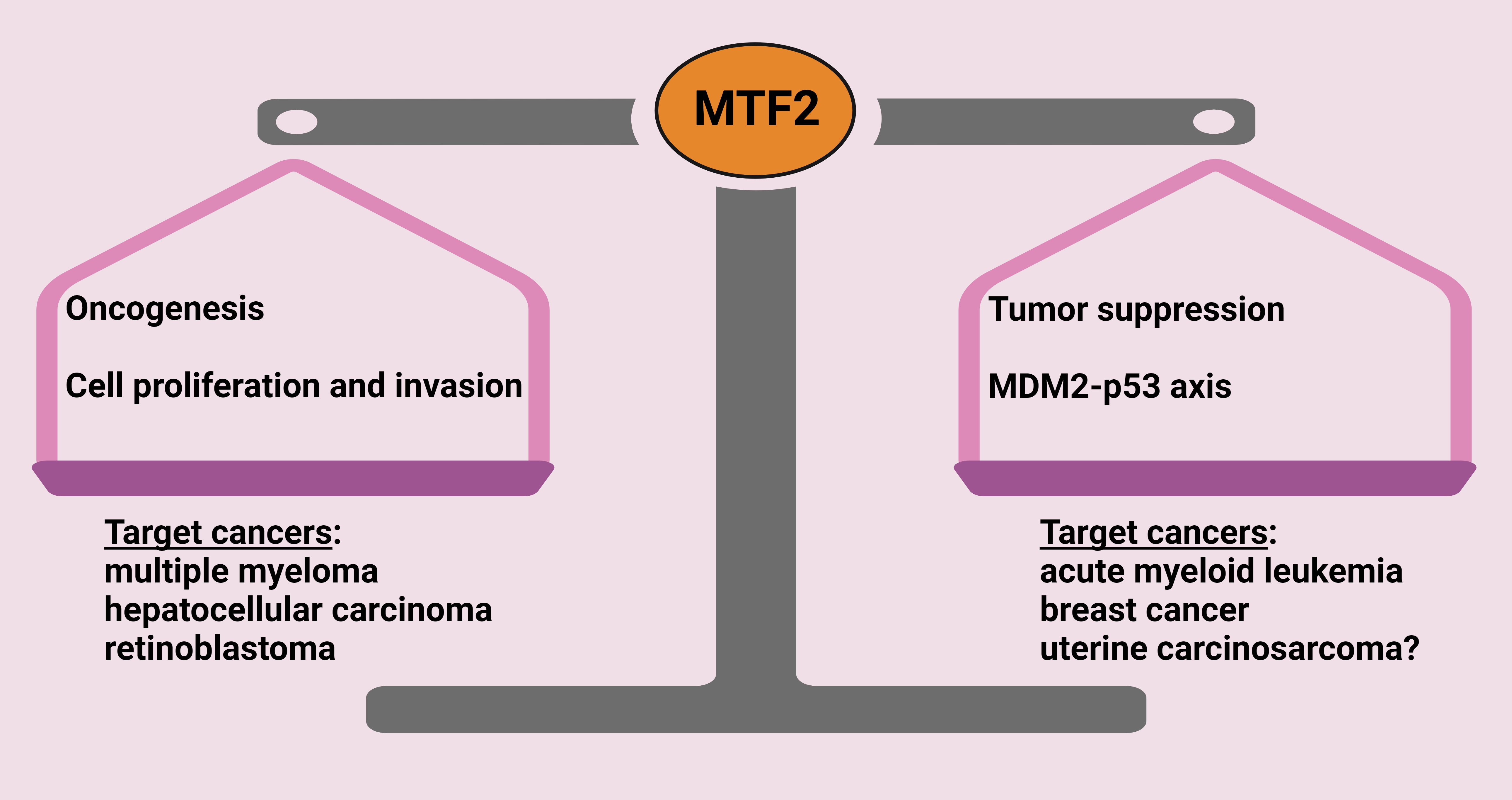
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. PRC2 Variant Complexes
PRC2.1 Gene Recruitment
3. MTF2 Structural Function
4. Regulation of ESC Fate by MTF2
5. Developmental Roles of MTF2
6. MTF2 Is Essential for Definitive Erythropoiesis and Regulating Wnt Signalling in Erythroid Cells
7. The Role of MTF2 in Acute Myeloid Leukaemia (AML)
8. A Role for MTF2 in Breast Cancer
9. Oncogenic Activity of MTF2
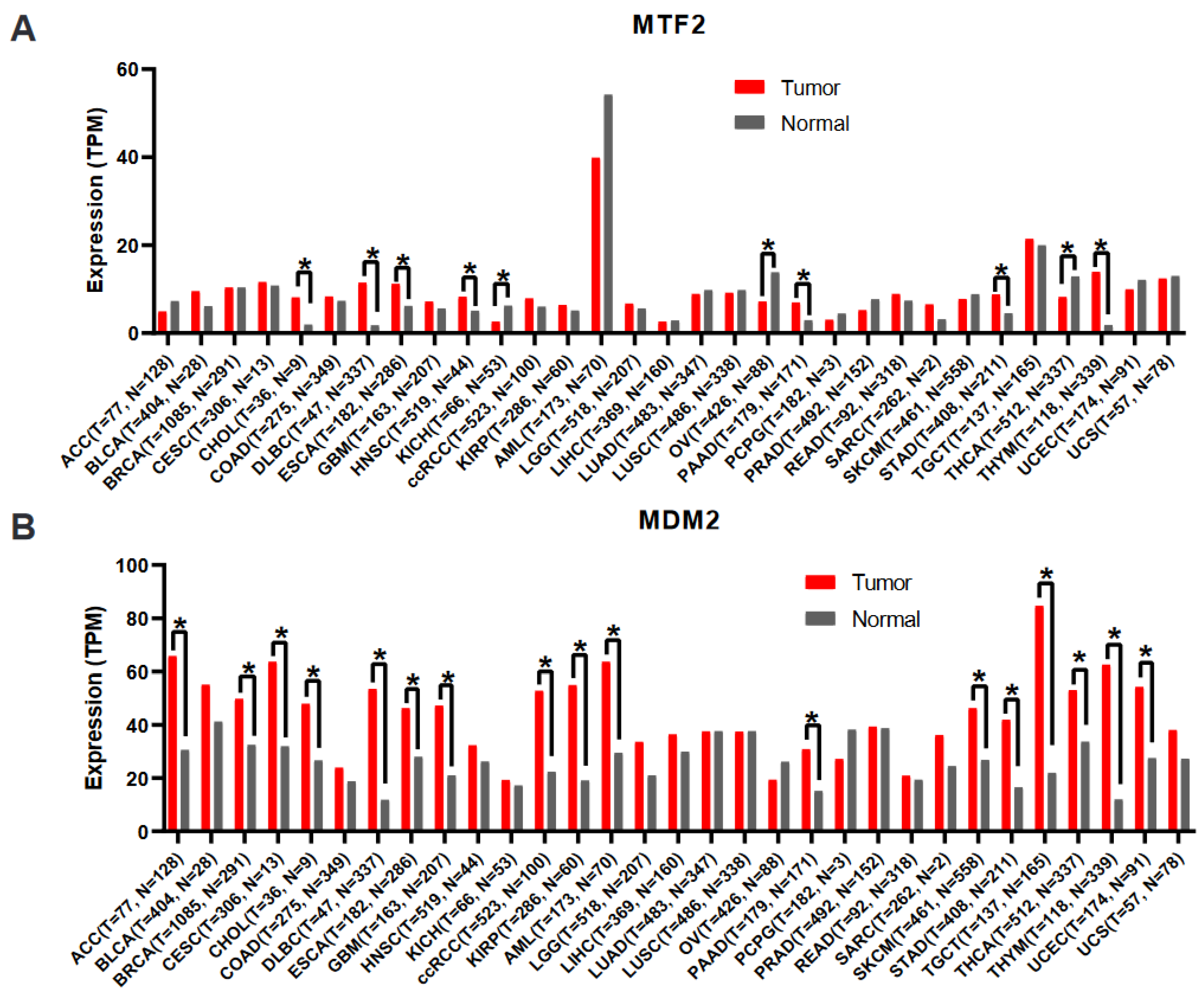
10. Bioinformatic Analyses of Public Databases
11. Future Directions
12. Materials and Methods
12.1. Gene Expression Analysis
12.2. Survival Analysis
12.3. Mutational Analysis
12.4. RNA-Seq Analysis
12.5. GO Term Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Shilatifard, A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 2021, 22, 326–345. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef] [PubMed]
- Duncan, I.M. Polycomblike: A gene that appears to be required for the normal expression of the bithorax and antennapedia gene complexes of Drosophila melanogaster. Genetics 1982, 102, 49–70. [Google Scholar] [CrossRef]
- Coulson, M.; Robert, S.; Eyre, H.J.; Saint, R. The identification and localization of a human gene with sequence similarity to Polycomblike of Drosophila melanogaster. Genomics 1998, 48, 381–383. [Google Scholar] [CrossRef]
- Kawakami, S.; Mitsunaga, K.; Kikuti, Y.Y.; Ando, A.; Inoko, H.; Yamamura, K.; Abe, K. Tctex3, related to Drosophila polycomblike, is expressed in male germ cells and mapped to the mouse t-complex. Mamm. Genome 1998, 9, 874–880. [Google Scholar] [CrossRef]
- Kitaguchi, T.; Nakata, K.; Nagai, T.; Aruga, J.; Mikoshiba, K. Xenopus Polycomblike 2 (XPcl2) controls anterior to posterior patterning of the neural tissue. Dev. Genes Evol. 2001, 211, 309–314. [Google Scholar] [CrossRef]
- Walker, E.; Chang, W.Y.; Hunkapiller, J.; Cagney, G.; Garcha, K.; Torchia, J.; Krogan, N.; Reiter, J.; Stanford, W.L. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell 2010, 6, 153–166. [Google Scholar] [CrossRef]
- Loh, C.H.; van Genesen, S.; Perino, M.; Bark, M.R.; Veenstra, G.J.C. Loss of PRC2 subunits primes lineage choice during exit of pluripotency. Nat. Commun. 2021, 12, 6985. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yu, X.; Zhang, T.; Zhang, X.; Zhang, Z.; Chen, Y. Chick Pcl2 regulates the left-right asymmetry by repressing Shh expression in Hensen’s node. Development 2004, 131, 4381–4391. [Google Scholar] [CrossRef] [PubMed]
- Maganti, H.B.; Jrade, H.; Cafariello, C.; Manias Rothberg, J.L.; Porter, C.J.; Yockell-Lelièvre, J.; Battaion, H.L.; Khan, S.T.; Li, Y.; Grzybowski, A.T.; et al. Targeting the MTF2-MDM2 axis sensitizes refractory acute myeloid leukemia to chemotherapy. Cancer Discov. 2018, 8, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Duy, C.; Melnick, A. Untangling the Role of Polycomb Complexes in Chemotherapy Resistance. Cancer Discov. 2018, 8, 1348–1351. [Google Scholar] [CrossRef]
- Wang, J.; He, C.; Gao, P.; Wang, S.; Lv, R.; Zhou, H.; Zhou, Q.; Zhang, K.; Sun, J.; Fan, C.; et al. HNF1B-mediated repression of SLUG is suppressed by EZH2 in aggressive prostate cancer. Oncogene 2020, 39, 1335–1346. [Google Scholar] [CrossRef]
- Mieczkowska, I.K.; Pantelaiou-Prokaki, G.; Prokakis, E.; Schmidt, G.E.; Muller-Kirschbaum, L.C.; Werner, M.; Sen, M.; Velychko, T.; Jannasch, K.; Dullin, C.; et al. Decreased PRC2 activity supports the survival of basal-like breast cancer cells to cytotoxic treatments. Cell Death Dis. 2021, 12, 1118. [Google Scholar] [CrossRef]
- Liang, Y.; Yang, Y.; Guo, R.; Gao, S.; Guo, X.; Li, D.; Wang, M.; Koseki, H.; Li, X. PCL2 regulates p53 stability and functions as a tumor suppressor in breast cancer. Sci. Bull. 2018, 63, 629–639. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, Y.; Hu, Y.; Zhong, J.; Jiang, C.; Zhang, H. LncRNA CCAT1 sponges miR-218-5p to promote EMT, cellular migration and invasion of retinoblastoma by targeting MTF2. Cell Signal 2021, 86, 110088. [Google Scholar] [CrossRef]
- Sun, F.; Cheng, Y.; Riordan, J.D.; Dupuy, A.; Dubois, W.; Pisano, M.; Dong, J.; Mock, B.; Zhan, F.; Hari, P.; et al. WDR26 and MTF2 are therapeutic targets in multiple myeloma. J. Hematol. Oncol. 2021, 14, 203. [Google Scholar] [CrossRef]
- Wu, T.T.; Cai, J.; Tian, Y.H.; Chen, J.F.; Cheng, Z.L.; Pu, C.S.; Shi, W.Z.; Suo, X.P.; Wu, X.J.; Dou, X.W.; et al. MTF2 Induces Epithelial-Mesenchymal Transition and Progression of Hepatocellular Carcinoma by Transcriptionally Activating Snail. Onco Targets Ther. 2019, 12, 11207–11220. [Google Scholar] [CrossRef]
- Fischer, S.; Liefke, R. Polycomb-like Proteins in Gene Regulation and Cancer. Genes 2023, 14, 938. [Google Scholar] [CrossRef]
- Kennison, J.A.; Tamkun, J.W. Dosage-dependent modifiers of polycomb and antennapedia mutations in Drosophila. Proc. Natl. Acad. Sci. USA 1988, 85, 8136–8140. [Google Scholar] [CrossRef]
- Strutt, H.; Paro, R. The polycomb group protein complex of Drosophila melanogaster has different compositions at different target genes. Mol. Cell Biol. 1997, 17, 6773–6783. [Google Scholar] [CrossRef]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Hou, J.; Feng, L.; Lv, A.; Ke, X.; Liang, H.; Wang, F.; Zhang, K.; Chen, K.; Cui, H. PHF19 promotes the proliferation, migration, and chemosensitivity of glioblastoma to doxorubicin through modulation of the SIAH1/β-catenin axis. Cell Death Dis. 2018, 9, 1049. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Ahn, J.H.; Liu, H.; Tsai, Y.H.; Bhanu, N.V.; Koss, B.; Allison, D.F.; Ma, A.; Storey, A.J.; Wang, P.; et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 2019, 134, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Muller, C.W.; Vermeulen, M.; Muller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Tamburri, S.; Lavarone, E.; Fernandez-Perez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856.e5. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Fursova, N.A.; Kelley, J.R.; Huseyin, M.K.; Feldmann, A.; Klose, R.J. PRC1 Catalytic Activity Is Central to Polycomb System Function. Mol. Cell 2020, 77, 857–874.e9. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Pengelly, A.R.; Copur, O.; Jackle, H.; Herzig, A.; Muller, J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 2013, 339, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Lavarone, E.; Barbieri, C.M.; Pasini, D. Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat. Commun. 2019, 10, 1679. [Google Scholar] [CrossRef] [PubMed]
- Swigut, T.; Wysocka, J. H3K27 demethylases, at long last. Cell 2007, 131, 29–32. [Google Scholar] [CrossRef]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef]
- van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef]
- Fischer, S.; Weber, L.M.; Liefke, R. Evolutionary adaptation of the Polycomb repressive complex 2. Epigenetics Chromatin 2022, 15, 7. [Google Scholar] [CrossRef]
- Beringer, M.; Pisano, P.; Di Carlo, V.; Blanco, E.; Chammas, P.; Vizan, P.; Gutierrez, A.; Aranda, S.; Payer, B.; Wierer, M.; et al. EPOP Functionally Links Elongin and Polycomb in Pluripotent Stem Cells. Mol. Cell 2016, 64, 645–658. [Google Scholar] [CrossRef]
- Liefke, R.; Karwacki-Neisius, V.; Shi, Y. EPOP Interacts with Elongin BC and USP7 to Modulate the Chromatin Landscape. Mol. Cell 2016, 64, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Alekseyenko, A.A.; Gorchakov, A.A.; Kharchenko, P.V.; Kuroda, M.I. Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc. Natl. Acad. Sci. USA 2014, 111, 2488–2493. [Google Scholar] [CrossRef] [PubMed]
- Chammas, P.; Mocavini, I.; Di Croce, L. Engaging chromatin: PRC2 structure meets function. Br. J. Cancer 2020, 122, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Healy, E.; Mucha, M.; Glancy, E.; Fitzpatrick, D.J.; Conway, E.; Neikes, H.K.; Monger, C.; Van Mierlo, G.; Baltissen, M.P.; Koseki, Y.; et al. PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol. Cell 2019, 76, 437–452.e6. [Google Scholar] [CrossRef] [PubMed]
- Glancy, E.; Wang, C.; Tuck, E.; Healy, E.; Amato, S.; Neikes, H.K.; Mariani, A.; Mucha, M.; Vermeulen, M.; Pasini, D.; et al. PRC2.1- and PRC2.2-specific accessory proteins drive recruitment of different forms of canonical PRC1. Mol. Cell 2023, 83, 1393–1411.e7. [Google Scholar] [CrossRef] [PubMed]
- Grijzenhout, A.; Godwin, J.; Koseki, H.; Gdula, M.R.; Szumska, D.; McGouran, J.F.; Bhattacharya, S.; Kessler, B.M.; Brockdorff, N.; Cooper, S. Functional analysis of AEBP2, a PRC2 Polycomb protein, reveals a Trithorax phenotype in embryonic development and in ESCs. Development 2016, 143, 2716–2723. [Google Scholar] [CrossRef]
- Chen, S.; Jiao, L.; Shubbar, M.; Yang, X.; Liu, X. Unique Structural Platforms of Suz12 Dictate Distinct Classes of PRC2 for Chromatin Binding. Mol. Cell 2018, 69, 840–852.e5. [Google Scholar] [CrossRef]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016, 17, 583–595. [Google Scholar] [CrossRef]
- Davidovich, C.; Zheng, L.; Goodrich, K.J.; Cech, T.R. Promiscuous RNA binding by Polycomb repressive complex 2. Nat. Struct. Mol. Biol. 2013, 20, 1250–1257. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Scelfo, A.; Piunti, A.; Pasini, D. The controversial role of the Polycomb group proteins in transcription and cancer: How much do we not understand Polycomb proteins? FEBS J. 2015, 282, 1703–1722. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Li, H.; Liefke, R.; Jiang, J.; Kurland, J.V.; Tian, W.; Deng, P.; Zhang, W.; He, Q.; Patel, D.J.; Bulyk, M.L.; et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 2017, 549, 287–291. [Google Scholar] [CrossRef]
- Smits, A.H.; Jansen, P.W.; Poser, I.; Hyman, A.A.; Vermeulen, M. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 2013, 41, e28. [Google Scholar] [CrossRef] [PubMed]
- Oliviero, G.; Brien, G.L.; Waston, A.; Streubel, G.; Jerman, E.; Andrews, D.; Doyle, B.; Munawar, N.; Wynne, K.; Crean, J.; et al. Dynamic Protein Interactions of the Polycomb Repressive Complex 2 during Differentiation of Pluripotent Cells. Mol. Cell. Proteom. MCP 2016, 15, 3450–3460. [Google Scholar] [CrossRef]
- Kloet, S.L.; Makowski, M.M.; Baymaz, H.I.; van Voorthuijsen, L.; Karemaker, I.D.; Santanach, A.; Jansen, P.; Di Croce, L.; Vermeulen, M. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 2016, 23, 682–690. [Google Scholar] [CrossRef]
- Hojfeldt, J.W.; Hedehus, L.; Laugesen, A.; Tatar, T.; Wiehle, L.; Helin, K. Non-core Subunits of the PRC2 Complex Are Collectively Required for Its Target-Site Specificity. Mol. Cell 2019, 76, 423–436.e3. [Google Scholar] [CrossRef]
- Cai, L.; Rothbart, S.B.; Lu, R.; Xu, B.; Chen, W.Y.; Tripathy, A.; Rockowitz, S.; Zheng, D.; Patel, D.J.; Allis, C.D.; et al. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 2013, 49, 571–582. [Google Scholar] [CrossRef]
- Kim, J.; Daniel, J.; Espejo, A.; Lake, A.; Krishna, M.; Xia, L.; Zhang, Y.; Bedford, M.T. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006, 7, 397–403. [Google Scholar] [CrossRef]
- Siomi, M.C.; Mannen, T.; Siomi, H. How does the royal family of Tudor rule the PIWI-interacting RNA pathway? Genes. Dev. 2010, 24, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Selenko, P.; Sprangers, R.; Stier, G.; Buhler, D.; Fischer, U.; Sattler, M. SMN tudor domain structure and its interaction with the Sm proteins. Nat. Struct. Biol. 2001, 8, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Ballare, C.; Lange, M.; Lapinaite, A.; Martin, G.M.; Morey, L.; Pascual, G.; Liefke, R.; Simon, B.; Shi, Y.; Gozani, O.; et al. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat. Struct. Mol. Biol. 2012, 19, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Gambero, G.; O’Connell, D.J.; Jerman, E.; Turner, S.A.; Egan, C.M.; Dunne, E.J.; Jurgens, M.C.; Wynne, K.; Piao, L.; et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 2012, 19, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Avvakumov, N.; Watanabe, R.; Abraham, C.G.; Lalonde, M.E.; Hong, Z.; Allen, C.; Roy, S.; Nunez, J.K.; Nickoloff, J.; et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat. Struct. Mol. Biol. 2012, 19, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Kycia, I.; Kudithipudi, S.; Tamas, R.; Kungulovski, G.; Dhayalan, A.; Jeltsch, A. The Tudor domain of the PHD finger protein 1 is a dual reader of lysine trimethylation at lysine 36 of histone H3 and lysine 27 of histone variant H3t. J. Mol. Biol. 2014, 426, 1651–1660. [Google Scholar] [CrossRef]
- Gatchalian, J.; Kingsley, M.C.; Moslet, S.D.; Rosas Ospina, R.D.; Kutateladze, T.G. An aromatic cage is required but not sufficient for binding of Tudor domains of the Polycomblike protein family to H3K36me3. Epigenetics 2015, 10, 467–473. [Google Scholar] [CrossRef]
- Bienz, M. The PHD finger, a nuclear protein-interaction domain. Trends Biochem. Sci. 2006, 31, 35–40. [Google Scholar] [CrossRef]
- Singh, N.; Reyes-Ordonez, A.; Compagnone, M.A.; Moreno, J.F.; Leslie, B.J.; Ha, T.; Chen, J. Redefining the specificity of phosphoinositide-binding by human PH domain-containing proteins. Nat. Commun. 2021, 12, 4339. [Google Scholar] [CrossRef]
- O’Connell, S.; Wang, L.; Robert, S.; Jones, C.A.; Saint, R.; Jones, R.S. Polycomblike PHD fingers mediate conserved interaction with enhancer of zeste protein. J. Biol. Chem. 2001, 276, 43065–43073. [Google Scholar] [CrossRef]
- Choi, J.; Bachmann, A.L.; Tauscher, K.; Benda, C.; Fierz, B.; Muller, J. DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nat. Struct. Mol. Biol. 2017, 24, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, J.; Yang, Y.; Qiu, R.; Zheng, Y.; Huang, W.; Zeng, Y.; Hou, Y.; Wang, S.; Leng, S.; et al. PHD finger protein 1 (PHF1) is a novel reader for histone H4R3 symmetric dimethylation and coordinates with PRMT5-WDR77/CRL4B complex to promote tumorigenesis. Nucleic Acids Res. 2018, 46, 6608–6626. [Google Scholar] [CrossRef] [PubMed]
- Perino, M.; van Mierlo, G.; Karemaker, I.D.; van Genesen, S.; Vermeulen, M.; Marks, H.; van Heeringen, S.J.; Veenstra, G.J.C. MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat. Genet. 2018, 50, 1002–1010. [Google Scholar] [CrossRef]
- Walker, E.; Stanford, W.L. Transcriptional Networks Regulating Embryonic Stem Cell Fate Decisions. In Regulatory Networks in Stem Cells; Rajasekhar, V.K., Vemuri, M.C., Eds.; Human Press: Totowa, NJ, USA, 2009; pp. 87–100. [Google Scholar]
- Walker, E.; Ohishi, M.; Davey, R.E.; Zhang, W.; Cassar, P.A.; Tanaka, T.S.; Der, S.D.; Morris, Q.; Hughes, T.R.; Zandstra, P.W.; et al. Prediction and Testing of Novel Transcriptional Networks Regulating Embryonic Stem Cell Self-Renewal and Commitment. Cell Stem Cell 2007, 1, 71–86. [Google Scholar] [CrossRef]
- Casanova, M.; Preissner, T.; Cerase, A.; Poot, R.; Yamada, D.; Li, X.; Appanah, R.; Bezstarosti, K.; Demmers, J.; Koseki, H.; et al. Polycomblike 2 facilitates the recruitment of PRC2 Polycomb group complexes to the inactive X chromosome and to target loci in embryonic stem cells. Development 2011, 138, 1471–1482. [Google Scholar] [CrossRef]
- Perino, M.; van Mierlo, G.; Loh, C.; Wardle, S.M.T.; Zijlmans, D.W.; Marks, H.; Veenstra, G.J.C. Two Functional Axes of Feedback-Enforced PRC2 Recruitment in Mouse Embryonic Stem Cells. Stem Cell Rep. 2020, 15, 1287–1300. [Google Scholar] [CrossRef]
- Petracovici, A.; Bonasio, R. Distinct PRC2 subunits regulate maintenance and establishment of Polycomb repression during differentiation. Mol. Cell 2021, 81, 2625–2639.e5. [Google Scholar] [CrossRef]
- Zhang, Z.; Jones, A.; Sun, C.W.; Li, C.; Chang, C.W.; Joo, H.Y.; Dai, Q.; Mysliwiec, M.R.; Wu, L.C.; Guo, Y.; et al. PRC2 complexes with JARID2, MTF2, and esPRC2p48 in ES cells to modulate ES cell pluripotency and somatic cell reprogramming. Stem Cells 2011, 29, 229–240. [Google Scholar] [CrossRef]
- Wang, S.; He, F.; Xiong, W.; Gu, S.; Liu, H.; Zhang, T.; Yu, X.; Chen, Y. Polycomblike-2-deficient mice exhibit normal left-right asymmetry. Dev. Dyn. 2007, 236, 853–861. [Google Scholar] [CrossRef]
- Li, X.; Isono, K.; Yamada, D.; Endo, T.A.; Endoh, M.; Shinga, J.; Mizutani-Koseki, Y.; Otte, A.P.; Casanova, M.; Kitamura, H.; et al. Mammalian polycomb-like Pcl2/Mtf2 is a novel regulatory component of PRC2 that can differentially modulate polycomb activity both at the HOX gene cluster and at Cdkn2a genes. Mol. Cell Biol. 2011, 31, 351–364. [Google Scholar] [CrossRef]
- Rothberg, J.L.M.; Maganti, H.B.; Jrade, H.; Porter, C.J.; Palidwor, G.A.; Cafariello, C.; Battaion, H.L.; Khan, S.T.; Perkins, T.J.; Paulson, R.F.; et al. Mtf2-PRC2 control of canonical Wnt signaling is required for definitive erythropoiesis. Cell Discov. 2018, 4, 21. [Google Scholar] [CrossRef]
- Motoyama, J.; Kitajima, K.; Kojima, M.; Kondo, S.; Takeuchi, T. Organogenesis of the liver, thymus and spleen is affected in jumonji mutant mice. Mech. Dev. 1997, 66, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Cloos, P.A.; Walfridsson, J.; Olsson, L.; Bukowski, J.P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Uehara, S.; Udagawa, N.; Takahashi, N. Regulation of bone metabolism by Wnt signals. J. Biochem. 2016, 159, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Oittinen, M.; Popp, A.; Kurppa, K.; Lindfors, K.; Maki, M.; Kaikkonen, M.U.; Viiri, K. Polycomb Repressive Complex 2 Enacts Wnt Signaling in Intestinal Homeostasis and Contributes to the Instigation of Stemness in Diseases Entailing Epithelial Hyperplasia or Neoplasia. Stem Cells 2017, 35, 445–457. [Google Scholar] [CrossRef]
- Yu, J.; Yu, J.; Rhodes, D.R.; Tomlins, S.A.; Cao, X.; Chen, G.; Mehra, R.; Wang, X.; Ghosh, D.; Shah, R.B.; et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007, 67, 10657–10663. [Google Scholar] [CrossRef]
- Hoffmann, F.; Niebel, D.; Aymans, P.; Ferring-Schmitt, S.; Dietrich, D.; Landsberg, J. H3K27me3 and EZH2 expression in melanoma: Relevance for melanoma progression and response to immune checkpoint blockade. Clin. Epigenetics 2020, 12, 24. [Google Scholar] [CrossRef]
- Cleven, A.H.; Sannaa, G.A.; Briaire-de Bruijn, I.; Ingram, D.R.; van de Rijn, M.; Rubin, B.P.; de Vries, M.W.; Watson, K.L.; Torres, K.E.; Wang, W.L.; et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 2016, 29, 582–590. [Google Scholar] [CrossRef]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Dolgin, E. The most popular genes in the human genome. Nature 2017, 551, 427–431. [Google Scholar] [CrossRef]
- Prat, A.; Fan, C.; Fernandez, A.; Hoadley, K.A.; Martinello, R.; Vidal, M.; Viladot, M.; Pineda, E.; Arance, A.; Munoz, M.; et al. Response and survival of breast cancer intrinsic subtypes following multi-agent neoadjuvant chemotherapy. BMC Med. 2015, 13, 303. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.J.; Schinke, C.; Eng, C.L.P.; Towfic, F.; Gruber, F.; Dervan, A.; White, B.S.; Pratapa, A.; Guan, Y.; Chen, H.; et al. Multiple Myeloma DREAM Challenge reveals epigenetic regulator PHF19 as marker of aggressive disease. Leukemia 2020, 34, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Rojanaporn, D.; Chawla, B.; Sundar, G.; Gopal, L.; Khetan, V. Retinoblastoma in Asia. Eye 2019, 33, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.Z.; Chu, B.F.; Zhang, Y.; Weng, M.Z.; Qin, Y.Y.; Gong, W.; Quan, Z.W. Long non-coding RNA CCAT1 promotes gallbladder cancer development via negative modulation of miRNA-218-5p. Cell Death Dis. 2015, 6, e1583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, H. Long non-coding RNA CCAT1/miR-218/ZFX axis modulates the progression of laryngeal squamous cell cancer. Tumour Biol. 2017, 39, 1010428317699417. [Google Scholar] [CrossRef]
- Han, C.; Li, X.; Fan, Q.; Liu, G.; Yin, J. CCAT1 promotes triple-negative breast cancer progression by suppressing miR-218/ZFX signaling. Aging 2019, 11, 4858–4875. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, L.; Wang, Y.; Ma, P. Long non-coding RNA CCAT1 enhances human non-small cell lung cancer growth through downregulation of microRNA-218. Oncol. Rep. 2020, 43, 1045–1052. [Google Scholar] [CrossRef]
- Wang, F.; Gao, Y.; Lv, Y.; Wu, Y.; Guo, Y.; Du, F.; Wang, S.; Yu, J.; Cao, X.; Li, P.A. Polycomb-like 2 regulates PRC2 components to affect proliferation in glioma cells. J. Neurooncol 2020, 148, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Hui, W.; Liu, S.; Zheng, J.; Fang, Z.; Ding, Q.; Feng, C. Nutlin-3a as a novel anticancer agent for adrenocortical carcinoma with CTNNB1 mutation. Cancer Med. 2018, 7, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Pairawan, S.; Akcakanat, A.; Kopetz, S.; Tapia, C.; Zheng, X.; Chen, H.; Ha, M.J.; Rizvi, Y.; Holla, V.; Wang, J.; et al. Combined MEK/MDM2 inhibition demonstrates antitumor efficacy in TP53 wild-type thyroid and colorectal cancers with MAPK alterations. Sci. Rep. 2022, 12, 1248. [Google Scholar] [CrossRef] [PubMed]
- Iwakuma, T.; Lozano, G. MDM2, an introduction. Mol. Cancer Res. 2003, 1, 993–1000. [Google Scholar]
- Zhao, Y.; Yu, H.; Hu, W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim Biophys Sin 2014, 46, 180–189. [Google Scholar] [CrossRef]
- Reyland, M.E.; Jones, D.N. Multifunctional roles of PKCdelta: Opportunities for targeted therapy in human disease. Pharmacol. Ther. 2016, 165, 1–13. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Xiong, J.; Lan, B.; Wang, X.; Liu, J.; Lin, J.; Fei, Z.; Zheng, X.; Chen, C. Role of PRKDC in cancer initiation, progression, and treatment. Cancer Cell Int. 2021, 21, 563. [Google Scholar] [CrossRef]
- Youmans, D.T.; Gooding, A.R.; Dowell, R.D.; Cech, T.R. Competition between PRC2.1 and 2.2 subcomplexes regulates PRC2 chromatin occupancy in human stem cells. Mol. Cell 2021, 81, 488–501.e9. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngubo, M.; Moradi, F.; Ito, C.Y.; Stanford, W.L. Tissue-Specific Tumour Suppressor and Oncogenic Activities of the Polycomb-like Protein MTF2. Genes 2023, 14, 1879. https://doi.org/10.3390/genes14101879
Ngubo M, Moradi F, Ito CY, Stanford WL. Tissue-Specific Tumour Suppressor and Oncogenic Activities of the Polycomb-like Protein MTF2. Genes. 2023; 14(10):1879. https://doi.org/10.3390/genes14101879
Chicago/Turabian StyleNgubo, Mzwanele, Fereshteh Moradi, Caryn Y. Ito, and William L. Stanford. 2023. "Tissue-Specific Tumour Suppressor and Oncogenic Activities of the Polycomb-like Protein MTF2" Genes 14, no. 10: 1879. https://doi.org/10.3390/genes14101879