
Transcriptome Analyses of Prophage in Mediating Persistent Methicillin-Resistant Staphylococcus aureus Endovascular Infection


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, and Growth Medium
2.2. RNA Isolation
2.3. RNA-Seq and Data Analyses
2.4. Verification of RNA-Seq Results by qRT-PCR
3. Results
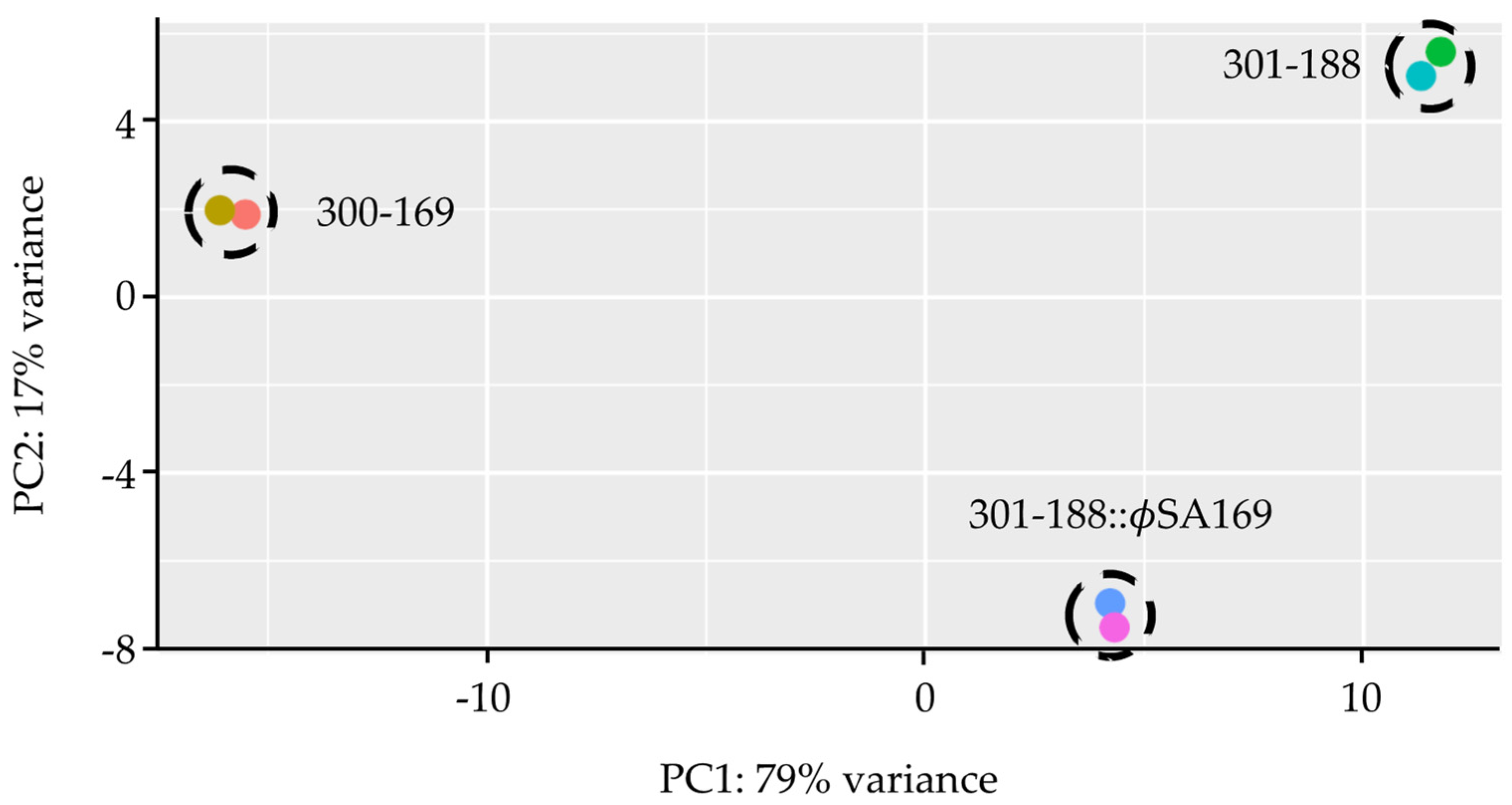
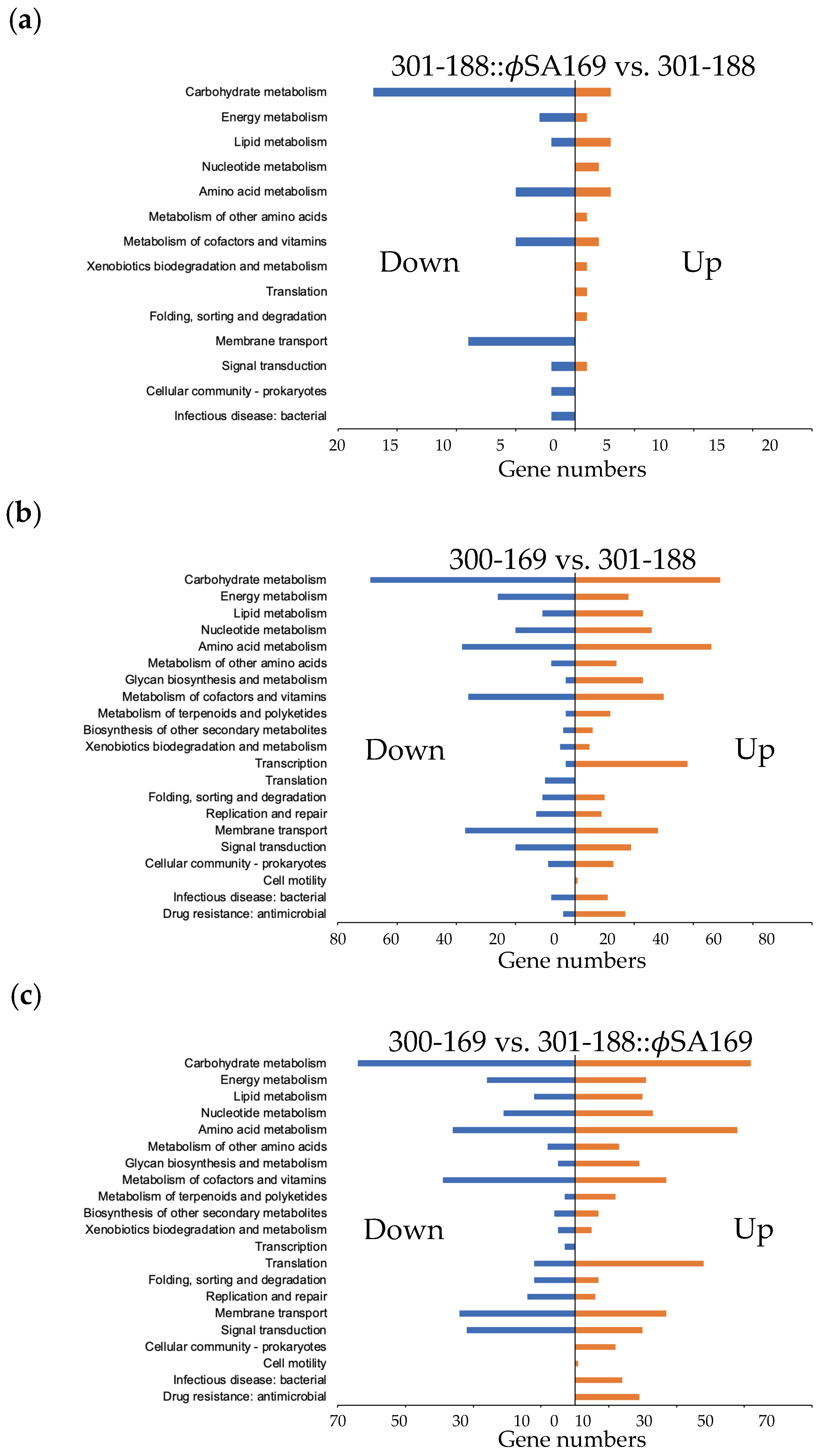
3.1. Global Analyses of Gene Expression
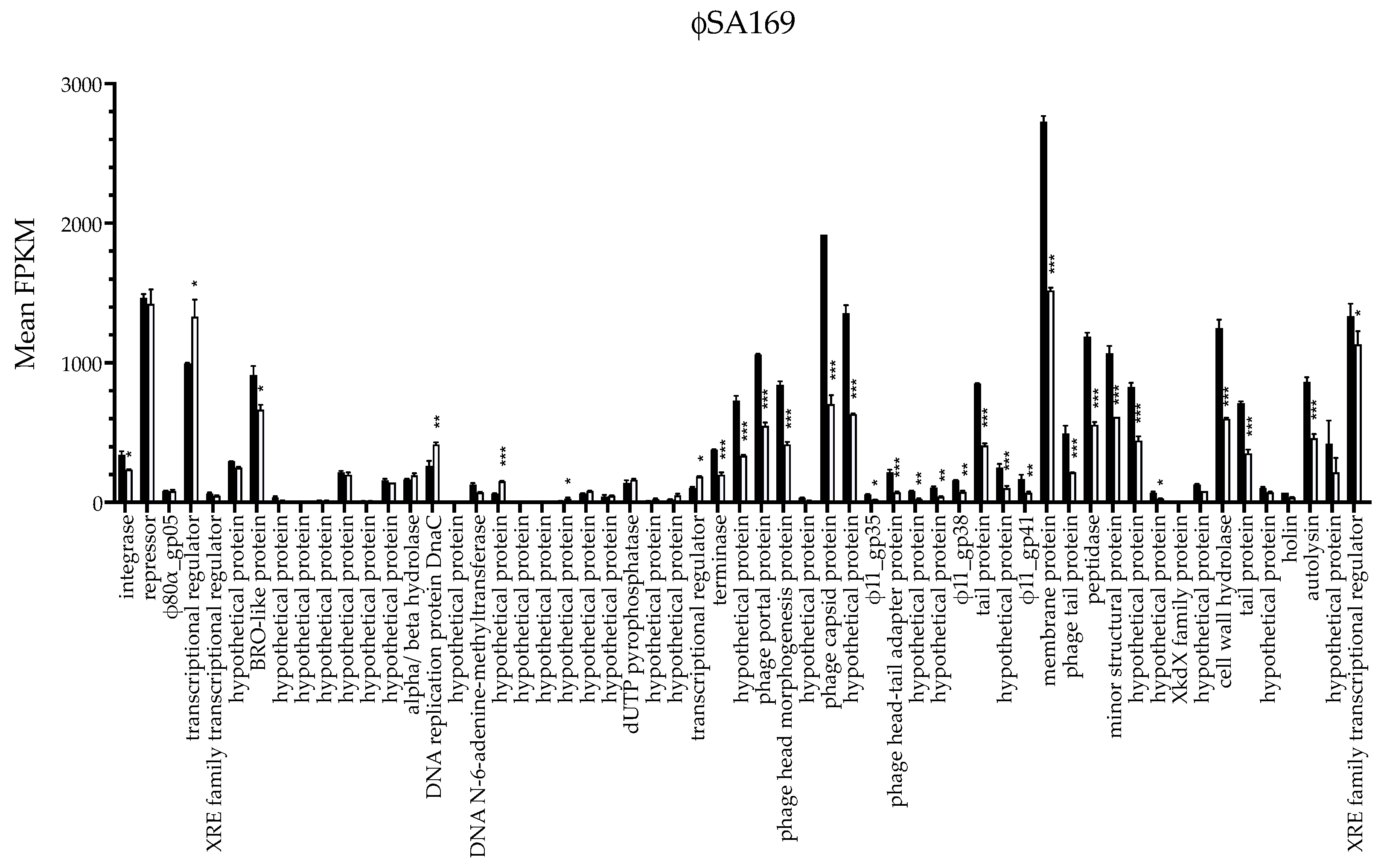
3.2. ϕSA169 Had Similar Transcriptional Profiles in 300-169 and 301-188::ϕSA169 Strains
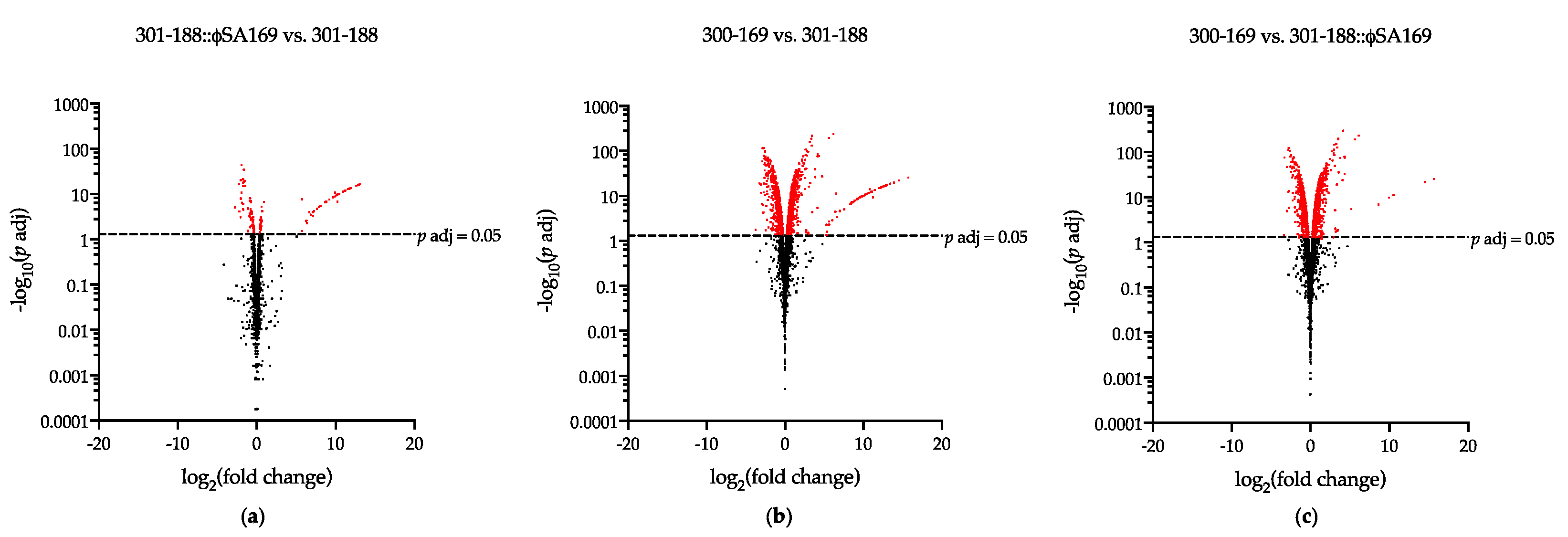
3.3. The Impact of ϕSA169 on Transcriptional Profiles
3.4. The Impact of MRSA Genetic Background on Transcriptional Profiles
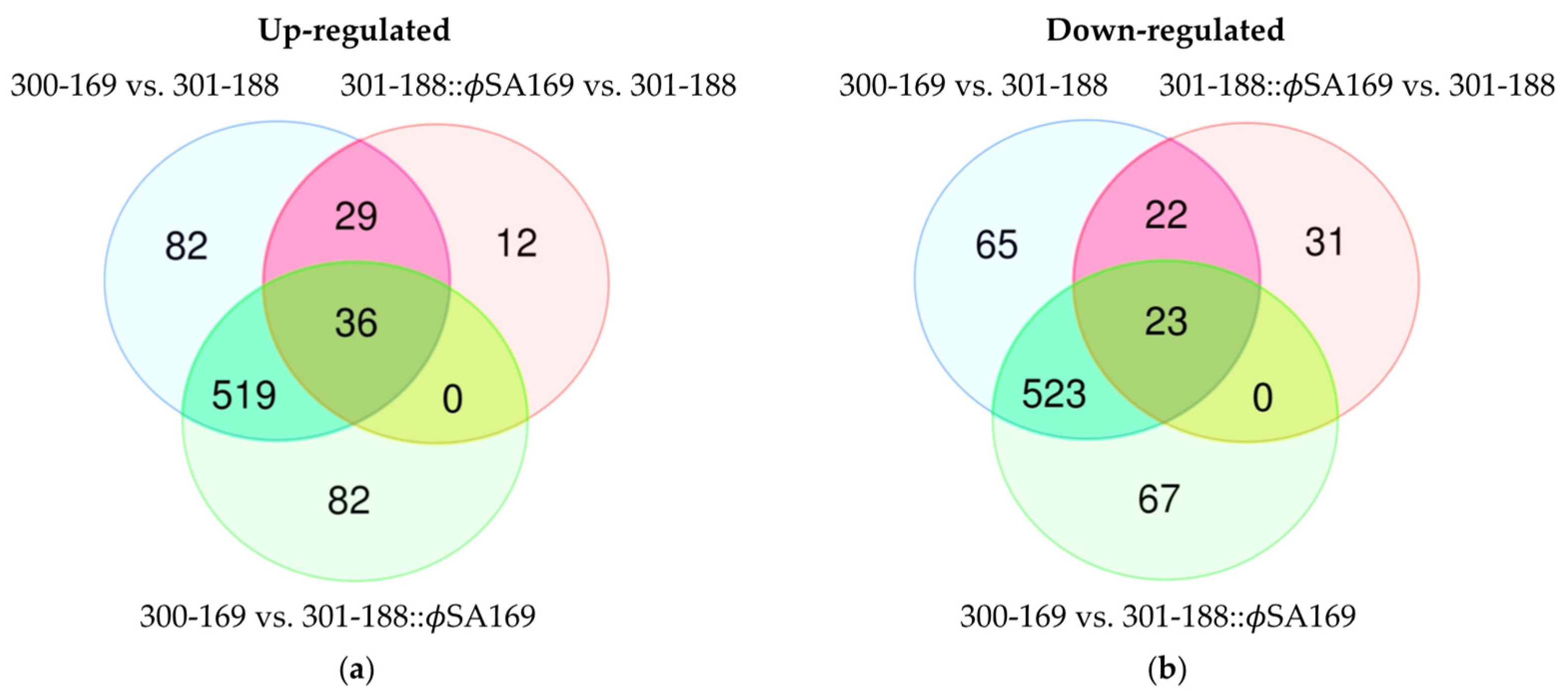
3.5. DEGs Impacted by Both ϕSA169 and MRSA Genetic Backgrounds
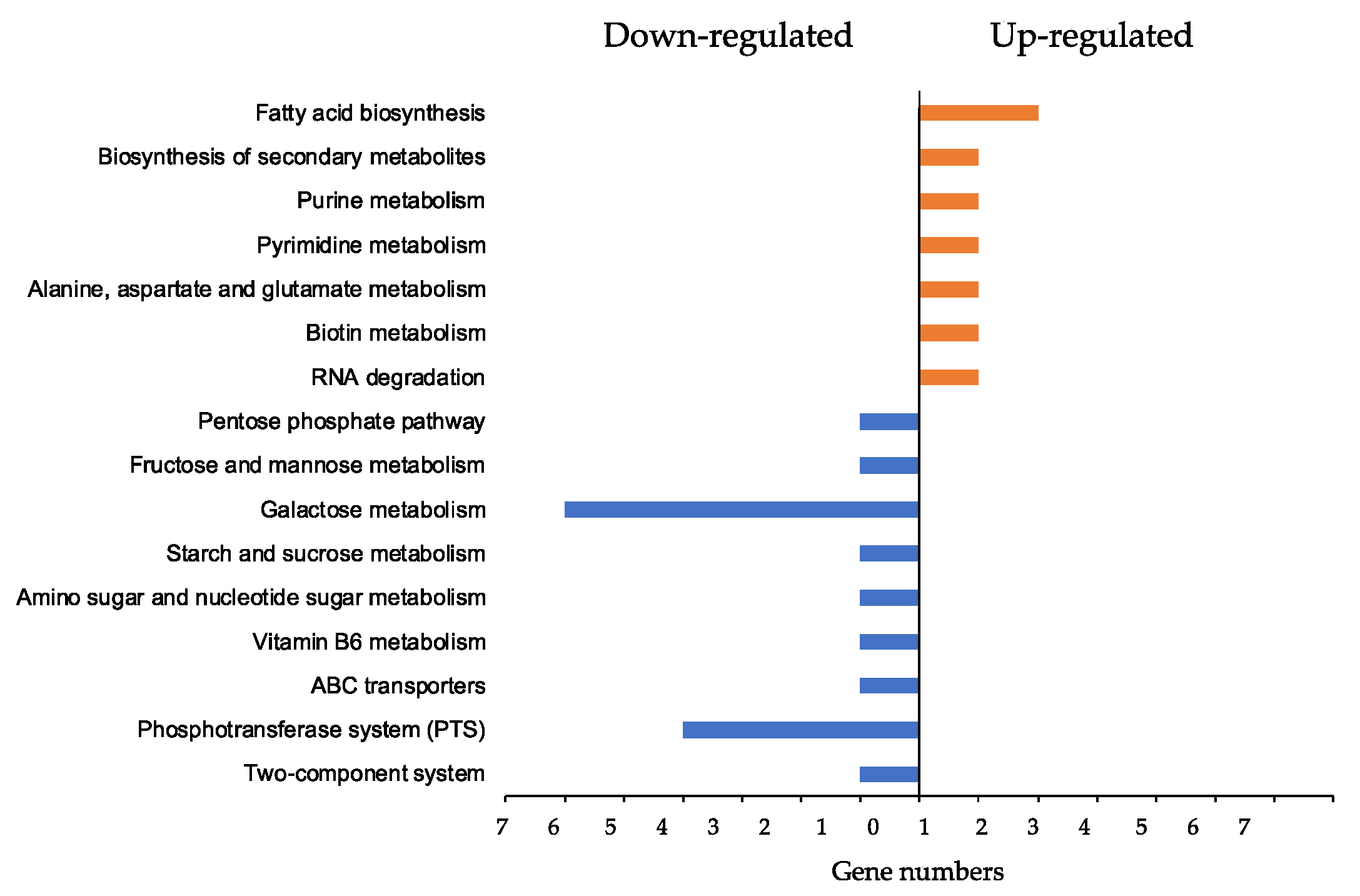
3.6. Global KEGG Analyses of DEG Profiles
3.7. ϕSA169-Specific KEGG Analyses
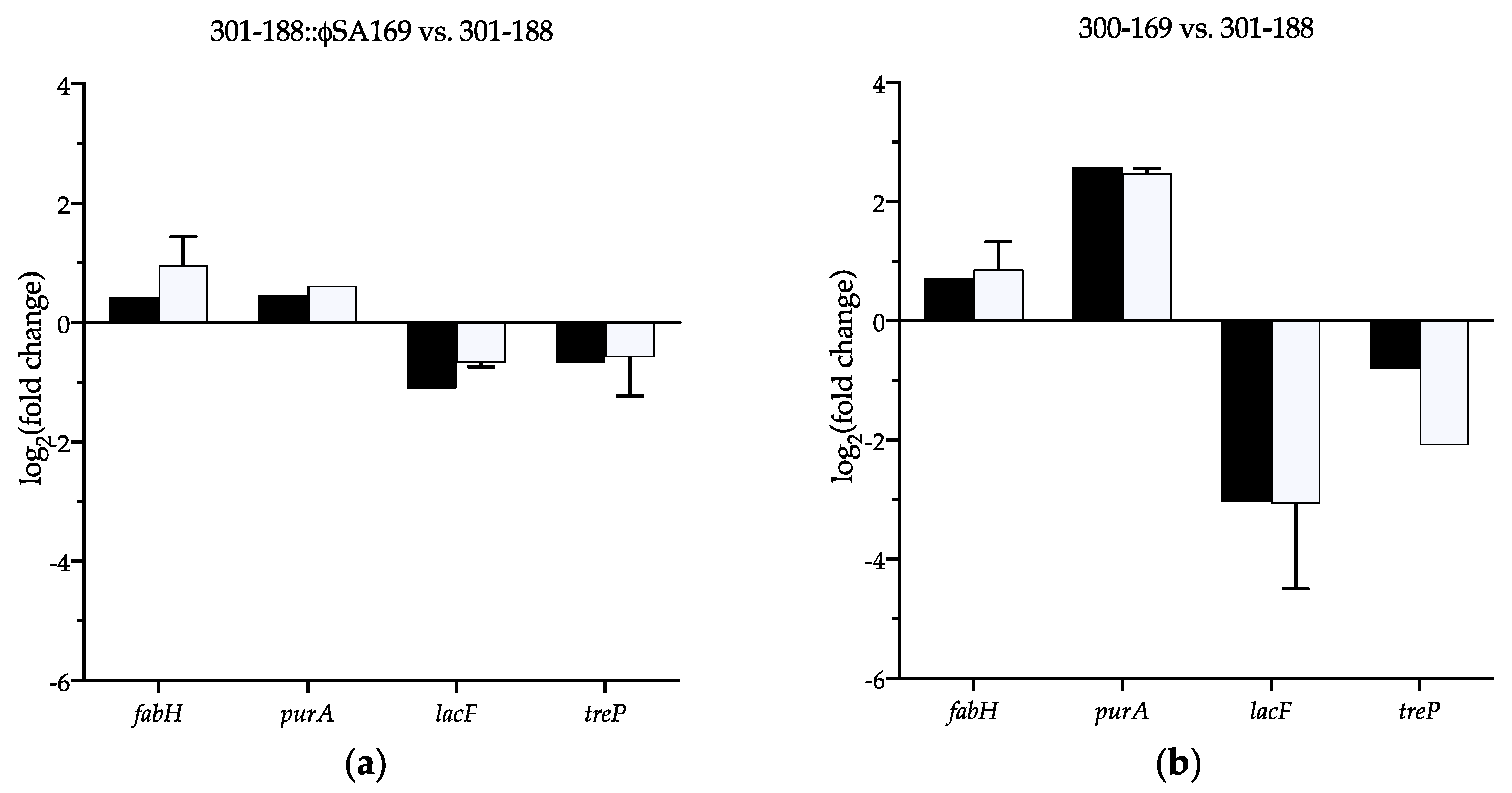
3.8. Verification of the Selected DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fowler, V.G., Jr.; Miro, J.M.; Hoen, B.; Cabell, C.H.; Abrutyn, E.; Rubinstein, E.; Corey, G.R.; Spelman, D.; Bradley, S.F.; Barsic, B.; et al. Staphylococcus aureus endocarditis: A consequence of medical progress. JAMA 2005, 293, 3012–3021. [Google Scholar] [CrossRef] [PubMed]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Fowler, V.G., Jr.; Sakoulas, G.; McIntyre, L.M.; Meka, V.G.; Arbeit, R.D.; Cabell, C.H.; Stryjewski, M.E.; Eliopoulos, G.M.; Reller, L.B.; Corey, G.R.; et al. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J. Infect. Dis. 2004, 190, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Q.; Fowler, V.G.; Yeaman, M.R.; Perdreau-Remington, F.; Kreiswirth, B.N.; Bayer, A.S. Phenotypic and genotypic characteristics of persistent methicillin-resistant Staphylococcus aureus bacteremia in vitro and in an experimental endocarditis model. J. Infect. Dis. 2009, 199, 201–208. [Google Scholar] [CrossRef]
- Seidl, K.; Chen, L.; Bayer, A.S.; Hady, W.A.; Kreiswirth, B.N.; Xiong, Y.Q. Relationship of agr expression and function with virulence and vancomycin treatment outcomes in experimental endocarditis due to methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 5631–5639. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yeaman, M.R.; Bayer, A.S.; Xiong, Y.Q. Phenotypic and genotypic characteristics of methicillin-resistant Staphylococcus aureus (MRSA) related to persistent endovascular infection. Antibiotics 2019, 8, 71. [Google Scholar] [CrossRef]
- Bae, T.; Baba, T.; Hiramatsu, K.; Schneewind, O. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 2006, 62, 1035–1047. [Google Scholar] [CrossRef]
- Fernandez, L.; Gonzalez, S.; Campelo, A.B.; Martinez, B.; Rodriguez, A.; Garcia, P. Low-level predation by lytic phage phiIPLA-RODI promotes biofilm formation and triggers the stringent response in Staphylococcus aureus. Sci. Rep. 2017, 7, 40965. [Google Scholar] [CrossRef]
- Fernandez, L.; Gonzalez, S.; Quiles-Puchalt, N.; Gutierrez, D.; Penades, J.R.; Garcia, P.; Rodriguez, A. Lysogenization of Staphylococcus aureus RN450 by phages ϕ11 and ϕ80α leads to the activation of the SigB regulon. Sci. Rep. 2018, 8, 12662. [Google Scholar] [CrossRef]
- El Haddad, L.; Moineau, S. Characterization of a novel Panton-Valentine leukocidin (PVL)-encoding staphylococcal phage and its naturally PVL-lacking variant. Appl. Environ. Microbiol. 2013, 79, 2828–2832. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, G.; Li, Y.; Francois, P.; Bayer, A.S.; Chen, L.; Seidl, K.; Cheung, A.; Xiong, Y.Q. Impact of the novel prophage ϕSA169 on persistent methicillin-resistant Staphylococcus aureus endovascular infection. mSystems 2020, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Seidl, K.; Corvaglia, A.R.; Bayer, A.S.; Xiong, Y.Q.; Francois, P. Genome sequences of sequence type 45 (ST45) persistent methicillin-resistant Staphylococcus aureus (MRSA) bacteremia strain 300-169 and ST45 resolving MRSA bacteremia strain 301-188. Genome Announc. 2014, 2, 2. [Google Scholar] [CrossRef]
- Li, L.; Wang, G.; Cheung, A.; Abdelhady, W.; Seidl, K.; Xiong, Y.Q. MgrA governs adherence, host cell interaction, and virulence in a murine model of bacteremia due to Staphylococcus aureus. J. Infect. Dis. 2019, 220, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Abdelhady, W.; Donegan, N.P.; Seidl, K.; Cheung, A.; Zhou, Y.F.; Yeaman, M.R.; Bayer, A.S.; Xiong, Y.Q. Role of purine biosynthesis in persistent methicillin-resistant Staphylococcus aureus infection. J. Infect. Dis. 2018, 218, 1367–1377. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Abdelhady, W.; Chen, L.; Bayer, A.S.; Seidl, K.; Yeaman, M.R.; Kreiswirth, B.N.; Xiong, Y.Q. Early agr activation correlates with vancomycin treatment failure in multi-clonotype MRSA endovascular infections. J. Antimicrob. Chemother. 2015, 70, 1443–1452. [Google Scholar] [CrossRef]
- Abdelhady, W.; Bayer, A.S.; Seidl, K.; Moormeier, D.E.; Bayles, K.W.; Cheung, A.; Yeaman, M.R.; Xiong, Y.Q. Impact of vancomycin on sarA-mediated biofilm formation: Role in persistent endovascular infections due to methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2014, 209, 1231–1240. [Google Scholar] [CrossRef]
- Novick, R.P. Mobile genetic elements and bacterial toxinoses: The superantigen-encoding pathogenicity islands of Staphylococcus aureus. Plasmid 2003, 49, 93–105. [Google Scholar] [CrossRef]
- Kondo, K.; Kawano, M.; Sugai, M. Distribution of antimicrobial resistance and virulence genes within the prophage-associated regions in nosocomial pathogens. mSphere 2021, 6, e0045221. [Google Scholar] [CrossRef]
- Payne, D.J.; Warren, P.V.; Holmes, D.J.; Ji, Y.; Lonsdale, J.T. Bacterial fatty-acid biosynthesis: A genomics-driven target for antibacterial drug discovery. Drug Discov. Today 2001, 6, 537–544. [Google Scholar] [CrossRef]
- Fujita, Y.; Matsuoka, H.; Hirooka, K. Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 2007, 66, 829–839. [Google Scholar] [CrossRef]
- Fozo, E.M.; Scott-Anne, K.; Koo, H.; Quivey, R.G., Jr. Role of unsaturated fatty acid biosynthesis in virulence of Streptococcus mutans. Infect. Immun. 2007, 75, 1537–1539. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Pargade, V.; Lamberet, G.; Gaudu, P.; Thomas, F.; Texereau, J.; Gruss, A.; Trieu-Cuot, P.; Poyart, C. The Group B Streptococcus NADH oxidase Nox-2 is involved in fatty acid biosynthesis during aerobic growth and contributes to virulence. Mol. Microbiol. 2006, 62, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Soisson, S.M.; Young, K.; Shoop, W.; Kodali, S.; Galgoci, A.; Painter, R.; Parthasarathy, G.; Tang, Y.S.; Cummings, R.; et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 2006, 441, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Sause, W.E.; Balasubramanian, D.; Irnov, I.; Copin, R.; Sullivan, M.J.; Sommerfield, A.; Chan, R.; Dhabaria, A.; Askenazi, M.; Ueberheide, B.; et al. The purine biosynthesis regulator PurR moonlights as a virulence regulator in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2019, 116, 13563–13572. [Google Scholar] [CrossRef]
- Lan, L.; Cheng, A.; Dunman, P.M.; Missiakas, D.; He, C. Golden pigment production and virulence gene expression are affected by metabolisms in Staphylococcus aureus. J. Bacteriol. 2010, 192, 3068–3077. [Google Scholar] [CrossRef]
- Goncheva, M.I.; Flannagan, R.S.; Sterling, B.E.; Laakso, H.A.; Friedrich, N.C.; Kaiser, J.C.; Watson, D.W.; Wilson, C.H.; Sheldon, J.R.; McGavin, M.J.; et al. Stress-induced inactivation of the Staphylococcus aureus purine biosynthesis repressor leads to hypervirulence. Nat. Commun. 2019, 10, 775. [Google Scholar] [CrossRef]
- Chai, Y.; Beauregard, P.B.; Vlamakis, H.; Losick, R.; Kolter, R. Galactose metabolism plays a crucial role in biofilm formation by Bacillus subtilis. mBio 2012, 3, e00184-12. [Google Scholar] [CrossRef]
- Miallau, L.; Hunter, W.N.; McSweeney, S.M.; Leonard, G.A. Structures of Staphylococcus aureus D-tagatose-6-phosphate kinase implicate domain motions in specificity and mechanism. J. Biol. Chem. 2007, 282, 19948–19957. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Isii, T.; Fukuda, M.; Ochiai, T.; Neoh, H.M.; Camargo, I.L.; Watanabe, Y.; Shoji, M.; Hishinuma, T.; Hiramatsu, K. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 5222–5233. [Google Scholar] [CrossRef] [PubMed]
- Deutscher, J.; Francke, C.; Postma, P.W. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 939–1031. [Google Scholar] [CrossRef] [PubMed]
- Gera, K.; Le, T.; Jamin, R.; Eichenbaum, Z.; McIver, K.S. The phosphoenolpyruvate phosphotransferase system in group A Streptococcus acts to reduce streptolysin S activity and lesion severity during soft tissue infection. Infect. Immun. 2014, 82, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Kok, M.; Bron, G.; Erni, B.; Mukhija, S. Effect of enzyme I of the bacterial phosphoenolpyruvate: Sugar phosphotransferase system (PTS) on virulence in a murine model. Microbiology 2003, 149, 2645–2652. [Google Scholar] [CrossRef]
- Egan, J.B.; Morse, M.L. Carbohydrate transport in Staphylococcus aureus I. Genetic and biochemical analysis of a pleiotropic transport mutant. Biochim. Biophys. Acta 1965, 97, 310–319. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. of Total DEGs | No. of Up-Regulated DEGs | No. of Down-Regulated DEGs | |
|---|---|---|---|
| 301-188::ϕSA169 vs. 301-188 | 153 | 77 | 76 |
| 300-169 vs. 301-188 | 1299 | 666 | 633 |
| 300-169 vs. 301-188::ϕSA169 | 1250 | 637 | 613 |
| Log2(Fold Change) | ||||
|---|---|---|---|---|
| Gene Locus | Group | 301-188::ϕSA169 vs. 301-188 | 300-169 vs. 301-188 | Products |
| AS94_02505 | host genes | 0.93 | 0.71 | MerR family transcriptional regulator |
| AS94_04115 | 0.42 | 0.72 | fabH, 3-oxoacyl-ACP synthase | |
| AS94_04120 | 0.58 | 1.09 | fabF, 3-oxoacyl-ACP synthase | |
| AS94_04780 | 0.51 | 1.29 | amino acid permease | |
| AS94_05160 | 0.69 | 0.86 | Na/Pi cotransporter | |
| AS94_05540 | 0.44 | 1.64 | glycine/betaine ABC transporter permease | |
| AS94_05860 | 0.56 | 0.68 | guanine permease | |
| AS94_06080 | 0.66 | 1.52 | hypothetical protein | |
| AS94_06090 | 0.50 | 0.70 | octopine dehydrogenase | |
| AS94_06310 | 0.48 | 1.09 | sodium:glutamate symporter | |
| AS94_07385 | 0.71 | 1.00 | transglycosylase | |
| AS94_08925 | 0.50 | 0.51 | DEAD/DEAH box helicase | |
| AS94_11275 | 0.47 | 2.59 | purA, adenylosuccinate synthetase | |
| AS94_11985 | 0.42 | 0.93 | multidrug ABC transporter ATP-binding protein | |
| AS94_12030 | 0.37 | 0.70 | general stress protein | |
| AS94_12410 | 0.42 | 1.23 | ribonuclease BN | |
| AS94_12040 | ϕSA169 genes | 7.11 | 8.46 | hypothetical protein |
| AS94_12045 | 12.65 | 12.89 | XRE family transcriptional regulator | |
| AS94_12050 | 10.26 | 11.22 | hypothetical protein | |
| AS94_12055 | 9.94 | 10.84 | autolysin | |
| AS94_12060 | 7.65 | 8.65 | holin | |
| AS94_12065 | 8.70 | 9.21 | hypothetical protein | |
| AS94_12070 | 10.97 | 11.98 | tail protein | |
| AS94_12075 | 11.73 | 12.79 | cell wall hydrolase | |
| AS94_12080 | 8.84 | 9.51 | hypothetical protein | |
| AS94_12090 | 7.30 | 8.65 | hypothetical protein | |
| AS94_12095 | 11.29 | 12.19 | hypothetical protein | |
| AS94_12100 | 11.76 | 12.57 | minor structural protein | |
| AS94_12105 | 11.63 | 12.72 | peptidase | |
| AS94_12110 | 10.25 | 11.46 | phage tail protein | |
| AS94_12115 | 13.07 | 13.92 | membrane protein | |
| AS94_12120 | 8.67 | 9.90 | ϕ11_gp41 | |
| AS94_12125 | 9.20 | 10.46 | hypothetical protein | |
| AS94_12130 | 11.18 | 12.24 | tail protein | |
| AS94_12135 | 8.78 | 9.81 | ϕ11_gp38 | |
| AS94_12140 | 7.89 | 9.24 | hypothetical protein | |
| AS94_12145 | 7.15 | 8.87 | hypothetical protein | |
| AS94_12150 | 8.69 | 10.26 | phage head-tail adapter protein | |
| AS94_12155 | 6.85 | 8.35 | ϕ11_gp35 | |
| AS94_12160 | 11.81 | 12.91 | hypothetical protein | |
| AS94_12165 | 11.97 | 13.41 | phage capsid protein | |
| AS94_12170 | 6.29 | 7.51 | hypothetical protein | |
| AS94_12175 | 11.21 | 12.22 | phage head morphogenesis protein | |
| AS94_12180 | 11.61 | 12.56 | phage portal protein | |
| AS94_12185 | 10.89 | 12.01 | hypothetical protein | |
| AS94_12190 | 10.14 | 11.07 | terminase | |
| AS94_12195 | 10.04 | 9.18 | transcriptional regulator | |
| AS94_12210 | 8.22 | 7.00 | hypothetical protein | |
| AS94_12215 | 7.15 | 5.65 | hypothetical protein | |
| AS94_12220 | 9.85 | 9.62 | dut, dUTP pyrophosphatase | |
| AS94_12230 | 6.69 | 6.41 | hypothetical protein | |
| AS94_12240 | 8.84 | 8.52 | hypothetical protein | |
| AS94_12270 | 5.76 | 6.56 | DNA N-6-adenine-methyltransferase | |
| AS94_12295 | 9.66 | 9.80 | hypothetical protein | |
| AS94_12320 | 5.72 | 5.64 | hypothetical protein | |
| AS94_12325 | 10.13 | 10.26 | hypothetical protein | |
| AS94_12330 | 6.36 | 6.05 | hypothetical protein | |
| AS94_12340 | 6.27 | 7.58 | hypothetical protein | |
| AS94_12345 | 11.88 | 12.34 | BRO-like protein | |
| AS94_12350 | 10.47 | 10.70 | hypothetical protein | |
| AS94_12355 | 8.09 | 8.50 | XRE family transcriptional regulator | |
| AS94_12360 | 12.88 | 12.46 | transcriptional regulator | |
| AS94_12365 | 8.86 | 8.90 | ϕ80α_gp05 | |
| AS94_12370 | 12.98 | 13.02 | repressor | |
| AS94_12375 | 10.38 | 10.92 | integrase | |
| Log2(Fold Change) | ||||
|---|---|---|---|---|
| Gene Locus | Group | 301-188::ϕSA169 vs. 301-188 | 300-169 vs. 301-188 | Products |
| AS94_03800 | host genes | −0.82 | −2.38 | cysteine protease |
| AS94_04675 | −0.39 | −0.50 | sdrD, hydrolase | |
| AS94_05575 | −0.74 | −2.33 | lacE, PTS lactose transporter subunit IIBC | |
| AS94_05580 | −1.11 | −3.04 | lacF, PTS lactose transporter subunit IIA | |
| AS94_05585 | −0.81 | −2.57 | lacD, tagatose-bisphosphate aldolase | |
| AS94_05590 | −0.85 | −2.37 | lacC, tagatose-6-phosphate kinase | |
| AS94_05595 | −1.09 | −2.30 | lacB, galactose-6-phosphate isomerase | |
| AS94_05600 | −0.76 | −2.60 | lacA, galactose-6-phosphate isomerase | |
| AS94_06915 | −0.54 | −0.84 | nikA, nickel ABC transporter substrate-binding protein | |
| AS94_07070 | −0.39 | −0.70 | gntk, gluconokinase | |
| AS94_08235 | −0.48 | −0.83 | pfkB, phosphofructokinase | |
| AS94_08280 | −0.48 | −0.40 | hypothetical protein | |
| AS94_09210 | −0.58 | −1.88 | general stress protein | |
| AS94_10090 | −0.86 | −0.78 | murein hydrolase regulator lrgA, LrgA | |
| AS94_10365 | −0.78 | −1.53 | sialic acid transporter | |
| AS94_10370 | −0.84 | −1.75 | nanA, N-acetylneuraminate lyase | |
| AS94_10375 | −0.37 | −0.87 | N-acetylmannosamine kinase | |
| AS94_11050 | −0.67 | −0.81 | treP, PTS ascorbate transporter subunit IIA | |
| AS94_11645 | −0.38 | −0.35 | pyridoxal biosynthesis protein | |
| AS94_12380 | −0.90 | −0.88 | hypothetical protein | |
| AS94_12875 | −0.43 | −0.72 | hld, delta-hemolysin | |
| AS94_13070 | the mutual prophage in 300-169 and 301-188 | −1.47 | −2.34 | autolysin |
| AS94_13075 | −1.69 | −3.25 | holin | |
| AS94_13080 | −1.68 | −2.51 | hypothetical protein | |
| AS94_13090 | −2.18 | −2.40 | hypothetical protein | |
| AS94_13095 | −1.70 | −2.35 | hypothetical protein | |
| AS94_13100 | −1.53 | −2.07 | minor structural protein | |
| AS94_13110 | −1.72 | −2.18 | peptidase | |
| AS94_13115 | −1.90 | −2.25 | holin | |
| AS94_13120 | −1.62 | −2.11 | tail protein | |
| AS94_13130 | −2.75 | −2.80 | hypothetical protein | |
| AS94_13135 | −1.42 | −2.07 | tail protein | |
| AS94_13140 | −2.01 | −2.12 | tail protein | |
| AS94_13150 | −1.85 | −1.72 | hypothetical protein | |
| AS94_13160 | −2.00 | −2.24 | hypothetical protein | |
| AS94_13165 | −1.93 | −2.10 | phage capsid protein | |
| AS94_13170 | −2.19 | −2.07 | ATP-dependent Clp protease ClpP | |
| AS94_13175 | −1.61 | −1.79 | portal protein | |
| AS94_13180 | −1.71 | −1.80 | terminase | |
| AS94_13185 | −1.74 | −1.59 | terminase | |
| AS94_13190 | −2.02 | −1.49 | HNH endonuclease | |
| AS94_13195 | −0.73 | −1.93 | transcriptional regulator | |
| AS94_13200 | −0.89 | −1.56 | helicase | |
| AS94_13205 | −0.75 | −1.44 | hypothetical protein | |
| AS94_13355 | −0.46 | −0.74 | antirepressor | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Chen, L.; Zhu, F.; Bayer, A.S.; Xiong, Y.Q. Transcriptome Analyses of Prophage in Mediating Persistent Methicillin-Resistant Staphylococcus aureus Endovascular Infection. Genes 2022, 13, 1527. https://doi.org/10.3390/genes13091527
Li Y, Chen L, Zhu F, Bayer AS, Xiong YQ. Transcriptome Analyses of Prophage in Mediating Persistent Methicillin-Resistant Staphylococcus aureus Endovascular Infection. Genes. 2022; 13(9):1527. https://doi.org/10.3390/genes13091527
Chicago/Turabian StyleLi, Yi, Liang Chen, Fengli Zhu, Arnold S. Bayer, and Yan Q. Xiong. 2022. "Transcriptome Analyses of Prophage in Mediating Persistent Methicillin-Resistant Staphylococcus aureus Endovascular Infection" Genes 13, no. 9: 1527. https://doi.org/10.3390/genes13091527