
Complete Mitochondrial Genome of Scolytoplatypodini Species (Coleoptera: Curculionidae: Scolytinae) and Phylogenetic Implications

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitogenome Sequencing and Assembly
2.3. Sequence Annotation and Analyses
2.4. Phylogenetic Analysis
3. Results and Discussion
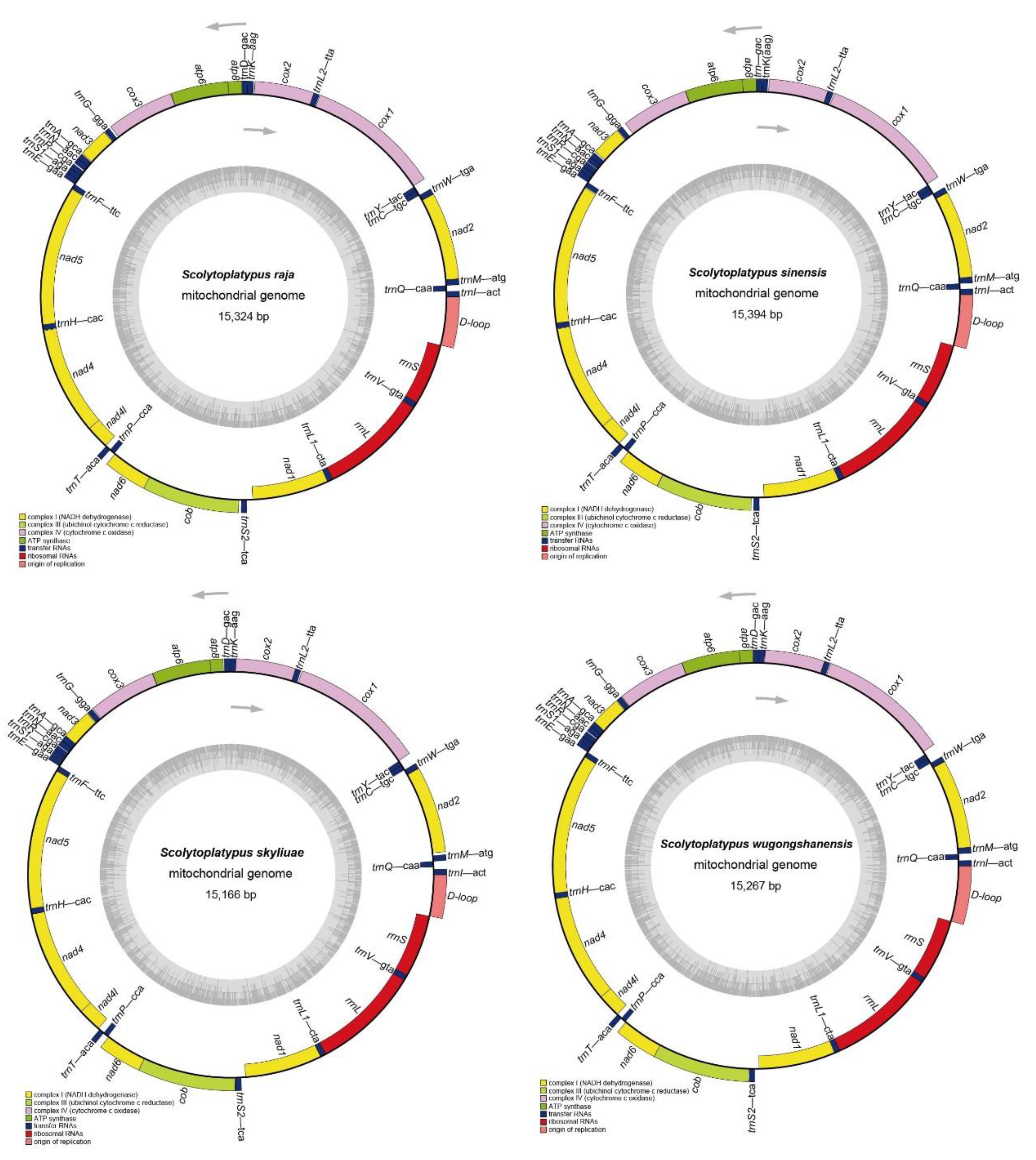
3.1. Mitogenome Organization and Nucleotide Composition
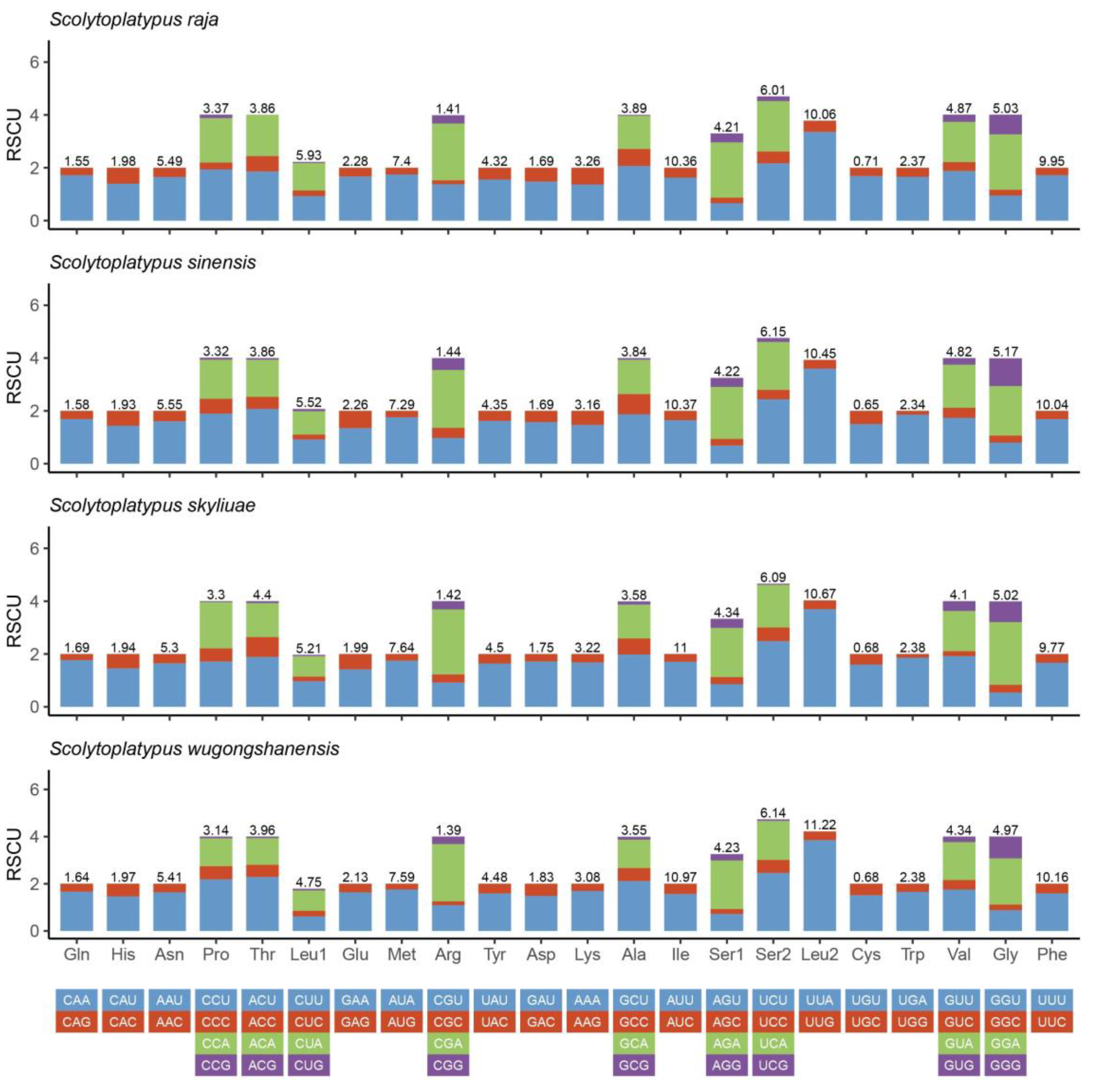
3.2. Protein-Coding Genes
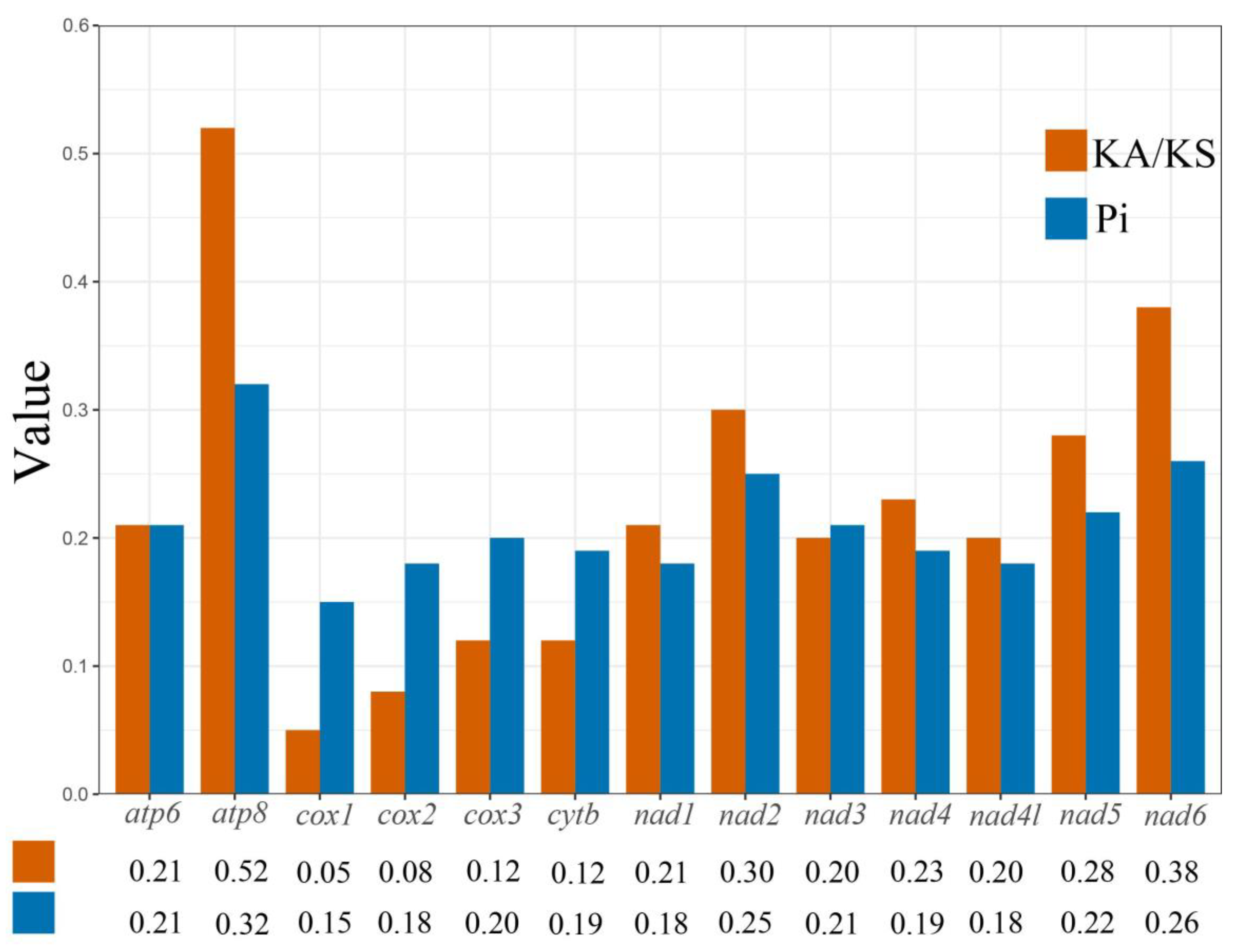
3.3. Nucleotide Diversity (Pi) and Nonsynonymous (Ka)/Synonymous (Ks) Mutation Rate Ratios
3.4. Gene Overlaps and Intergenic Spacers
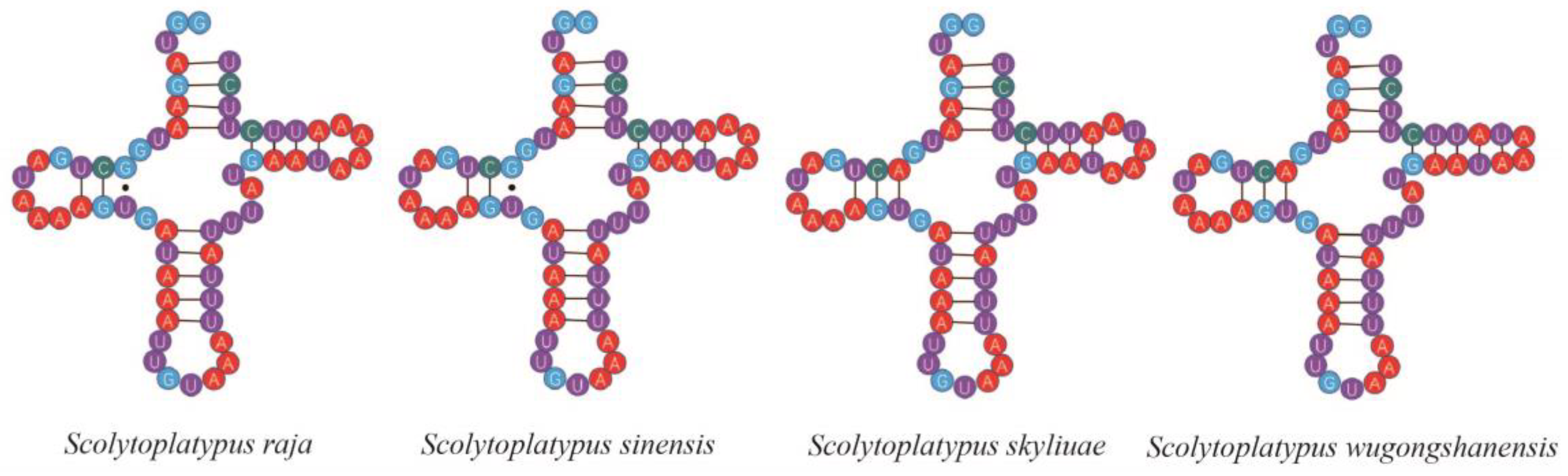
3.5. Transfer RNA, Ribosomal RNA Genes, and Non-Coding Regions
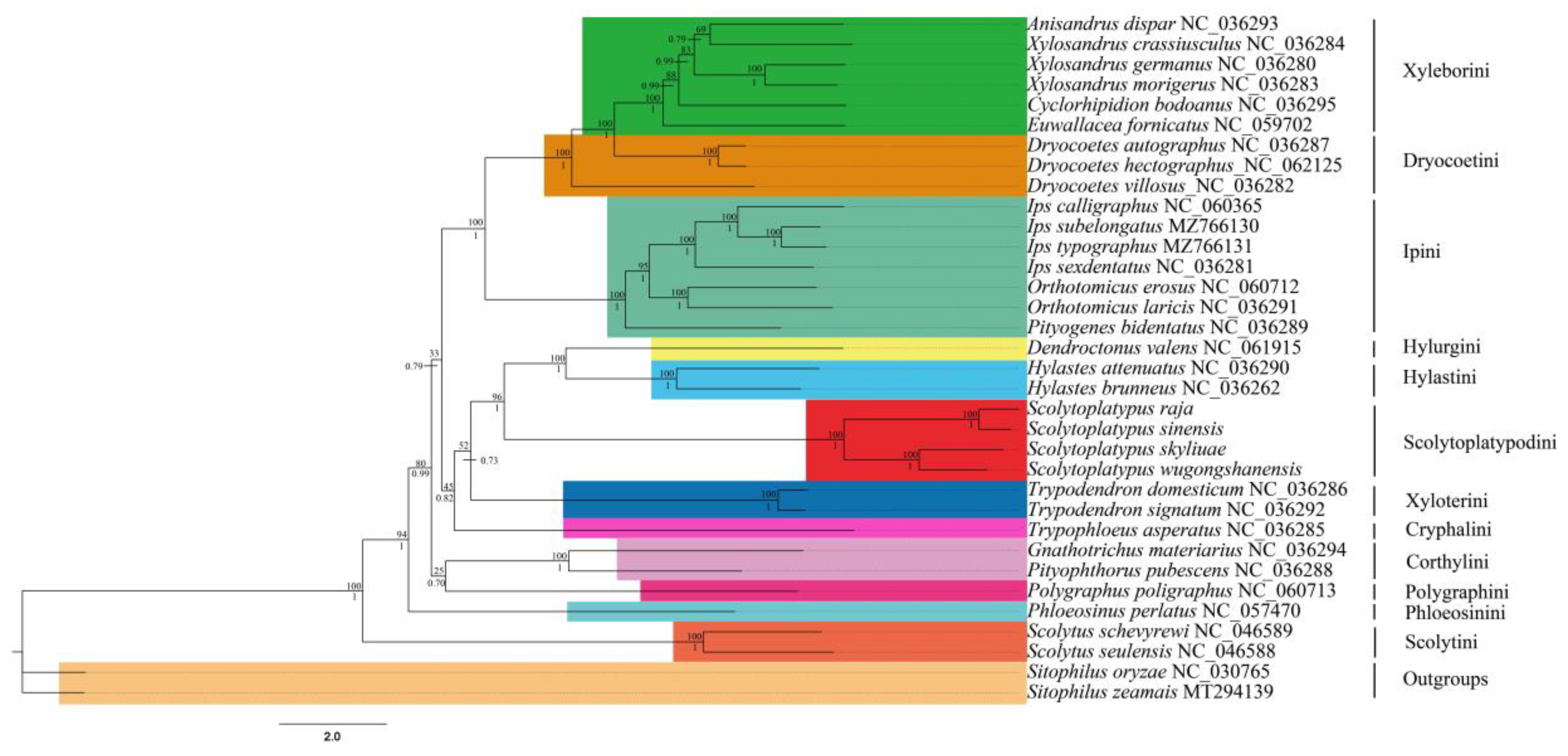
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liao, S.; Lai, S.; Beaver, R.A.; Gebhardt, H.; Wang, J. New species and new records of Scolytoplatypus Schaufuss (Curculionidae, Scolytinae) from China, and resurrection of Scolytoplatypus sinensis (Tsai & Huang, 1965) as a distinct species. ZooKeys 2022, 1082, 27–50. [Google Scholar] [PubMed]
- Gebhardt, H.; Beaver, R.A.; Allgaier, C. Three new species of Scolytoplatypus Schaufuss from China, and notes on the movement and functions of the prosternal processes (Coleoptera: Curculionidae: Scolytinae). Zootaxa 2021, 5082, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Mayers, C.G.; Harrington, T.C.; Masuya, H.; Jordal, B.H.; McNew, D.L.; Shih, H.H.; Roets, F.; Kietzka, G.J. Patterns of coevolution between ambrosia beetle mycangia and the Ceratocystidaceae, with five new fungal genera and seven new species. Persoonia 2020, 44, 41–66. [Google Scholar] [CrossRef]
- Beaver, R.A.; Gebhardt, H. A review of the Oriental species of Scolytoplatypus Schaufuss (Coleoptera, Curculionidae, Scolytinae). Dtsch. Entomol. Z. 2006, 53, 155–178. [Google Scholar] [CrossRef]
- Bhalla, O.; Sharma, P. A new record of shot-holeborer, scolytoplatypus raja blandaf. (scolytidae: Col.) of apple trees in H.P. Curr. Sci. 1963, 32, 86. [Google Scholar]
- Jordal, B.H. Deep phylogenetic divergence between Scolytoplatypus and Remansus, a new genus of Scolytoplatypodini from Madagascar (Coleoptera, Curculionidae, Scolytinae). ZooKeys 2013, 352, 9–33. [Google Scholar] [CrossRef]
- Gillett, C.P.; Crampton-Platt, A.; Timmermans, M.J.; Jordal, B.H.; Emerson, B.C.; Vogler, A.P. Bulk de novo mitogenome assembly from pooled total DNA elucidates the phylogeny of weevils (Coleoptera: Curculionoidea). Mol. Biol. Evol. 2014, 31, 2223–2237. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhang, W.; Ma, Z.; Zhou, C. Novel gene rearrangement pattern in the mitochondrial genomes of Torleya mikhaili and Cincticostella fusca (Ephemeroptera: Ephemerellidae). Int. J. Biol. Macromol. 2020, 165, 3106–3114. [Google Scholar] [CrossRef]
- Sharma, A.; Siva, C.; Ali, S.; Sahoo, P.K.; Nath, R.; Laskar, M.A.; Sarma, D. The complete mitochondrial genome of the medicinal fish, Cyprinion semiplotum: Insight into its structural features and phylogenetic implications. Int. J. Biol. Macromol. 2020, 164, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Barr, C.M.; Neiman, M.; Taylor, D.R. Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 2005, 168, 39–50. [Google Scholar] [CrossRef]
- Haran, J.; Timmermans, M.J.; Vogler, A.P. Mitogenome sequences stabilize the phylogenetics of weevils (Curculionoidea) and establish the monophyly of larval ectophagy. Mol. Phylogenet. Evol. 2013, 67, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Fang, J.; Shi, X.; Zhang, S.; Liu, F.; Yu, C.; Zhang, Z.; Kong, X. Comparative Analysis of Eight Mitogenomes of Bark Beetles and Their Phylogenetic Implications. Insects 2021, 12, 949. [Google Scholar] [CrossRef]
- Li, J.Y.; Li, W.X.; Wang, A.T.; Yu, Z. MitoFlex: An efficient, high-performance toolkit for animal mitogenome assembly, annotation, and visualization. Bioinformatics 2021, 37, 3001–3003. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.J.; Hsu, M.H.; Liu, T.Y.; Lin, M.Y.; Sung, C.H. Characterization of the complete mitochondrial genome of Euwallacea fornicatus (Eichhoff, 1868) (Coleoptera: Curculionidae: Scolytinae) and its phylogenetic implications. Mitochondrial DNA B 2020, 5, 3502–3504. [Google Scholar] [CrossRef]
- Xu, M.F.; Meng, R.; Shui, K.J.; Cai, B.; Lin, W. The complete mitochondrial genome of Ips calligraphus (Germar 1824) (Coleoptera: Curculionidae: Scolytinae). Mitochondrial DNA B 2021, 6, 2494–2495. [Google Scholar] [CrossRef]
- Zhang, L.; Wen, Y.; Lai, S.; He, P.; Li, T.; Zhou, Q.; Wang, J. The complete mitochondrial genome of Scolytus schevyrewi Semenov (Coleoptera: Curculionidae). Mitochondrial DNA Part B 2020, 5, 1841–1842. [Google Scholar] [CrossRef] [Green Version]
- Ojo, J.A.; Valero, M.C.; Sun, W.; Coates, B.S.; Omoloye, A.A.; Pittendrigh, B.R. Comparison of full mitochondrial genomes for the rice weevil, Sitophilus oryzae and the maize weevil, Sitophilus zeamais (Coleoptera: Curculionidae). Agri Gene 2016, 2, 29–37. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486. [Google Scholar] [CrossRef]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNA(serAGN) that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [Green Version]
- Masta, S.E.; Boore, J.L. The Complete Mitochondrial Genome Sequence of the Spider Habronattus oregonensis Reveals Rearranged and Extremely Truncated tRNAs. Mol. Biol. Evol. 2004, 21, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Ballard, J.W. Comparative genomics of mitochondrial DNA in members of the Drosophila melanogaster subgroup. J. Mol. Evol. 2000, 51, 48–63. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Pistone, D.; Gohli, J.; Jordal, B.H. Molecular phylogeny of bark and ambrosia beetles (Curculionidae: Scolytinae) based on 18 molecular markers. Syst. Entomol. 2017, 43, 387–406. [Google Scholar] [CrossRef]
- Jordal, B.H.; Cognato, A.I. Molecular phylogeny of bark and ambrosia beetles reveals multiple origins of fungus farming during periods of global warming. BMC Evol. Biol. 2012, 12, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.M.; Cognato, A.I. A taxonomic monograph of Nearctic Scolytus Geoffroy (Coleoptera, Curculionidae, Scolytinae). ZooKeys 2014, 450, 1–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmermans, M.J.; Vogler, A.P. Phylogenetically informative rearrangements in mitochondrial genomes of Coleoptera, and monophyly of aquatic elateriform beetles (Dryopoidea). Mol. Phylogenet. Evol. 2012, 63, 299–304. [Google Scholar] [CrossRef]
- Liu, N.; Fang, L.; Zhang, Y. The Complete Mitochondrial Genomes of Four Species in the Subfamily Limenitidinae (Lepidoptera, Nymphalidae) and a Phylogenetic Analysis. Insects 2021, 13, 16. [Google Scholar] [CrossRef]
- Zou, Z.; Min, Q.; Cheng, S.; Xin, T.; Xia, B. The complete mitochondrial genome of Thitarodes sejilaensis (Lepidoptera: Hepialidae), a host insect of Ophiocordyceps sinensis and its implication in taxonomic revision of Hepialus adopted in China. Gene 2017, 601, 44–55. [Google Scholar] [CrossRef]
- Mugu, S.; Pistone, D.; Jordal, B. New molecular markers resolve the phylogenetic position of the enigmatic wood-boring weevils Platypodinae (Coleoptera: Curculionidae). Arthropod Syst. Phylogeny 2018, 76, 45–58. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specimens | Location | Geographic Info. | Date of Collection |
|---|---|---|---|
| S. raja | Gaoligong National Nature Reserve, Yunnan | 25.29 N, 98.80 E | 27 July 2019 |
| S. sinensis | Leibo County, Sichuan | 28.41 N, 103.77 E | 5 August 2021 |
| S. skyliuae | Wuyishan National Nature Reserve, Jiangxi | 27.88 N, 117.78 E | 17 July 2017 |
| S. wugongshanensis | Wugong Mountain, Jiangxi | 27.58 N, 114.23 E | 27 September 2017 |
| Subfamily | Species | Length (bp) | GenBank Accession No. | References |
|---|---|---|---|---|
| Scolytinae | Anisandrus dispar | 16,665 | NC_036293 | Unpublished |
| Scolytinae | Xylosandrus crassiusculus | 16,875 | NC_036284 | Unpublished |
| Scolytinae | Xylosandrus germanus | 16,099 | NC_036280 | Unpublished |
| Scolytinae | Xylosandrus morigerus | 16,246 | NC_036283 | Unpublished |
| Scolytinae | Cyclorhipidion bodoanus | 15,899 | NC_036295 | Unpublished |
| Scolytinae | Euwallacea fornicates | 15,745 | NC_059702 | [27] |
| Scolytinae | Dryocoetes autographus | 17,055 | NC_036287 | Unpublished |
| Scolytinae | Dryocoetes hectographus | 16,040 | NC_062125 | [15] |
| Scolytinae | Dryocoetes villosus | 15,859 | NC_036282 | Unpublished |
| Scolytinae | Ips calligraphus | 19,144 | NC_060365 | [28] |
| Scolytinae | Ips subelongatus | 16,040 | MZ766130 | [15] |
| Scolytinae | Ips typographus | 16,793 | MZ766131 | [15] |
| Scolytinae | Ips sexdentatus | 18,579 | NC_036281 | Unpublished |
| Scolytinae | Orthotomicus erosus | 16,753 | NC_060712 | [15] |
| Scolytinae | Orthotomicus laricis | 18,887 | NC_036291 | Unpublished |
| Scolytinae | Pityogenes bidentatus | 18,781 | NC_036289 | Unpublished |
| Scolytinae | Dendroctonus valens | 16,547 | NC_061915 | Unpublished |
| Scolytinae | Hylastes attenuates | 17,409 | NC_036290 | Unpublished |
| Scolytinae | Hylastes brunneus | 15,774 | NC_036262 | Unpublished |
| Scolytinae | Scolytoplatypus raja | 15,324 | OP719285 | This study |
| Scolytinae | Scolytoplatypus skyliuae | 15,166 | OP719283 | This study |
| Scolytinae | Scolytoplatypus sinensis | 15,394 | OP719284 | This study |
| Scolytinae | Scolytoplatypus wugongshanensis | 15,267 | OP712675 | This study |
| Scolytinae | Trypodendron domesticum | 16,986 | NC_036286 | Unpublished |
| Scolytinae | Trypodendron signatum | 16,909 | NC_036292 | Unpublished |
| Scolytinae | Trypophloeus asperatus | 17,039 | NC_036285 | Unpublished |
| Scolytinae | Gnathotrichus materiarius | 16,871 | NC_036294 | Unpublished |
| Scolytinae | Pityophthorus pubescens | 17,316 | NC_036288 | Unpublished |
| Scolytinae | Polygraphus poligraphus | 17,434 | NC_060713 | [15] |
| Scolytinae | Phloeosinus perlatus | 17,054 | NC_057470 | Unpublished |
| Scolytinae | Scolytus schevyrewi | 15,891 | NC_046589 | [29] |
| Scolytinae | Scolytus seulensis | 16,396 | NC_046588 | Unpublished |
| Dryophthorinae | Sitophilus oryzae | 17,602 | NC_030765 | [30] |
| Dryophthorinae | Sitophilus zeamais | 18,531 | MT294139 | [30] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, G.; Lai, S.; Liao, S.; Cao, Y.; Li, W.; Long, C.; Tarno, H.; Wang, J. Complete Mitochondrial Genome of Scolytoplatypodini Species (Coleoptera: Curculionidae: Scolytinae) and Phylogenetic Implications. Genes 2023, 14, 162. https://doi.org/10.3390/genes14010162
Yu G, Lai S, Liao S, Cao Y, Li W, Long C, Tarno H, Wang J. Complete Mitochondrial Genome of Scolytoplatypodini Species (Coleoptera: Curculionidae: Scolytinae) and Phylogenetic Implications. Genes. 2023; 14(1):162. https://doi.org/10.3390/genes14010162
Chicago/Turabian StyleYu, Guangyu, Shengchang Lai, Song Liao, Yufeng Cao, Weijun Li, Chengpeng Long, Hagus Tarno, and Jianguo Wang. 2023. "Complete Mitochondrial Genome of Scolytoplatypodini Species (Coleoptera: Curculionidae: Scolytinae) and Phylogenetic Implications" Genes 14, no. 1: 162. https://doi.org/10.3390/genes14010162