
Polycomb-like Proteins in Gene Regulation and Cancer

1
Institute of Molecular Biology and Tumor Research (IMT), Philipps University of Marburg, 35043 Marburg, Germany
2
Department of Hematology, Oncology and Immunology, University Hospital Giessen and Marburg, 35043 Marburg, Germany
*
Author to whom correspondence should be addressed.
Genes 2023, 14(4), 938; https://doi.org/10.3390/genes14040938
Submission received: 19 March 2023
/
Revised: 14 April 2023
/
Accepted: 17 April 2023
/
Published: 18 April 2023
(This article belongs to the Special Issue Molecular Mechanisms of the Polycomb Repressive Complex 2 (PRC2) and Its Role in Human Cancer)
Abstract
:Polycomb-like proteins (PCLs) are a crucial group of proteins associated with the Polycomb repressive complex 2 (PRC2) and are responsible for setting up the PRC2.1 subcomplex. In the vertebrate system, three homologous PCLs exist: PHF1 (PCL1), MTF2 (PCL2), and PHF19 (PCL3). Although the PCLs share a similar domain composition, they differ significantly in their primary sequence. PCLs play a critical role in targeting PRC2.1 to its genomic targets and regulating the functionality of PRC2. However, they also have PRC2-independent functions. In addition to their physiological roles, their dysregulation has been associated with various human cancers. In this review, we summarize the current understanding of the molecular mechanisms of the PCLs and how alterations in their functionality contribute to cancer development. We particularly highlight the nonoverlapping and partially opposing roles of the three PCLs in human cancer. Our review provides important insights into the biological significance of the PCLs and their potential as therapeutic targets for cancer treatment.
Keywords:
Polycomb-like; PHF1; MTF2; PHF19; cancer; Polycomb repressive complex 2; chromatin; transcription; epigenetics; leukemia1. Introduction
Failures in chromatin and gene regulatory mechanisms can be the source of various human diseases [1]. Polycomb repressive complex 2 (PRC2) is a key component of the chromatin regulatory machinery of the cell [2,3]. The primary function of PRC2 is to add methyl groups to lysine 27 of histone H3 (H3K27) to establish H3K27 trimethylation (H3K27me3), which induces gene repression. PRC2 is particularly relevant for maintaining the correct gene expression pattern during development [2] and is often associated with cell differentiation [3].
Given its central role in gene regulation, dysregulation of PRC2 is commonly observed in various cancer types [2,4,5]. In solid tumors, such as prostate cancer, bladder cancer, and melanoma, high PRC2 activity is typically associated with aggressive and advanced disease progression [5]. Thereby, overexpression of PRC2 leads to the repression of tumor suppressor genes, which can contribute to the development and progression of these cancers [5]. In contrast, in some cancer types, such as in T-ALL (T-cell acute lymphoblastic leukemia) [6,7] and breast cancer [8], PRC2 activity is reduced, leading to enhanced expression of oncogenes.
In some cancer types, loss- or gain-of-function mutations of the core components of PRC2 are observed. Many mutations affect the histone methyltransferase EZH2, such as in hematologic malignancies [7,9,10,11], which globally influence the chromatin landscape [7,12]. Furthermore, the target of PRC2, the amino acid K27 of histone 3, can be altered. In pediatric glioblastoma, a mutation to methionine (H3K27M) is commonly observed [13,14] and prevents the efficient spread of H3K27me3, a requirement for tumorigenesis in this cancer type [15].
The activity of PRC2 is mainly determined by the expression of its core components, which facilitate the enzymatic deposition of the H3K27me3 mark. However, not only the PRC2 core determines the activity of PRC2 but also the associated factors influence the composition, recruitment, and functionality of PRC2. Here, we describe and discuss the role of the Polycomb-like proteins (PCLs) in regulating PRC2 and how their dysregulation contributes to the development and progression of human cancer.
2. Polycomb-like Proteins Are Key Components of the PRC2.1 Subcomplex
Research in recent years has demonstrated that mammalian PRC2 represents not a single complex but a variety of alternative complexes that possess a common core complex but differ in the accessory factors [16]. The number of PRC2 components has also considerably increased during evolution, leading to a greater variety of possible compositions of the mammalian PRC2 compared to nonvertebrate PRC2 [17].
The catalytic core of PRC2 consists of four components: The histone methyltransferase EZH2 is responsible for the deposition of the majority of the H3K27me3 histone mark. EZH1, the homolog of EZH2, is structurally similar to EZH2 but is less essential for H3K27me3 deposition [18]. Instead, EZH1 may play a role in chromatin compaction by establishing dimeric PRC2 [19]. EZH2 alone cannot perform efficient H3K27me3 methylation but requires additional factors [20]. Another critical component of PRC2 is SUZ12, which acts as a scaffold that brings all necessary proteins together [16,21]. It allows the binding of EZH2, EED, and one of the two highly homologous proteins, RBBP4 and RBBP7 [21]. RBBP4/7 are structural components of the PRC2 complex, but are not required for its enzymatic activity [20]. EED also has a structural function [22]; additionally, it also recognizes the substrate of PRC2, H3K27me3, which is vital for the activity of PRC2 on chromatin [23]. Only the combination of EZH2, SUZ12, and EED establishes the necessary structural conformation that allows efficient methylation of histone proteins [16,22].
The PRC2 core complex is associated with additional proteins, establishing alternative subcomplexes [16,17]. The two major PRC2 subcomplexes are named PRC2.1 and PRC2.2 and are defined based on the proteins that are associated with the PRC2 core. The PRC2.2 subcomplex possesses JARID2 and AEBP2 [16,17] and will not be further addressed here. The defining characteristic of PRC2.1 is the presence of one of the PCL proteins, which is mutually exclusive to the binding of JARID2 and AEBP2 [17,24]. In the mammalian system, three PCL proteins exist: PHF1, MTF2, or PHF19 (alternative names are PCL1, PCL2, and PCL3, respectively). These three PCL proteins have evolved from the same ancestor protein, called Polycomb-like (Pcl) [25], and consequently have a similar domain composition [17]. They contain five different conserved domains (Figure 1A,B). In the central part of the protein, the PCLs possess a large globular region that consists of a Tudor domain, two PHD fingers, and a winged helix (WH) domain. This large domain is the main chromatin-binding module of the PCL proteins [26]. In addition to this sizeable globular domain in the center, the PCLs possess a conserved domain at the very C-terminus, which holds distant similarity with chromodomains [27]. This domain has been demonstrated to be essential for interaction with the PRC2 core [21,27].
Although the PCL proteins are the key features defining the PRC2.1 subcomplex, they are not the only associated proteins found explicitly with PRC2.1. In addition to the PCLs, PRC2.1 can also be associated with either EPOP [28,29] or PALI1 [30] or their homologous proteins, SKIDA1 and PALI2, respectively [17,24]. The specific role of these proteins regarding PRC2 and how they may work together with the PCLs in the context of PRC2.1 are not fully understood and will not be addressed further in this review.
Figure 1.
(A) General domain structure of the three PCLs. (B) Amino acid homology between the three human PCLs. (C) Model of the functional role of the PCLs for recruiting PRC2. PCL interacts via the winged helix domain with unmethylated CpG islands and recruits PRC2 by interaction of the C-terminal chromo-like domain with the PRC2 core. The histone methyltransferase EZH2 facilitates H3K27me3 methylation. EED interacts with H3K27me3 and contributes to PRC2 recruitment, as well. (D) Predicted aligned error of human PHF1, MTF2, and PHF19 obtained from the AlphaFold database [31]. Green rectangles indicate predicted associations of the unstructured regions with the chromatin-binding internal domains. (E) AlphaFold2 predicted the association of the amino acids 443–454 with the Tudor domain of PHF1. Amino acids forming the hydrophobic pocket are marked yellow. Structure obtained from the AlphaFold database of human PHF1 (O43189, version 2022-11-01) [31].
Figure 1.
(A) General domain structure of the three PCLs. (B) Amino acid homology between the three human PCLs. (C) Model of the functional role of the PCLs for recruiting PRC2. PCL interacts via the winged helix domain with unmethylated CpG islands and recruits PRC2 by interaction of the C-terminal chromo-like domain with the PRC2 core. The histone methyltransferase EZH2 facilitates H3K27me3 methylation. EED interacts with H3K27me3 and contributes to PRC2 recruitment, as well. (D) Predicted aligned error of human PHF1, MTF2, and PHF19 obtained from the AlphaFold database [31]. Green rectangles indicate predicted associations of the unstructured regions with the chromatin-binding internal domains. (E) AlphaFold2 predicted the association of the amino acids 443–454 with the Tudor domain of PHF1. Amino acids forming the hydrophobic pocket are marked yellow. Structure obtained from the AlphaFold database of human PHF1 (O43189, version 2022-11-01) [31].

3. Polycomb-like Proteins in PRC2 Chromatin Recruitment and Mammalian Gene Regulation
PCLs are crucial in targeting PRC2 to chromatin and, thus, important for gene repression (Figure 1C). Deleting all three PCL homologs substantially reduces the chromatin association of PRC2 [32,33], suggesting that PRC2 chromatin binding strongly depends on the PCLs.
The PRC2 recruitment function of the PCLs particularly relies on the DNA-binding ability of their winged helix (WH) domains (Figure 1C). The WH domain recognizes explicitly unmethylated CpG motifs via a positively charged region that reaches deeply into the major groove of the DNA [26]. The presence of methylated cytosine leads to a steric clash, preventing DNA binding [26]. This binding property is consistent with the common presence of PRC2 at unmethylated CpG islands in the genome [34]. Notably, the three PCLs proteins differ in their ability to interact with DNA. While MTF2 and PHF1 can strongly bind to DNA, the DNA binding function of PHF19 is more subtle [26]. Consistently, in mouse embryonic stem cells, the recruitment of PRC2 to CpG islands is particularly dependent on MTF2 [32], the most strongly expressed PCL protein in these cells. Consequently, mutation of the DNA-binding ability of MTF2 substantially reduces the PRC2 recruitment function of MTF2 in these cells [26]. It has also been proposed that the MTF2 DNA-binding function is linked to a specific helical confirmation of the DNA [35]. Thus, these findings suggest that the DNA-binding ability constitutes a major feature of the PCLs by which they are involved in recruiting PRC2 to its genomic targets.
The role of the other domains is less well understood. The Tudor domains of all three mammalian PCLs form a hydrophobic cage and have a high affinity for trimethylated lysine 36 of histone H3 (H3K36me3) [36,37,38,39]. PHF1 and PHF19 can also recognize the K27 trimethylation of the testis-specific histone variant H3t [40,41]. However, the binding strength of the Tudor domain to this modification is substantially lower than that for H3K36me3, suggesting that H3K36me3 is the preferred target of the Tudor domain. Nonetheless, the relevance of the H3K36me3 binding function of the Tudor domain remains controversial, given that H3K36me3 is a transcription elongation mark found in the gene body, and PRC2 typically does not colocalize with H3K36me3 in the genome. Thus, it is unclear whether this histone-binding ability is important for the chromatin recruitment of PRC2 or whether it may have another function related to H3K36me3. Given that H3K36me3 has been found to inhibit the enzymatic activity of PRC2 [42,43], the H3K36me3-binding function of the Tudor domain could potentially also play a role in inferring with the activity of PRC2 at active genes. In addition, the PHD finger 1 of PHF1 has been proposed to interact with symmetrically methylated arginine at position 3 of histone 4 (H4R3me2s) [44]. Whether the PHD fingers of PHF19 and MTF2 have merely a structural function or whether they are also directly involved in chromatin binding has not yet been determined.
To recruit PRC2 to its target genes, the PCL must tightly interact with the PRC2 core complex. This function is facilitated via the very C-terminal chromo-like domain of the PCLs, (Figure 1A), which is necessary and sufficient for interaction with PRC2 [21,27]. The C-terminal domain is sequence-wise and similar between MTF2 and PHF19, consists of one helix and two β sheets, and shares some similarities with the chromodomains of HP1 (heterochromatin-protein 1) [27]. The C-terminal domain of PHF1 lacks the α helix [27], which may suggest some alternative structural functionality. For PHF19 and MTF2, it has been shown that the domain directly associates with the N-terminal part of SUZ12 [45] and that this association enhances the establishment of a dimeric state of PRC2, which increases its affinity for CpG islands and chromatin [45].
Notably, the region between the chromatin-binding internal domain, comprising the Tudor, PHD fingers, and the winged helix domain and the C-terminal PRC2-interacting domain consists of a long unstructured stretch, which does not appear to establish any globular domain (Figure 1D). This region differs substantially among the three PCLs in their amino acid sequences and shows almost no homology (Figure 1B). Furthermore, AlphaFold2 [31,46] predicts that this unstructured region may contact the internal domain (Figure 1D), which could potentially be important for modulating the chromatin-binding function. For example, in the case of PHF1, but not for MTF2 and PHF19, this predicted association is close to the hydrophobic pocket of the Tudor domain (Figure 1E). This raises the possibility that the histone reader function of the Tudor domain could be influenced by such an association.
In summary, the PCLs link the PRC2.1 subcomplex to chromatin by interacting with the PRC2 core via the C-terminal chromo-like domain [27,45] and by interacting with DNA [26] and histones [27,37,38,39] via their internal domain (Figure 1C). However, the functions of the domains vary in detail, suggesting that PCLs may have nonredundant roles in regulating PRC2.
4. Polycomb-like Proteins in Human Cancer
An investigation of the role of the PCLs in cancer using publicly available databases suggests that the three PCLs have nonoverlapping roles in cancer (Figure 2). First, the expression changes in cancer versus normal tissue considerably differ between the proteins (Figure 2A). PHF1 is predominantly downregulated in cancer, while PHF19 is largely upregulated [28], implicating a potentially opposing role of these two proteins. Indeed, when looking at all cancer types, high PHF1 expression is associated with a better prognosis, while high PHF19 expression is typically linked to a worse outcome (Figure 2B). MTF2 expression is not strongly affected in most cancer types, and its expression does not correlate with patient survival (Figure 2A,B). Notably, however, the pattern of the gene expression changes, and the correlation with patient survival is variable and dependent on the individual cancer type and on the investigated PCL protein (Figure 2A,C). For example, a high PHF1 expression is linked to a worse prognosis in colon adenocarcinoma (COAD) but a better prognosis in pancreatic cancer. In addition, only a few cancer types, such as ovarian cancer (OV) and thymoma (THYM), show a similar pattern for all three PCLs (Figure 2A,C). In most other cancer types, the role of the PCLs is highly individual. In addition to changes in gene expression, all three PCL proteins are also commonly mutated in cancer (Figure 2D) [47]. Currently, the specific consequences of these mutations are largely unknown. Interestingly, for PHF1 and MTF2 frameshift or nonsense mutations, which lead to truncated proteins, are often observed. Especially the recurrent frameshift at the amino acid R6 of PHF1 (Figure 2D) suggests that abolishing the function of PHF1 is advantageous for cancer progression, consistent with commonly observed reduced gene expression of PHF1 (Figure 2A).
Thus, although these data globally support a tumor-suppressive role of PHF1, an oncogenic role of PHF19, and a relatively neutral role of MTF2, the three PCL proteins appear to have individual impacts dependent on the cancer type. In the following, we will address the known roles of PHF1, MTF2, and PHF19 in cancer and the possible mechanisms involved.
4.1. Tumor-Suppressive and Oncogenic Roles of PHF1
Public data suggest that PHF1 is typically downregulated in many cancer types (Figure 2A), and its low expression is often associated with a poorer prognosis (Figure 2B). In addition, in some cancer types, such as carcinomas [47], frameshift mutations are commonly observed (Figure 2D). This indicates a more tumor-suppressive role of PHF1. Indeed, several publications support this idea, and it has been shown that the tumor-suppressive function of PHF1 is linked to its ability to regulate p53-dependent pathways in cancer [51]. P53 is one of the most common tumor-suppressor genes in cancer, and inactivating mutations are often crucial for cancer development [52]. The interaction of PHF1 stabilizes p53 by preventing MDM2-dependent ubiquitination and proteasomal degradation [51]. Thus, the downregulation of PHF1 in cancer cells could be involved in reducing the protein level of p53 and thereby impairing its tumor-suppressor activity. Additional work suggests that the link between PHF1 and p53 depends on two unique serines in the N-terminal PHD finger of PHF1. These serines are not present in MTF2 or PHF19 [53], explaining why the interaction with p53 is restricted to PHF1. The PHF1–p53 axis is essential for inducing cellular quiescence and to prevent uncontrolled cell growth [53]. One mechanism that regulates PHF1 expression levels in cancer involves the RNA-binding protein FTO, which stabilizes the PHF1 mRNA [54]. PHF1 has also been shown to be involved in DNA repair mechanisms via interaction with the Ku70/Ku80 heterodimer [38,55]. Therefore, PHF1′s tumor-suppressive role may also be linked to its ability to suppress DNA damage and thereby prevent harmful chromosome rearrangement.
Although PHF1 appears to function mostly as a tumor suppressor, in the context of breast cancer cells, PHF1 has also been described as promoting cellular proliferation [44]. Here, PHF1 was found to interact not only with the PRC2 complex but also with the PRMT5/WDR77/CRL4B complex, which is involved in histone arginine methylation and functions as an H2AK119ub reader. Like the association with p53, this interaction appears to be specific for PHF1.
4.2. PHF1 in Gene Fusions
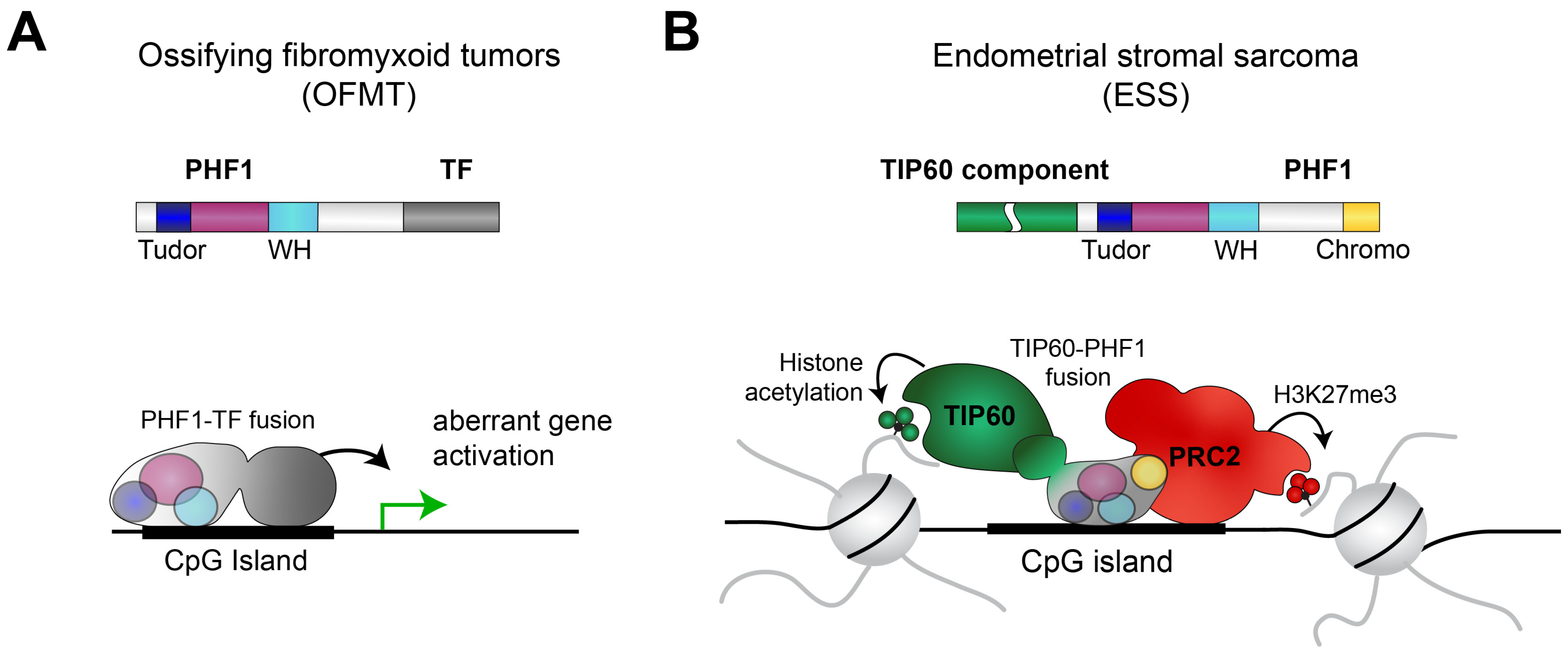
In addition to the role of PHF1 itself, PHF1 is also commonly involved in gene rearrangements in some cancer types. To date, eleven in-frame gene fusions have been described (Table 1, Figure 3). Most of these rearrangements were identified in ossifying fibromyxoid tumor (OFMT) or endometrial stromal sarcoma (ESS), both relatively rare cancer diseases.
OFMT is a soft-tissue tumor characterized by bone-like tissue formation within the tumor [57]. In approximately 50% percent of the OFMT cases, the PHF1 gene is rearranged [58], often with a transcription factor gene, such as FOXR1 [59], FOXR2 [59], or TFE3 [60]. In these cases, the N-terminal chromatin-binding region of PHF1 is translocated to the transcription factor in frame, resulting in a transcription factor that possesses an additional chromatin-binding function. These fusion proteins lack the C-terminal PRC2-interacting domain of PHF1, suggesting that these fusion proteins act in a PRC2-independent manner. One could speculate that the fusion protein can be recruited to places normally targeted by PHF1, such as CpG islands [26]. The displaced transcription factor recruitment to these locations may lead to the dysregulation of target genes relevant to the development of OFMT. This idea is supported by the observed gene expression changes upon ectopic expression of a PHF1-TFE3 fusion protein [61].
Figure 3.
(A) Common PHF1 rearrangements in OFMT consist of the N-terminal part of PHF1 and transcription factors (TF), such as FOXR1, FOXR2, and TFE3. Consequently, the resulting fusion protein may be aberrantly recruited to the wrong locations in the genome, leading to dysregulated gene expression. (B) In ESS, full-length PHF1 is recurrently fused to components of the TIP60 complex, such as TEAF1, JAZF1, EPC1, and EPC2 (see also Table 1). The resulting protein can interact with both the TIP60 and the PRC2 complexes [62], leading to aberrant chromatin states in cancer cells.
Figure 3.
(A) Common PHF1 rearrangements in OFMT consist of the N-terminal part of PHF1 and transcription factors (TF), such as FOXR1, FOXR2, and TFE3. Consequently, the resulting fusion protein may be aberrantly recruited to the wrong locations in the genome, leading to dysregulated gene expression. (B) In ESS, full-length PHF1 is recurrently fused to components of the TIP60 complex, such as TEAF1, JAZF1, EPC1, and EPC2 (see also Table 1). The resulting protein can interact with both the TIP60 and the PRC2 complexes [62], leading to aberrant chromatin states in cancer cells.

ESS is a relatively rare malignant tumor of the uterus, accounting for approximately 0.2% of all uterine malignancies [63]. In ESS, most known translocation partners of PHF1 are linked to the NuA6/TIP60 histone acetyltransferase complex [62]. This includes EPC1, EPC2, EP400, MEAF6, MBTD1, ING3, BRD8, and JAZF1. In most cases, the entire PHF1 is fused to one of the TIP60 components. The EPC1-PHF1 fusion protein has been demonstrated to assemble a supercomplex consisting of both the TIP60 complex and PRC2 [62]. Expression of the EPC1-PHF1 fusion proteins leads to dysregulation of gene expression, particularly of the PRC2 target genes, which become upregulated [62]. This finding can be explained by the observation that the EPC1-PHF1 fusion protein is recruited to PRC2 target genes and enhances the histone acetylation level, likely due to the activity of TIP60 [62]. Similar supercomplexes are likely also formed by the other PHF1 fusion proteins in ESS, as well as by other rearrangements, such as the JAZF1-SUZ12 translocation. These observations suggest that bringing together TIP60 and PRC2 via a gene fusion is a common event in ESS [62].

{kind=link}
{kind=link}
{kind=link}
Table 1.
Recurrent PHF1 gene rearrangements in cancer.
| Translocation | Cancer Type | Reference |
|---|---|---|
| PHF1-FOXR1 | ossifying fibromyxoid tumors | [59] |
| PHF1-FOXR2 | ossifying fibromyxoid tumors | [59] |
| PHF1-TFE3 | ossifying fibromyxoid tumors, malignant chondroid syringoma | [60,64,65] |
| MBTD1-PHF1 | endometrial stromal sarcoma | [66] |
| JAZF1-PHF1 | endometrial stromal sarcoma cardiac ossifying sarcoma | [67,68,69] |
| EPC1-PHF1 | endometrial stromal sarcoma ossifying fibromyxoid tumors | [62,67,70] |
| EPC2-PHF1 | endometrial stromal sarcoma | [71] |
| EP400-PHF1 | ossifying fibromyxoid tumors | [72,73] |
| MEAF6-PHF1 | ossifying fibromyxoid tumors endometrial stromal sarcoma | [70,72,74,75,76] |
| ING3-PHF1 | endometrial stromal sarcoma | [77] |
| BRD8-PHF1 | endometrial stromal sarcoma | [78,79] |
4.3. A Predominantly Tumor-Suppressive Role of MTF2 in Cancer
To date, the physiological function of MTF2 has been predominantly linked to its role in stem cell maintenance and differentiation [26,80,81] and during hematopoiesis [82]. The cancer-related role of MTF2 appears rather versatile and can both promote and inhibit tumor growth.
In acute myeloid leukemia, a reduced level of MTF2 has been associated with increased chemotherapy resistance [83], thus resulting in a worse prognosis. In AML cells, it has been demonstrated that MTF2, together with PRC2, inhibits the expression of MDM2, a key regulator of the tumor suppressor p53. Consequently, it has been proposed that either overexpression of MTF2 or inhibition of MDM2 sensitizes the leukemic cells to chemotherapeutics [83], offering a potential new strategy for treating AML patients. A similar link between low MTF2 expression and increased resistance to chemotherapeutics has been shown in basal-like breast cancer cells [84]. Here, reduced activity of PRC2 led to the derepression of Nfat1c, which is important for epithelial–mesenchymal transition (EMT) and resistance to cytotoxic treatments. Another study suggests that MTF2 may influence the MDM2-p53 axis in breast cancer cells, similar to AML cells [56]. Low expression of MTF2 triggered by miR-218-5p has also been linked to increased epithelial–mesenchymal transition, cell migration, and invasion in retinoblastoma [85]. Thus, most studies propose a more tumor-suppressive role of MTF2.
However, some studies also suggest an oncogenic role of MTF2. MTF2 was identified as a candidate gene in myeloma using Myc-transgenic mice in an unbiased genetic forward screen [86]. Indeed, a high expression of MTF2 is linked to a poorer patient prognosis in this cancer type, and a depletion of MTF2 leads to reduced proliferation of cells in vitro and in xenograft experiments [86], supporting a putative oncogenic role of MTF2 in this context. In hepatocellular carcinoma (HCC), the high expression of MTF2 has been linked to the activation of EMT-related pathways, whereby Snail was identified as an important downstream effector of MTF2 [87], further supporting that MTF2 can act as an oncogene in some cancer types.
4.4. The Oncogenic Role of PHF19
In contrast to PHF1 and MTF2, PHF19 has primarily been described as an oncogene in multiple cancer types [88]. Of note, in humans, but not in mice, PHF19 has a shorter isoform, which contains the Tudor domain and the first PHD finger but lacks the second PHD finger, the winged helix domain, and the PRC2-interacting chromo-like domain [89]. Consistently, this isoform does not interact with PRC2 [90], suggesting that this isoform has a distinct functionality from full-length PHF19. Currently, many studies regarding PHF19 in cancer do not fully distinguish between these two isoforms, making the conclusion about the role of PHF19 in cancer less defined.
Initially, PHF19 was identified as a gene similar to Drosophila PCL, which shows elevated expression levels in cancer [89]. This upregulation is particularly evident for the short isoform but also measurable for the long isoform. The first functional study in cancer about PHF19 was performed in melanoma cells, where depletion of PHF19 leads to reduced proliferation but enhances invasiveness [91], suggesting a nontrivial role of PHF19. A similar role in regulating invasiveness was subsequently found in mammary tumors [92].
To date, the most compelling role of PHF19 in cancer has been found in multiple myomas (MM) [93]. In this cancer type, PHF19 is required for setting up PRC2-dependent broad H3K27me3 domains, which is important for the optimal silencing of PRC2 target genes [94]. The expression of PHF19 in multiple myoma cells is negatively regulated by miRNA-15a [95]. The relevance of PHF19 in this cancer type is also supported by computational approaches [89,96], functional assays [94,97], and patient data [98].
In the context of prostate cancer, a study demonstrated that the depletion of the full-length PHF19 unexpectedly leads to increased chromatin binding of PRC2 [90]. An enhanced chromatin binding of MTF2 in the absence of PHF19 explains this finding [90]. Consequently, the deletion of PHF19 leads to the substantial deregulation of multiple pathways, including those involved in cell growth, metastasis, and blood vessel formation. Interestingly, the absence of PHF19 changes the properties of the cells to a less proliferative but, at the same time to a more aggressive phenotype, similar to the effect in melanomas [91]. Another study suggests that the short isoform of PHF19 is also important for the proliferation and migration of prostate cancer cells, suggesting a sophisticated role of PHF19 in this cancer type [99].
A study in hepatocellular carcinoma (HCC) suggests that a high expression of PHF19 promotes cell migration and invasiveness but also proliferation. The expression of PHF19 has here been linked to the expression of the tumor-suppressive miRNA hsa-miR-195-5p [100]. A similar link between an miRNA and a PHF19 has been proposed in ovarian carcinoma, where PHF19 expression is regulated by miR-211 [101].
In colorectal cancer, the PHF19 gene appears to possess an oncogenic super-enhancer [102], a key feature in gene regulation [103,104]. The super-enhancer at PHF19 likely drives a high expression of PHF19 in this cancer type, required for efficient tumor growth [102]. An oncogenic role of PHF19 has also been demonstrated in Ewing Sarcomas, where the PHF19 gene is downstream of the EWS/ETS fusion proteins that typically drive the oncogenic program in this cancer type [105]. Further studies suggest a role of PHF19 in other cancer types, such as ovarian and gastric cancer, for which PHF19 has mostly been described to function as an oncogene (Table 2).
PHF19 is relevant not only in the cancer cells themselves but also in cells involved in the fight against cancer. Namely, a high expression of PHF19, triggered by miR-155, in CD8+ T cells supported an antitumor response [106]. Notably, this function depends on the chromatin-binding ability of the Tudor domain of PHF19 [27,36,106], supporting the biological relevance of the chromatin-binding ability of the Tudor domain.
In summary, most studies on the role of PCL proteins in cancer refer to the oncogenic role of PHF19 in a variety of cancers and PHF1 gene translocations in endometrial stromal sarcoma and ossifying fibromyxoid tumors. Given that human PCL proteins have just emerged as interesting proteins in cancer, many more insights can be expected in the future.
Table 2.
Roles of the PCLs in cancer.
| Cancer Type | Expression Regulated by | Tumor Role | Reference |
|---|---|---|---|
| PHF1 | |||
| breast cancer | n.d. | mixed | [44,51] |
| lung adenocarcinoma | FTO | tumor-suppressor | [54] |
| MTF2 | |||
| acute myeloid leukemia | n.d. | tumor-suppressor | [83] |
| basal-like breast cancer | n.d. | tumor-suppressor | [84] |
| breast cancer | n.d. | tumor-suppressor | [56] |
| glioma | n.d. | oncogene | [107] |
| hepatocellular carcinoma | n.d. | oncogene | [87] |
| myeloma | n.d. | oncogene | [86] |
| retinoblastoma | n.d. | tumor suppressor | [85] |
| PHF19 | |||
| chronic myeloid leukemia | n.d. | oncogene | [108] |
| colorectal cancer | n.d. | oncogene | [102,109] |
| Ewing sarcoma | EWS/ETS fusion TFs | oncogene | [105] |
| gastric cancer | n.d. | oncogene | [110] |
| glioblastoma | n.d. | oncogene | [111,112] |
| glioma | miR-124a | oncogene | [113] |
| hepatocellular carcinoma | miR-195-5p | oncogene | [100,114] |
| mammary tumor | n.d. | mixed | [92] |
| melanoma | n.d. | mixed | [91] |
| multiple myeloma | miR-15a | oncogene | [93,94,95,96,97,98] |
| ovarian carcinoma | miR-211 | oncogene | [101,115] |
| prostate cancer | n.d. | mixed | [90,99] |
n.d. means not determined.
5. Conclusions
Polycomb-like proteins are key components of the Polycomb repressive complex 2 and are the defining factors for the PRC2.1 subcomplex. They are essential for targeting PRC2 to chromatin via the histone-binding Tudor [27,36,37] and the DNA-binding winged helix domains [26]. Although the three PCL proteins have a similar domain composition, their biological roles are not identical and differ in physiological and nonphysiological settings.
The difference in functionality can partly be explained by their distinct ability to interact with other proteins beyond PRC2. In particular, PHF1 has been described to interact with PRC2-unrelated proteins, including p53 [53] and Ku70/Ku80 [55]. This unique interaction of PHF1 allows it to participate in PRC2-independent biological pathways. PHF19 has been described to interact with the histone lysine demethylase RIOX1 (NO66) [36], while for MTF2, no PRC2-unrelated interacting partners have been described so far, suggesting that their functions are likely mostly linked to their chromatin function. Another reason for their distinct functionality is that the DNA-binding winged helix and the histone-binding Tudor domain differ in their binding strength [26,39]. Consequently, the ratio of the three PCL proteins in a specific cell may be critical to determine which PCL preferentially binds to the PRC2 core, which in turn influences the extent and where PRC2 is recruited. Beyond these known differences, the variation in the primary amino acid sequence of the PCLs likely contributes to other currently unknown differences in their functionality.
Thus, it is unsurprising that each of the three PCL proteins has a distinct role in cancer. While PHF1 has a predominantly tumor-suppressive role linked to its ability to interact with p53, PHF19 acts mainly as an oncogene (Table 2). Furthermore, PHF1 is recurrently translocated in two rare cancer types: ossifying fibromyxoid tumors and endometrial stromal sarcoma (Table 1). The highly specific occurrence of these translocations further emphasizes the context dependency of the PCLs in cancer.
Taken together, PCLs play complex and diverse roles in cancer, and their precise functions can vary depending on the specific type of cancer and the stage of the disease. Further research is needed to fully understand the mechanisms by which the PCLs contribute to cancer development and progression and to identify strategies to influence their functions in cancer.
Author Contributions
Conceptualization, S.F. and R.L.; Writing—Original Draft Preparation, R.L.; Writing—Review and Editing, S.F. and R.L.; Visualization, S.F. and R.L.; Supervision, R.L.; Project Administration, R.L; Funding Acquisition, R.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, 416910386, 516068166) and the Fritz Thyssen Foundation (10.20.1.005MN).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used in this article were obtained from the UniProt (www.uniprot.org (accessed on 19 March 2023)), GePIA (http://gepia.cancer-pku.cn/ (accessed on 19 March 2023)) and AlphaFold databases (https://alphafold.ebi.ac.uk/ (accessed on 19 March 2023)).
Acknowledgments
We thank Bastian Stielow for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Janssen, S.M.; Lorincz, M.C. Interplay between chromatin marks in development and disease. Nat. Rev. Genet. 2022, 23, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026575. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Wassef, M.; Margueron, R. The Multiple Facets of PRC2 Alterations in Cancers. J. Mol. Biol. 2017, 429, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Tsirigos, A.; van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Wassef, M.; Rodilla, V.; Teissandier, A.; Zeitouni, B.; Gruel, N.; Sadacca, B.; Irondelle, M.; Charruel, M.; Ducos, B.; Michaud, A.; et al. Impaired PRC2 activity promotes transcriptional instability and favors breast tumorigenesis. Genes Dev. 2015, 29, 2547–2562. [Google Scholar] [CrossRef]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.-W.G.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, Q.; Zi, J.; Ma, J.; Song, H.; Tian, Y.; McGrath, M.; Song, C.; Ge, Z. Mutations in EZH2 are associated with poor prognosis for patients with myeloid neoplasms. Genes Dis. 2019, 6, 276–281. [Google Scholar] [CrossRef]
- Bödör, C.; O’Riain, C.; Wrench, D.; Matthews, J.; Iyengar, S.; Tayyib, H.; Calaminici, M.; Clear, A.; Iqbal, S.; Quentmeier, H.; et al. EZH2 Y641 mutations in follicular lymphoma. Leukemia 2011, 25, 726–729. [Google Scholar] [CrossRef]
- Donaldson-Collier, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; de Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Chammas, P.; Mocavini, I.; Di Croce, L. Engaging chromatin: PRC2 structure meets function. Br. J. Cancer 2020, 122, 315–328. [Google Scholar] [CrossRef]
- Fischer, S.; Weber, L.M.; Liefke, R. Evolutionary adaptation of the Polycomb repressive complex 2. Epigenet. Chromatin 2022, 15, 7. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef]
- Grau, D.; Zhang, Y.; Lee, C.-H.; Valencia-Sánchez, M.; Zhang, J.; Wang, M.; Holder, M.; Svetlov, V.; Tan, D.; Nudler, E.; et al. Structures of monomeric and dimeric PRC2:EZH1 reveal flexible modules involved in chromatin compaction. Nat. Commun. 2021, 12, 714. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef]
- Chen, S.; Jiao, L.; Shubbar, M.; Yang, X.; Liu, X. Unique Structural Platforms of Suz12 Dictate Distinct Classes of PRC2 for Chromatin Binding. Mol. Cell 2018, 69, 840–852.e5. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Liu, X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015, 350, aac4383. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Drury, W.J.; Voigt, P.; Martin, S.R.; Taylor, W.R.; de Marco, V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016, 17, 583–595. [Google Scholar] [CrossRef]
- Tie, F.; Prasad-Sinha, J.; Birve, A.; Rasmuson-Lestander, A.; Harte, P.J. A 1-megadalton ESC/E(Z) complex from Drosophila that contains polycomblike and RPD3. Mol. Cell. Biol. 2003, 23, 3352–3362. [Google Scholar] [CrossRef]
- Li, H.; Liefke, R.; Jiang, J.; Kurland, J.V.; Tian, W.; Deng, P.; Zhang, W.; He, Q.; Patel, D.J.; Bulyk, M.L.; et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 2017, 549, 287–291. [Google Scholar] [CrossRef]
- Ballaré, C.; Lange, M.; Lapinaite, A.; Martin, G.M.; Morey, L.; Pascual, G.; Liefke, R.; Simon, B.; Shi, Y.; Gozani, O.; et al. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat. Struct. Mol. Biol. 2012, 19, 1257–1265. [Google Scholar] [CrossRef]
- Liefke, R.; Karwacki-Neisius, V.; Shi, Y. EPOP Interacts with Elongin BC and USP7 to Modulate the Chromatin Landscape. Mol. Cell 2016, 64, 659–672. [Google Scholar] [CrossRef]
- Beringer, M.; Pisano, P.; Di Carlo, V.; Blanco, E.; Chammas, P.; Vizán, P.; Gutiérrez, A.; Aranda, S.; Payer, B.; Wierer, M.; et al. EPOP Functionally Links Elongin and Polycomb in Pluripotent Stem Cells. Mol. Cell 2016, 64, 645–658. [Google Scholar] [CrossRef]
- Conway, E.; Jerman, E.; Healy, E.; Ito, S.; Holoch, D.; Oliviero, G.; Deevy, O.; Glancy, E.; Fitzpatrick, D.J.; Mucha, M.; et al. A Family of Vertebrate-Specific Polycombs Encoded by the LCOR/LCORL Genes Balance PRC2 Subtype Activities. Mol. Cell 2018, 70, 408–421.e8. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Højfeldt, J.W.; Hedehus, L.; Laugesen, A.; Tatar, T.; Wiehle, L.; Helin, K. Non-core Subunits of the PRC2 Complex Are Collectively Required for Its Target-Site Specificity. Mol. Cell 2019, 76, 423–436.e3. [Google Scholar] [CrossRef]
- Healy, E.; Mucha, M.; Glancy, E.; Fitzpatrick, D.J.; Conway, E.; Neikes, H.K.; Monger, C.; van Mierlo, G.; Baltissen, M.P.; Koseki, Y.; et al. PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol. Cell 2019, 76, 437–452.e6. [Google Scholar] [CrossRef]
- Mendenhall, E.M.; Koche, R.P.; Truong, T.; Zhou, V.W.; Issac, B.; Chi, A.S.; Ku, M.; Bernstein, B.E. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010, 6, e1001244. [Google Scholar] [CrossRef]
- Perino, M.; van Mierlo, G.; Karemaker, I.D.; van Genesen, S.; Vermeulen, M.; Marks, H.; van Heeringen, S.J.; Veenstra, G.J.C. MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat. Genet. 2018, 50, 1002–1010. [Google Scholar] [CrossRef]
- Brien, G.L.; Gambero, G.; O’Connell, D.J.; Jerman, E.; Turner, S.A.; Egan, C.M.; Dunne, E.J.; Jurgens, M.C.; Wynne, K.; Piao, L.; et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 2012, 19, 1273–1281. [Google Scholar] [CrossRef]
- Cai, L.; Rothbart, S.B.; Lu, R.; Xu, B.; Chen, W.-Y.; Tripathy, A.; Rockowitz, S.; Zheng, D.; Patel, D.J.; Allis, C.D.; et al. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 2013, 49, 571–582. [Google Scholar] [CrossRef]
- Musselman, C.A.; Avvakumov, N.; Watanabe, R.; Abraham, C.G.; Lalonde, M.-E.; Hong, Z.; Allen, C.; Roy, S.; Nuñez, J.K.; Nickoloff, J.; et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat. Struct. Mol. Biol. 2012, 19, 1266–1272. [Google Scholar] [CrossRef]
- Qin, S.; Guo, Y.; Xu, C.; Bian, C.; Fu, M.; Gong, S.; Min, J. Tudor domains of the PRC2 components PHF1 and PHF19 selectively bind to histone H3K36me3. Biochem. Biophys. Res. Commun. 2013, 430, 547–553. [Google Scholar] [CrossRef]
- Dong, C.; Nakagawa, R.; Oyama, K.; Yamamoto, Y.; Zhang, W.; Dong, A.; Li, Y.; Yoshimura, Y.; Kamiya, H.; Nakayama, J.-I.; et al. Structural basis for histone variant H3tK27me3 recognition by PHF1 and PHF19. Elife 2020, 9, e58675. [Google Scholar] [CrossRef]
- Kycia, I.; Kudithipudi, S.; Tamas, R.; Kungulovski, G.; Dhayalan, A.; Jeltsch, A. The Tudor domain of the PHD finger protein 1 is a dual reader of lysine trimethylation at lysine 36 of histone H3 and lysine 27 of histone variant H3t. J. Mol. Biol. 2014, 426, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Finogenova, K.; Bonnet, J.; Poepsel, S.; Schäfer, I.B.; Finkl, K.; Schmid, K.; Litz, C.; Strauss, M.; Benda, C.; Müller, J. Structural basis for PRC2 decoding of active histone methylation marks H3K36me2/3. Elife 2020, 9, e61964. [Google Scholar] [CrossRef]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stützer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S.; et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 2011, 42, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, J.; Yang, Y.; Qiu, R.; Zheng, Y.; Huang, W.; Zeng, Y.; Hou, Y.; Wang, S.; Leng, S.; et al. PHD finger protein 1 (PHF1) is a novel reader for histone H4R3 symmetric dimethylation and coordinates with PRMT5-WDR77/CRL4B complex to promote tumorigenesis. Nucleic Acids Res. 2018, 46, 6608–6626. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Jiao, L.; Liu, X.; Yang, X.; Liu, X. A Dimeric Structural Scaffold for PRC2-PCL Targeting to CpG Island Chromatin. Mol. Cell 2020, 77, 1265–1278.e7. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, C.; Zhang, P.; Gao, K.; Wang, D.; Yu, H.; Zhang, T.; Jiang, S.; Hexige, S.; Hong, Z.; et al. Polycomb group protein PHF1 regulates p53-dependent cell growth arrest and apoptosis. J. Biol. Chem. 2013, 288, 529–539. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Brien, G.L.; Healy, E.; Jerman, E.; Conway, E.; Fadda, E.; O’Donovan, D.; Krivtsov, A.V.; Rice, A.M.; Kearney, C.J.; Flaus, A.; et al. A chromatin-independent role of Polycomb-like 1 to stabilize p53 and promote cellular quiescence. Genes Dev. 2015, 29, 2231–2243. [Google Scholar] [CrossRef]
- Ning, J.; Wang, F.; Bu, J.; Zhu, K.; Liu, W. Down-regulated m6A reader FTO destabilizes PHF1 that triggers enhanced stemness capacity and tumor progression in lung adenocarcinoma. Cell Death Discov. 2022, 8, 354. [Google Scholar] [CrossRef]
- Hong, Z.; Jiang, J.; Lan, L.; Nakajima, S.; Kanno, S.; Koseki, H.; Yasui, A. A polycomb group protein, PHF1, is involved in the response to DNA double-strand breaks in human cell. Nucleic Acids Res. 2008, 36, 2939–2947. [Google Scholar] [CrossRef]
- Liang, Y.; Yang, Y.; Guo, R.; Gao, S.; Guo, X.; Li, D.; Wang, M.; Koseki, H.; Li, X. PCL2 regulates p53 stability and functions as a tumor suppressor in breast cancer. Sci. Bull. 2018, 63, 629–639. [Google Scholar] [CrossRef]
- Miettinen, M.; Finnell, V.; Fetsch, J.F. Ossifying fibromyxoid tumor of soft parts—A clinicopathologic and immunohistochemical study of 104 cases with long-term follow-up and a critical review of the literature. Am. J. Surg. Pathol. 2008, 32, 996–1005. [Google Scholar] [CrossRef]
- Graham, R.P.; Weiss, S.W.; Sukov, W.R.; Goldblum, J.R.; Billings, S.D.; Dotlic, S.; Folpe, A.L. PHF1 rearrangements in ossifying fibromyxoid tumors of soft parts: A fluorescence in situ hybridization study of 41 cases with emphasis on the malignant variant. Am. J. Surg. Pathol. 2013, 37, 1751–1755. [Google Scholar] [CrossRef]
- Srivastava, P.; Zilla, M.L.; Naous, R.; Marker, D.; Khoshnoodi, P.; Burgess, M.; Herradura, A.; Wu, J.; Surrey, L.F.; John, I. Expanding the molecular signatures of malignant ossifying fibromyxoid tumours with two novel gene fusions: PHF1:FOXR1 and PHF1:FOXR2. Histopathology 2023, 82, 946–952. [Google Scholar] [CrossRef]
- Suurmeijer, A.J.H.; Song, W.; Sung, Y.-S.; Zhang, L.; Swanson, D.; Fletcher, C.D.M.; Dickson, B.C.; Antonescu, C.R. Novel recurrent PHF1-TFE3 fusions in ossifying fibromyxoid tumors. Genes Chromosomes Cancer 2019, 58, 643–649. [Google Scholar] [CrossRef]
- Hofvander, J.; Jo, V.Y.; Fletcher, C.D.M.; Puls, F.; Flucke, U.; Nilsson, J.; Magnusson, L.; Mertens, F. PHF1 fusions cause distinct gene expression and chromatin accessibility profiles in ossifying fibromyxoid tumors and mesenchymal cells. Mod. Pathol. 2020, 33, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Sudarshan, D.; Avvakumov, N.; Lalonde, M.-E.; Alerasool, N.; Joly-Beauparlant, C.; Jacquet, K.; Mameri, A.; Lambert, J.-P.; Rousseau, J.; Lachance, C.; et al. Recurrent chromosomal translocations in sarcomas create a megacomplex that mislocalizes NuA4/TIP60 to Polycomb target loci. Genes Dev. 2022, 36, 664–683. [Google Scholar] [CrossRef] [PubMed]
- Puliyath, G.; Nair, M.K. Endometrial stromal sarcoma: A review of the literature. Indian J. Med. Paediatr. Oncol. 2012, 33, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Gorunova, L.; Lund-Iversen, M.; Bassarova, A.; Heim, S. Fusion of the Genes PHF1 and TFE3 in Malignant Chondroid Syringoma. Cancer Genom. Proteom. 2019, 16, 345–351. [Google Scholar] [CrossRef]
- Zou, C.; Ru, G.-Q.; Zhao, M. A PHF1-TFE3 fusion atypical ossifying fibromyxoid tumor with prominent collagenous rosettes: Case report with a brief review. Exp. Mol. Pathol. 2021, 123, 104686. [Google Scholar] [CrossRef]
- Han, L.; Liu, Y.J.; Ricciotti, R.W.; Mantilla, J.G. A novel MBTD1-PHF1 gene fusion in endometrial stromal sarcoma: A case report and literature review. Genes Chromosomes Cancer 2020, 59, 428–432. [Google Scholar] [CrossRef]
- Micci, F.; Panagopoulos, I.; Bjerkehagen, B.; Heim, S. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res. 2006, 66, 107–112. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Mertens, F.; Griffin, C.A. An endometrial stromal sarcoma cell line with the JAZF1/PHF1 chimera. Cancer Genet. Cytogenet. 2008, 185, 74–77. [Google Scholar] [CrossRef]
- Schoolmeester, J.K.; Sukov, W.R.; Maleszewski, J.J.; Bedroske, P.P.; Folpe, A.L.; Hodge, J.C. JAZF1 rearrangement in a mesenchymal tumor of nonendometrial stromal origin: Report of an unusual ossifying sarcoma of the heart demonstrating JAZF1/PHF1 fusion. Am. J. Surg. Pathol. 2013, 37, 938–942. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Sung, Y.-S.; Chen, C.-L.; Zhang, L.; Chen, H.-W.; Singer, S.; Agaram, N.P.; Sboner, A.; Fletcher, C.D. Novel ZC3H7B-BCOR, MEAF6-PHF1, and EPC1-PHF1 fusions in ossifying fibromyxoid tumors—Molecular characterization shows genetic overlap with endometrial stromal sarcoma. Genes Chromosomes Cancer 2014, 53, 183–193. [Google Scholar] [CrossRef]
- Brunetti, M.; Gorunova, L.; Davidson, B.; Heim, S.; Panagopoulos, I.; Micci, F. Identification of an EPC2-PHF1 fusion transcript in low-grade endometrial stromal sarcoma. Oncotarget 2018, 9, 19203–19208. [Google Scholar] [CrossRef]
- Gebre-Medhin, S.; Nord, K.H.; Möller, E.; Mandahl, N.; Magnusson, L.; Nilsson, J.; Jo, V.Y.; Vult von Steyern, F.; Brosjö, O.; Larsson, O.; et al. Recurrent rearrangement of the PHF1 gene in ossifying fibromyxoid tumors. Am. J. Pathol. 2012, 181, 1069–1077. [Google Scholar] [CrossRef]
- Endo, M.; Kohashi, K.; Yamamoto, H.; Ishii, T.; Yoshida, T.; Matsunobu, T.; Iwamoto, Y.; Oda, Y. Ossifying fibromyxoid tumor presenting EP400-PHF1 fusion gene. Hum. Pathol. 2013, 44, 2603–2608. [Google Scholar] [CrossRef]
- Micci, F.; Gorunova, L.; Gatius, S.; Matias-Guiu, X.; Davidson, B.; Heim, S.; Panagopoulos, I. MEAF6/PHF1 is a recurrent gene fusion in endometrial stromal sarcoma. Cancer Lett. 2014, 347, 75–78. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Micci, F.; Thorsen, J.; Gorunova, L.; Eibak, A.M.; Bjerkehagen, B.; Davidson, B.; Heim, S. Novel fusion of MYST/Esa1-associated factor 6 and PHF1 in endometrial stromal sarcoma. PLoS ONE 2012, 7, e39354. [Google Scholar] [CrossRef]
- Nomura, Y.; Tamura, D.; Horie, M.; Sato, M.; Sasaki, S.; Yamamoto, Y.; Kudo-Asabe, Y.; Umakoshi, M.; Koyama, K.; Makino, K.; et al. Detection of MEAF6-PHF1 translocation in an endometrial stromal nodule. Genes Chromosomes Cancer 2020, 59, 702–708. [Google Scholar] [CrossRef]
- Dermawan, J.K.; Dashti, N.; Chiang, S.; Turashvili, G.; Dickson, B.C.; Ellenson, L.H.; Kirchner, M.; Stenzinger, A.; Mechtersheimer, G.; Agaimy, A.; et al. Expanding the molecular spectrum of gene fusions in endometrial stromal sarcoma: Novel subunits of the chromatin remodeling complexes PRC2 and NuA4/TIP60 as alternative fusion partners. Genes Chromosomes Cancer 2023, 62, 152–160. [Google Scholar] [CrossRef]
- Micci, F.; Brunetti, M.; Dal Cin, P.; Nucci, M.R.; Gorunova, L.; Heim, S.; Panagopoulos, I. Fusion of the genes BRD8 and PHF1 in endometrial stromal sarcoma. Genes Chromosomes Cancer 2017, 56, 841–845. [Google Scholar] [CrossRef]
- Chen, P.C.-H.; Huang, H.-Y.; Chung, M.-Y.; Pan, C.-C. Ossifying low grade endometrial stromal sarcoma with PHF1-BRD8 fusion. Cancer Genet. 2021, 256–257, 81–85. [Google Scholar] [CrossRef]
- Loh, C.H.; van Genesen, S.; Perino, M.; Bark, M.R.; Veenstra, G.J.C. Loss of PRC2 subunits primes lineage choice during exit of pluripotency. Nat. Commun. 2021, 12, 6985. [Google Scholar] [CrossRef]
- Petracovici, A.; Bonasio, R. Distinct PRC2 subunits regulate maintenance and establishment of Polycomb repression during differentiation. Mol. Cell 2021, 81, 2625–2639.e5. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.L.M.; Maganti, H.B.; Jrade, H.; Porter, C.J.; Palidwor, G.A.; Cafariello, C.; Battaion, H.L.; Khan, S.T.; Perkins, T.J.; Paulson, R.F.; et al. Mtf2-PRC2 control of canonical Wnt signaling is required for definitive erythropoiesis. Cell Discov. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Maganti, H.B.; Jrade, H.; Cafariello, C.; Manias Rothberg, J.L.; Porter, C.J.; Yockell-Lelièvre, J.; Battaion, H.L.; Khan, S.T.; Howard, J.P.; Li, Y.; et al. Targeting the MTF2-MDM2 Axis Sensitizes Refractory Acute Myeloid Leukemia to Chemotherapy. Cancer Discov. 2018, 8, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Mieczkowska, I.K.; Pantelaiou-Prokaki, G.; Prokakis, E.; Schmidt, G.E.; Müller-Kirschbaum, L.C.; Werner, M.; Sen, M.; Velychko, T.; Jannasch, K.; Dullin, C.; et al. Decreased PRC2 activity supports the survival of basal-like breast cancer cells to cytotoxic treatments. Cell Death Dis. 2021, 12, 1118. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, Y.; Hu, Y.; Zhong, J.; Jiang, C.; Zhang, H. LncRNA CCAT1 sponges miR-218-5p to promote EMT, cellular migration and invasion of retinoblastoma by targeting MTF2. Cell. Signal. 2021, 86, 110088. [Google Scholar] [CrossRef]
- Sun, F.; Cheng, Y.; Riordan, J.D.; Dupuy, A.; Dubois, W.; Pisano, M.; Dong, J.; Mock, B.; Zhan, F.; Hari, P.; et al. WDR26 and MTF2 are therapeutic targets in multiple myeloma. J. Hematol. Oncol. 2021, 14, 203. [Google Scholar] [CrossRef]
- Wu, T.-T.; Cai, J.; Tian, Y.-H.; Chen, J.-F.; Cheng, Z.-L.; Pu, C.-S.; Shi, W.-Z.; Suo, X.-P.; Wu, X.-J.; Dou, X.-W.; et al. MTF2 Induces Epithelial-Mesenchymal Transition and Progression of Hepatocellular Carcinoma by Transcriptionally Activating Snail. Onco. Targets. Ther. 2019, 12, 11207–11220. [Google Scholar] [CrossRef]
- Zhu, Z.; Tang, N.; Wang, M.; Zhou, J.; Wang, J.; Ren, H.; Shi, X. Comprehensive Pan-Cancer Genomic Analysis Reveals PHF19 as a Carcinogenic Indicator Related to Immune Infiltration and Prognosis of Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 781087. [Google Scholar] [CrossRef]
- Wang, S.; Robertson, G.P.; Zhu, J. A novel human homologue of Drosophila polycomblike gene is up-regulated in multiple cancers. Gene 2004, 343, 69–78. [Google Scholar] [CrossRef]
- Jain, P.; Ballare, C.; Blanco, E.; Vizan, P.; Di Croce, L. PHF19 mediated regulation of proliferation and invasiveness in prostate cancer cells. Elife 2020, 9, e51373. [Google Scholar] [CrossRef]
- Ghislin, S.; Deshayes, F.; Middendorp, S.; Boggetto, N.; Alcaide-Loridan, C. PHF19 and Akt control the switch between proliferative and invasive states in melanoma. Cell Cycle 2012, 11, 1634–1645. [Google Scholar] [CrossRef]
- Callahan, R.; Mudunur, U.; Bargo, S.; Raafat, A.; McCurdy, D.; Boulanger, C.; Lowther, W.; Stephens, R.; Luke, B.T.; Stewart, C.; et al. Genes affected by mouse mammary tumor virus (MMTV) proviral insertions in mouse mammary tumors are deregulated or mutated in primary human mammary tumors. Oncotarget 2012, 3, 1320–1334. [Google Scholar] [CrossRef]
- Ghamlouch, H.; Boyle, E.M.; Blaney, P.; Wang, Y.; Choi, J.; Williams, L.; Bauer, M.; Auclair, D.; Bruno, B.; Walker, B.A.; et al. Insights into high-risk multiple myeloma from an analysis of the role of PHF19 in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 380. [Google Scholar] [CrossRef]
- Ren, Z.; Ahn, J.H.; Liu, H.; Tsai, Y.-H.; Bhanu, N.V.; Koss, B.; Allison, D.F.; Ma, A.; Storey, A.J.; Wang, P.; et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 2019, 134, 1176–1189. [Google Scholar] [CrossRef]
- Yu, T.; Du, C.; Ma, X.; Sui, W.; Yu, Z.; Liu, L.; Zhao, L.; Li, Z.; Xu, J.; Wei, X.; et al. Polycomb-like Protein 3 Induces Proliferation and Drug Resistance in Multiple Myeloma and Is Regulated by miRNA-15a. Mol. Cancer Res. 2020, 18, 1063–1073. [Google Scholar] [CrossRef]
- Mason, M.J.; Schinke, C.; Eng, C.L.P.; Towfic, F.; Gruber, F.; Dervan, A.; White, B.S.; Pratapa, A.; Guan, Y.; Chen, H.; et al. Multiple Myeloma DREAM Challenge reveals epigenetic regulator PHF19 as marker of aggressive disease. Leukemia 2020, 34, 1866–1874. [Google Scholar] [CrossRef]
- Schinke, C.D.; Bird, J.T.; Qu, P.; Yaccoby, S.; Lyzogubov, V.V.; Shelton, R.; Ling, W.; Boyle, E.M.; Deshpande, S.; Byrum, S.D.; et al. PHF19 inhibition as a therapeutic target in multiple myeloma. Curr. Res. Transl. Med. 2021, 69, 103290. [Google Scholar] [CrossRef]
- Li, Y.; Gong, J.; Zhang, L. PHD finger protein 19 expression in multiple myeloma: Association with clinical features, induction therapy outcome, disease progression, and survival. J. Clin. Lab. Anal. 2021, 35, e23910. [Google Scholar] [CrossRef]
- Abdelfettah, S.; Boulay, G.; Dubuissez, M.; Spruyt, N.; Garcia, S.P.; Rengarajan, S.; Loison, I.; Leroy, X.; Rivera, M.N.; Leprince, D. hPCL3S promotes proliferation and migration of androgen-independent prostate cancer cells. Oncotarget 2020, 11, 1051–1074. [Google Scholar] [CrossRef]
- Xu, H.; Hu, Y.-W.; Zhao, J.-Y.; Hu, X.-M.; Li, S.-F.; Wang, Y.-C.; Gao, J.-J.; Sha, Y.-H.; Kang, C.-M.; Lin, L.; et al. MicroRNA-195-5p acts as an anti-oncogene by targeting PHF19 in hepatocellular carcinoma. Oncol. Rep. 2015, 34, 175–182. [Google Scholar] [CrossRef]
- Tao, F.; Tian, X.; Ruan, S.; Shen, M.; Zhang, Z. miR-211 sponges lncRNA MALAT1 to suppress tumor growth and progression through inhibiting PHF19 in ovarian carcinoma. FASEB J. 2018, 32, 6330–6343. [Google Scholar] [CrossRef]
- Li, Q.-L.; Lin, X.; Yu, Y.-L.; Chen, L.; Hu, Q.-X.; Chen, M.; Cao, N.; Zhao, C.; Wang, C.-Y.; Huang, C.-W.; et al. Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers. Nat. Commun. 2021, 12, 6407. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Liefke, R.; Bokelmann, K.; Ghadimi, B.M.; Dango, S. Enhancer-driven transcriptional regulation is a potential key determinant for human visceral and subcutaneous adipocytes. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Gollavilli, P.N.; Pawar, A.; Wilder-Romans, K.; Natesan, R.; Engelke, C.G.; Dommeti, V.L.; Krishnamurthy, P.M.; Nallasivam, A.; Apel, I.J.; Xu, T.; et al. EWS/ETS-Driven Ewing Sarcoma Requires BET Bromodomain Proteins. Cancer Res. 2018, 78, 4760–4773. [Google Scholar] [CrossRef]
- Ji, Y.; Fioravanti, J.; Zhu, W.; Wang, H.; Wu, T.; Hu, J.; Lacey, N.E.; Gautam, S.; Le Gall, J.B.; Yang, X.; et al. miR-155 harnesses Phf19 to potentiate cancer immunotherapy through epigenetic reprogramming of CD8+ T cell fate. Nat. Commun. 2019, 10, 2157. [Google Scholar] [CrossRef]
- Wang, F.; Gao, Y.; Lv, Y.; Wu, Y.; Guo, Y.; Du, F.; Wang, S.; Yu, J.; Cao, X.; Li, P.A. Polycomb-like 2 regulates PRC2 components to affect proliferation in glioma cells. J. Neurooncol. 2020, 148, 259–271. [Google Scholar] [CrossRef]
- García-Montolio, M.; Ballaré, C.; Blanco, E.; Gutiérrez, A.; Aranda, S.; Gómez, A.; Kok, C.H.; Yeung, D.T.; Hughes, T.P.; Vizán, P.; et al. Polycomb Factor PHF19 Controls Cell Growth and Differentiation Toward Erythroid Pathway in Chronic Myeloid Leukemia Cells. Front. Cell Dev. Biol. 2021, 9, 655201. [Google Scholar] [CrossRef]
- Li, P.; Sun, J.; Ruan, Y.; Song, L. High PHD Finger Protein 19 (PHF19) expression predicts poor prognosis in colorectal cancer: A retrospective study. PeerJ 2021, 9, e11551. [Google Scholar] [CrossRef]
- Wang, H.; Xu, P.; Sun, G.; Lv, J.; Cao, J.; Xu, Z. Downregulation of PHF19 inhibits cell growth and migration in gastric cancer. Scand. J. Gastroenterol. 2020, 55, 687–693. [Google Scholar] [CrossRef]
- Deng, Q.; Hou, J.; Feng, L.; Lv, A.; Ke, X.; Liang, H.; Wang, F.; Zhang, K.; Chen, K.; Cui, H. PHF19 promotes the proliferation, migration, and chemosensitivity of glioblastoma to doxorubicin through modulation of the SIAH1/β-catenin axis. Cell Death Dis. 2018, 9, 1049. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Warden, C.; Zou, Z.; Neman, J.; Krueger, J.S.; Jain, A.; Jandial, R.; Chen, M. Altered expression of polycomb group genes in glioblastoma multiforme. PLoS ONE 2013, 8, e80970. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ji, H.; Tang, H.; Xu, Z. microRNA-124a suppresses PHF19 over-expression, EZH2 hyper-activation, and aberrant cell proliferation in human glioma. Biochem. Biophys. Res. Commun. 2018, 503, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Xiaoyun, S.; Yuyuan, Z.; Jie, X.; Yingjie, N.; Qing, X.; Yuezhen, D.; Haiguang, X. PHF19 activates hedgehog signaling and promotes tumorigenesis in hepatocellular carcinoma. Exp. Cell Res. 2021, 406, 112690. [Google Scholar] [CrossRef]
- Ruan, S.; Zhang, H.; Tian, X.; Zhang, Z.; Huang, H.; Shi, C.; Liu, W.; Jiang, X.; Huang, D.; Tao, F. PHD Finger Protein 19 Enhances the Resistance of Ovarian Cancer Cells to Compound Fuling Granule by Protecting Cell Growth, Invasion, Migration, and Stemness. Front. Pharmacol. 2020, 11, 150. [Google Scholar] [CrossRef]
Figure 2.
(A) Comparison of gene expression of the PCL proteins in normal (N) and tumor (T) tissue. Red and blue circles indicate significantly increased or decreased gene expression in tumor versus normal samples, respectively. (B) Kaplan–Meier plot of pancancer patient survival (overall survival) dependent on the expression of PHF1, MTF2, and PHF19. The data were split based on median expression. (C) Survival plots showing the hazard ratio (HR) for the three PCLs for individual cancer types. RFS = relapse free survival and OS = overall survival. Cancers with rectangles indicate a significant difference in survival between low and high expression. Data from TCGA [48] and visualized by GePIA [49]. (D) Mutations of the three PCL proteins, based on the COSMIC database [47]. Visualized using ProteinPaint [50]. * = mutation to a stop codon, fs = frameshift mutation. The pie charts show the relative distribution of missense, nonsense, and frameshift mutations for each PCL protein.
Figure 2.
(A) Comparison of gene expression of the PCL proteins in normal (N) and tumor (T) tissue. Red and blue circles indicate significantly increased or decreased gene expression in tumor versus normal samples, respectively. (B) Kaplan–Meier plot of pancancer patient survival (overall survival) dependent on the expression of PHF1, MTF2, and PHF19. The data were split based on median expression. (C) Survival plots showing the hazard ratio (HR) for the three PCLs for individual cancer types. RFS = relapse free survival and OS = overall survival. Cancers with rectangles indicate a significant difference in survival between low and high expression. Data from TCGA [48] and visualized by GePIA [49]. (D) Mutations of the three PCL proteins, based on the COSMIC database [47]. Visualized using ProteinPaint [50]. * = mutation to a stop codon, fs = frameshift mutation. The pie charts show the relative distribution of missense, nonsense, and frameshift mutations for each PCL protein.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fischer, S.; Liefke, R. Polycomb-like Proteins in Gene Regulation and Cancer. Genes 2023, 14, 938. https://doi.org/10.3390/genes14040938
AMA Style
Fischer S, Liefke R. Polycomb-like Proteins in Gene Regulation and Cancer. Genes. 2023; 14(4):938. https://doi.org/10.3390/genes14040938
Chicago/Turabian StyleFischer, Sabrina, and Robert Liefke. 2023. "Polycomb-like Proteins in Gene Regulation and Cancer" Genes 14, no. 4: 938. https://doi.org/10.3390/genes14040938
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.