
Occurrence of vanHAX and Related Genes beyond the Actinobacteria Phylum

Abstract
:1. Introduction
2. Methods
2.1. van Genes Discovery
2.2. Mapping MGE-Related Genes
2.3. Phylogenetic Reconstruction
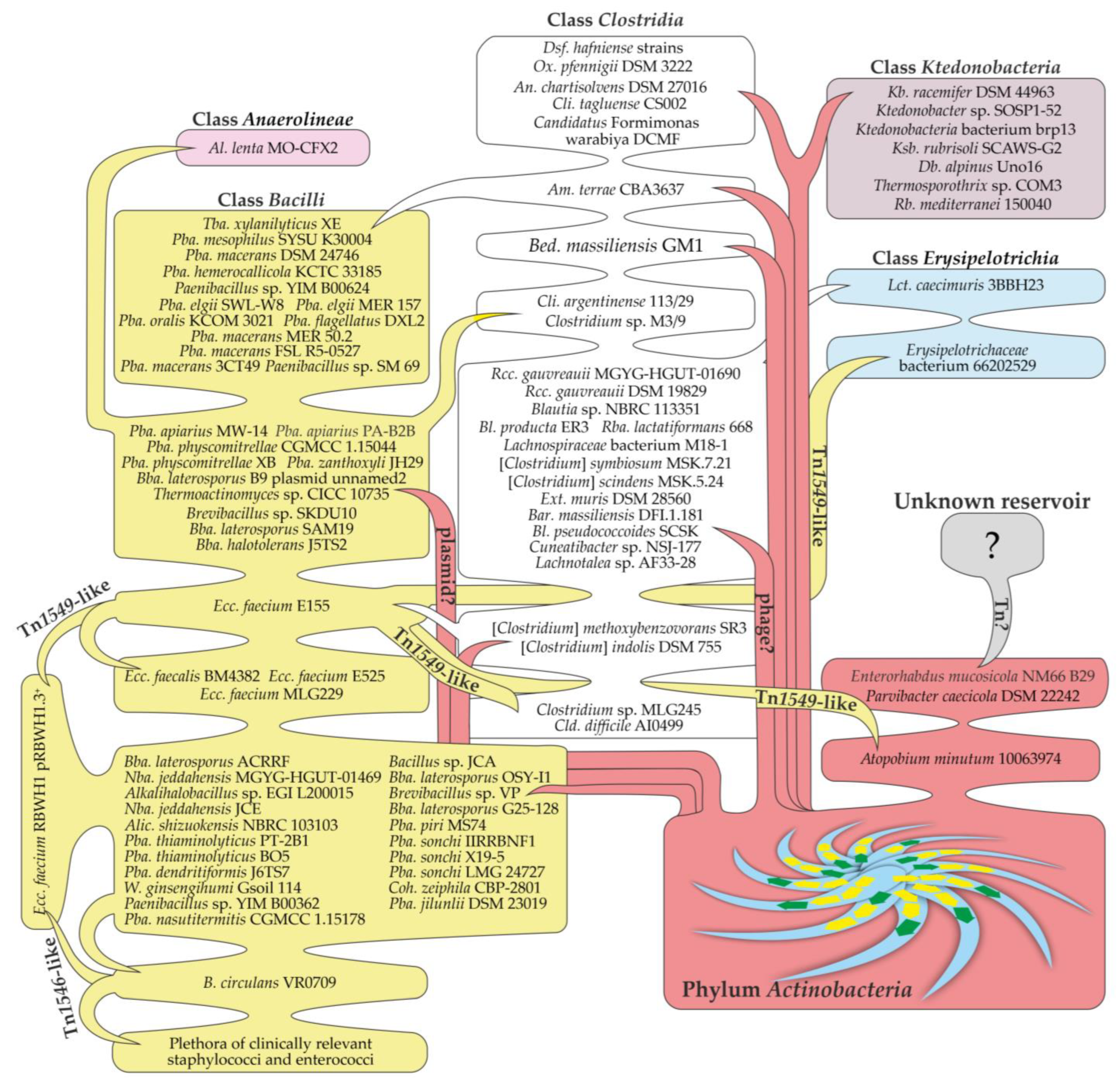
3. Results
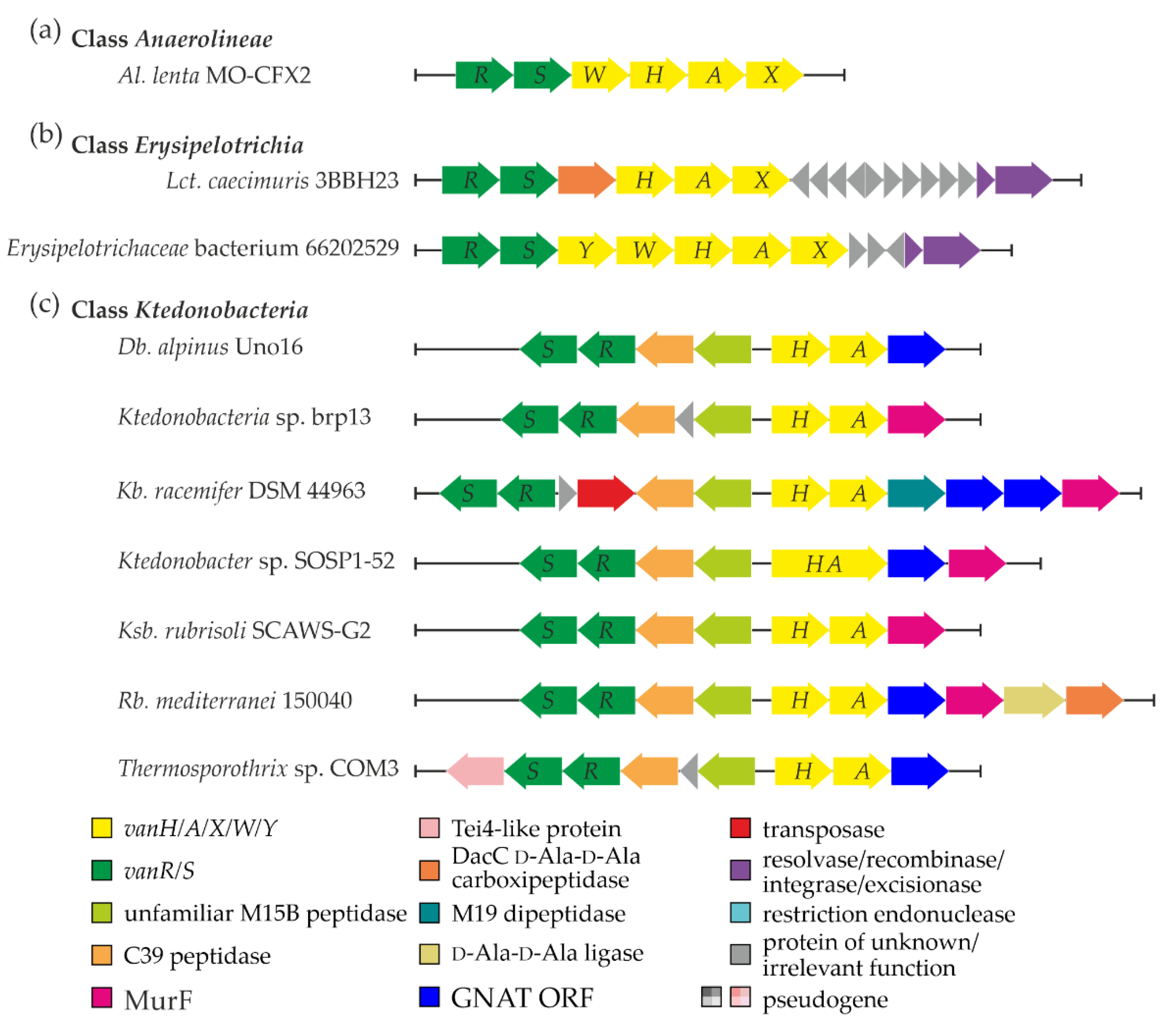
3.1. van Genes in Class Anaerolineae
3.2. van Genes in Class Erysipelotrichia
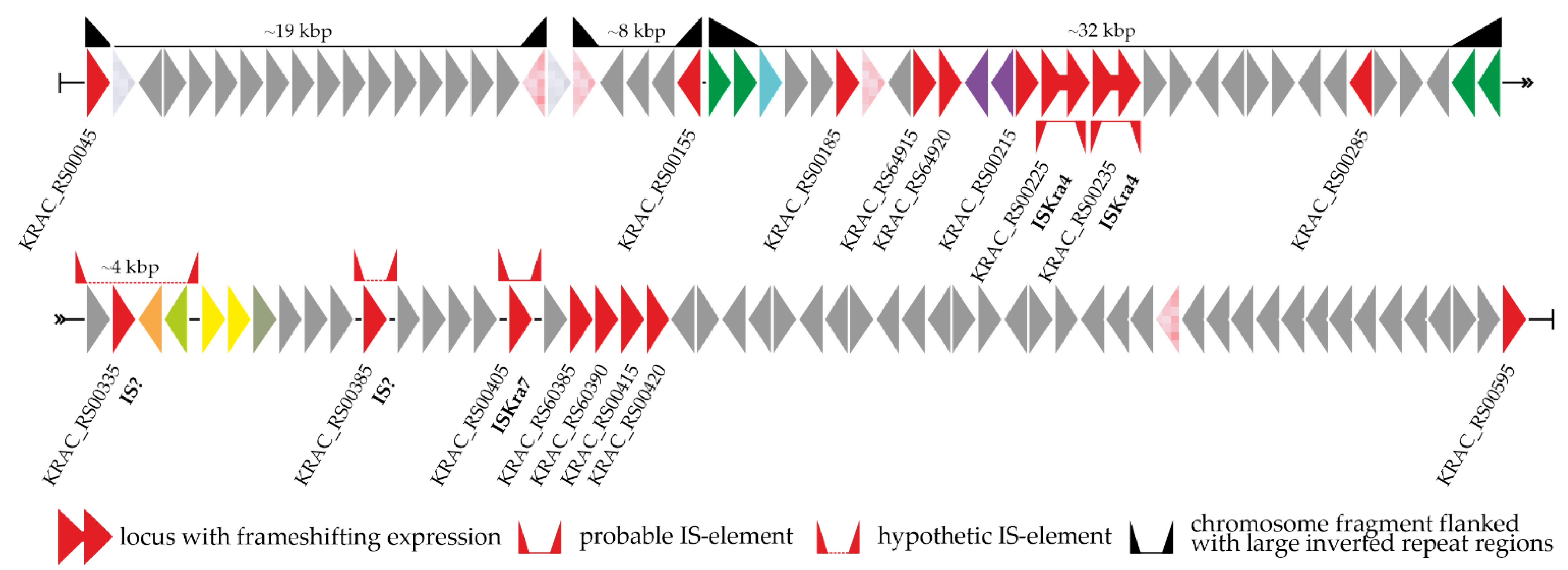
3.3. Peculiarities of Organization and Genetic Context of van Genes in Class Ktedonobacteria
3.4. Updates on vanHAXRS Gene Distribution in Bacilli Class
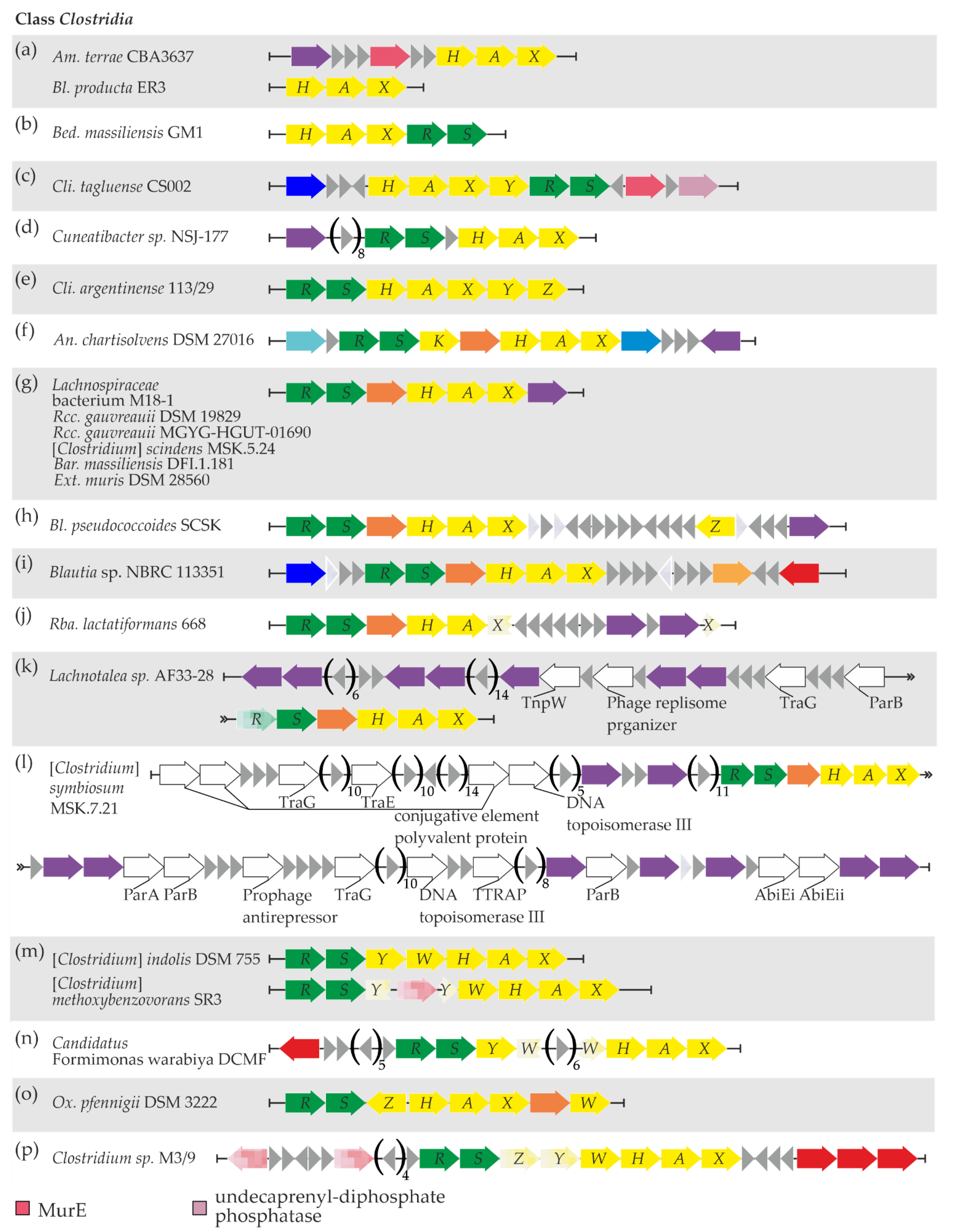
3.5. Updating the Picture of vanHAXRS Gene Distribution in Clostridia Class
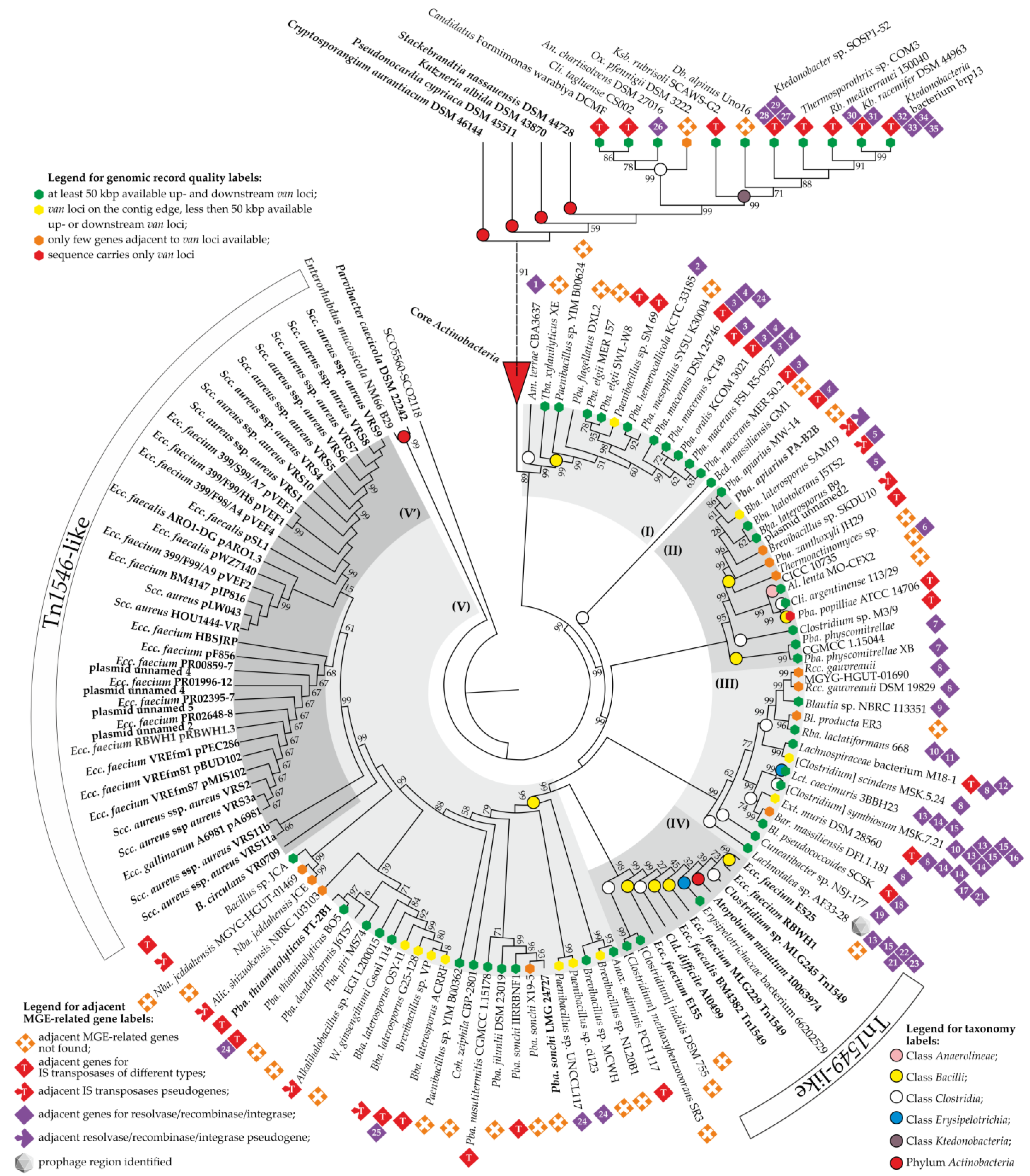
3.6. Building a Consensus Scheme for Phylogenetic Relations between Newly Discovered van Proteins and Those from Phylum Actinobacteria
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicolaou, K.C.; Boddy, C.N.C.; Bräse, S.; Winssinger, N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. 1999, 38, 2096–2152. [Google Scholar] [CrossRef]
- Parenti, F.; Cavalleri, B. Proposal to name the vancomycin-ristocetin like glycopeptides as dalbaheptides. J. Antibiot. 1989, 42, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Kaniusaite, M.; Tailhades, J.; Kittilä, T.; Fage, C.D.; Goode, R.J.A.; Schittenhelm, R.B.; Cryle, M.J. Understanding the early stages of peptide formation during the biosynthesis of teicoplanin and related glycopeptide antibiotics. FEBS J. 2021, 288, 507–529. [Google Scholar] [CrossRef]
- Stegmann, E.; Frasch, H.J.; Wohlleben, W. Glycopeptide biosynthesis in the context of basic cellular functions. Curr. Opin. Microbiol. 2010, 13, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Thaker, M.N.; Wright, G.D. Opportunities for synthetic biology in antibiotics: Expanding glycopeptide chemical diversity. ACS Synth. Biol. 2015, 4, 195–206. [Google Scholar] [CrossRef]
- Yushchuk, O.; Ostash, B. Glycopeptide antibiotics: Genetics, chemistry, and new screening approaches. In Natural Products from Actinomycetes; Springer: Singapore, 2022; pp. 411–444. [Google Scholar] [CrossRef]
- Yushchuk, O.; Zhukrovska, K.; Berini, F.; Fedorenko, V.; Marinelli, F. Genetics behind the glycosylation patterns in the biosynthesis of dalbaheptides. Front. Chem. 2022, 10, 858708. [Google Scholar] [CrossRef]
- Marcone, G.L.; Binda, E.; Berini, F.; Marinelli, F. Old and new glycopeptide antibiotics: From product to gene and back in the post-genomic era. Biotechnol. Adv. 2018, 36, 534–554. [Google Scholar] [CrossRef]
- Marschall, E.; Cryle, M.J.; Tailhades, J. Biological, chemical, and biochemical strategies for modifying glycopeptide antibiotics. J. Biol. Chem. 2019, 294, 18769–18783. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.; Klöckner, A.; Schneider, T. Targeting a cell wall biosynthesis hot spot. Nat. Prod. Rep. 2017, 34, 909–932. [Google Scholar] [CrossRef]
- Jovetic, S.; Zhu, Y.; Marcone, G.L.; Marinelli, F.; Tramper, J. β-Lactam and glycopeptide antibiotics: First and last line of defense? Trends Biotechnol. 2010, 28, 596–604. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Calic, D.; Schweizer, F.; Zelenitsky, S.; Adam, H.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; Karlowsky, J.A. New lipoglycopeptides: A comparative review of dalbavancin, oritavancin and telavancin. Drugs 2010, 70, 859–886. [Google Scholar] [CrossRef] [PubMed]
- Binda, E.; Marinelli, F.; Marcone, G.L. Old and new glycopeptide antibiotics: Action and resistance. Antibiotics 2014, 3, 572–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, P.; Yarlagadda, V.; Ghosh, C.; Haldar, J. A review on cell wall synthesis inhibitors with an emphasis on glycopeptide antibiotics. Medchemcomm 2017, 8, 516–533. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitanai, Y.; Kikuchi, T.; Kakoi, K.; Hanamaki, S.; Fujisawa, I.; Aoki, K. Crystal structures of the complexes between vancomycin and cell-wall precursor analogs. J. Mol. Biol. 2009, 385, 1422–1432. [Google Scholar] [CrossRef]
- Williams, D.H. The glycopeptide story—How to kill the deadly “superbugs. ” Nat. Prod. Rep. 1996, 13, 469–477. [Google Scholar] [CrossRef]
- Binda, E.; Marcone, G.L.; Pollegioni, L.; Marinelli, F. Characterization of VanYn, a novel d,d-peptidase/d,d-carboxypeptidase involved in glycopeptide antibiotic resistance in Nonomuraea sp. ATCC 39727. FEBS J. 2012, 279, 3203–3213. [Google Scholar] [CrossRef]
- Marcone, G.L.; Beltrametti, F.; Binda, E.; Carrano, L.; Foulston, L.; Hesketh, A.; Bibb, M.; Marinelli, F. Novel mechanism of glycopeptide resistance in the A40926 producer Nonomuraea sp. ATCC 39727. Antimicrob. Agents Chemother. 2010, 54, 2465–2472. [Google Scholar] [CrossRef] [Green Version]
- Yushchuk, O.; Homoniuk, V.; Ostash, B.; Marinelli, F.; Fedorenko, V. Genetic insights into the mechanism of teicoplanin self-resistance in Actinoplanes teichomyceticus. J. Antibiot. 2020, 73, 255–259. [Google Scholar] [CrossRef]
- Kilian, R.; Frasch, H.J.; Kulik, A.; Wohlleben, W.; Stegmann, E. The VanRS homologous two-component system VnlRSAb of the glycopeptide producer Amycolatopsis balhimycina activates transcription of the vanHAXSc genes in Streptomyces coelicolor, but not in A. balhimycina. Microb. Drug Resist. 2016, 22, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Yushchuk, O.; Binda, E.; Marinelli, F. Glycopeptide antibiotic resistance genes: Distribution and function in the producer actinomycetes. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, E.; Frasch, H.J.; Kilian, R.; Pozzi, R. Self-resistance mechanisms of actinomycetes producing lipid II-targeting antibiotics. Int. J. Med. Microbiol. 2015, 305, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.J.; Hutchings, M.I.; Neu, J.M.; Wright, G.D.; Paget, M.S.B.; Buttner, M.J. Characterization of an inducible vancomycin resistance system in Streptomyces coelicolor reveals a novel gene (vanK) required for drug resistance. Mol. Microbiol. 2004, 52, 1107–1121. [Google Scholar] [CrossRef] [PubMed]
- Andreo-Vidal, A.; Binda, E.; Fedorenko, V.; Marinelli, F.; Yushchuk, O. Genomic insights into the distribution and phylogeny of glycopeptide resistance determinants within the actinobacteria phylum. Antibiotics 2021, 10, 1533. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.G.; Broadhead, G.; Leskiw, B.K.; Wright, G.D. d-Ala-d-Ala ligases from glycopeptide antibiotic-producing organisms are highly homologous to the enterococcal vancomycin-resistance ligases VanA and VanB. Proc. Natl. Acad. Sci. USA 1997, 94, 6480–6483. [Google Scholar] [CrossRef] [Green Version]
- Rushton-Green, R.; Darnell, R.L.; Taiaroa, G.; Carter, G.P.; Cook, G.M.; Morgan, X.C. Agricultural origins of a highly persistent lineage of vancomycin-resistant Enterococcus faecalis in New Zealand. Appl. Environ. Microbiol. 2019, 85, e00137-19. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.K.; Tanimoto, K.; Tomita, H.; Ike, Y. Pheromone-responsive conjugative vancomycin resistance plasmids in Enterococcus faecalis isolates from humans and chicken feces. Appl. Environ. Microbiol. 2006, 72, 6544–6553. [Google Scholar] [CrossRef] [Green Version]
- Mello, S.S.; Van Tyne, D.; Lebreton, F.; Silva, S.Q.; Nogueira, M.C.L.; Gilmore, M.S.; Camargo, I.L.B.C. A mutation in the glycosyltransferase gene lafB causes daptomycin hypersusceptibility in Enterococcus faecium. J. Antimicrob. Chemother. 2020, 75, 36–45. [Google Scholar] [CrossRef]
- Ballard, S.A.; Pertile, K.K.; Lim, M.; Johnson, P.D.R.; Grayson, M.L. Molecular characterization of vanB elements in naturally occurring gut anaerobes. Antimicrob. Agents Chemother. 2005, 49, 1688–1694. [Google Scholar] [CrossRef]
- Kinnear, C.L.; Hansen, E.; Morley, V.J.; Tracy, K.C.; Forstchen, M.; Read, A.F.; Woods, R.J. Daptomycin treatment impacts resistance in off-target populations of vancomycin-resistant Enterococcus faecium. PLoS Biol. 2020, 18, e3000987. [Google Scholar] [CrossRef]
- Bohlmann, L.; De Oliveira, D.M.P.; El-Deeb, I.M.; Brazel, E.B.; Harbison-Price, N.; Ong, C.L.Y.; Rivera-Hernandez, T.; Ferguson, S.A.; Cork, A.J.; Phan, M.D.; et al. Chemical synergy between ionophore PBT2 and zinc reverses antibiotic resistance. MBio 2018, 9, e02391-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Sung, K.; Marasa, B.; Min, S.; Kweon, O.; Nawaz, M.; Cerniglia, C. Draft genome sequence of multidrug-resistant Enterococcus faecium clinical isolate VRE3, with a sequence type 16 pattern and novel structural arrangement of Tn1546. Genome Announc. 2015, 3, 1000000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melegh, S.; Nyul, A.; Kovács, K.; Kovács, T.; Ghidán, Á.; Dombrádi, Z.; Szabó, J.; Berta, B.; Lesinszki, V.; Pászti, J.; et al. Dissemination of VanA-type Enterococcus faecium isolates in Hungary. Microb. Drug Resist. 2018, 24, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Eshaghi, A.; Shahinas, D.; Li, A.; Kariyawasam, R.; Banh, P.; Desjardins, M.; Melano, R.G.; Patel, S.N. Characterization of an Enterococcus gallinarum isolate carrying a dual vanA and vanB cassette. J. Clin. Microbiol. 2015, 53, 2225–2229. [Google Scholar] [CrossRef] [Green Version]
- Zaheer, R.; Cook, S.R.; Barbieri, R.; Goji, N.; Cameron, A.; Petkau, A.; Polo, R.O.; Tymensen, L.; Stamm, C.; Song, J.; et al. Surveillance of Enterococcus spp. reveals distinct species and antimicrobial resistance diversity across a One-Health continuum. Sci. Rep. 2020, 10, 3937. [Google Scholar] [CrossRef] [Green Version]
- Weigel, L.M.; Clewell, D.B.; Gill, S.R.; Clark, N.C.; McDougal, L.K.; Flannagan, S.E.; Kolonay, J.F.; Shetty, J.; Killgore, G.E.; Tenover, F.C. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571. [Google Scholar] [CrossRef] [PubMed]
- Panesso, D.; Planet, P.J.; Diaz, L.; Hugonnet, J.E.; Tran, T.T.; Narechania, A.; Munita, J.M.; Rincon, S.; Carvajal, L.P.; Reyes, J.; et al. Methicillin-susceptible, vancomycin-resistant Staphylococcus aureus, Brazil. Emerg. Infect. Dis. 2015, 21, 1844–1848. [Google Scholar] [CrossRef]
- Zhu, W.; Murray, P.R.; Huskins, W.C.; Jernigan, J.A.; McDonald, L.C.; Clark, N.C.; Anderson, K.F.; McDougal, L.K.; Hageman, J.C.; Olsen-Rasmussen, M.; et al. Dissemination of an Enterococcus Inc18-like vanA plasmid associated with vancomycin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 4314–4320. [Google Scholar] [CrossRef] [Green Version]
- Kos, V.N.; Desjardins, C.A.; Griggs, A.; Cerqueira, G.; Van Tonder, A.; Holden, M.T.G.; Godfrey, P.; Palmer, K.L.; Bodi, K.; Mongodin, E.F.; et al. Comparative genomics of vancomycin-resistant Staphylococcus aureus strains and their positions within the clade most commonly associated with methicillin-resistant S. aureus hospital-acquired infection in the United States. MBio 2012, 3, e00112-12. [Google Scholar] [CrossRef]
- Garnier, F.; Taourit, S.; Glaser, P.; Courvalin, P.; Galimand, M. Characterization of transposon Tn1549, conferring VanB-type resistance in Enterococcus spp. Microbiology 2000, 146, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- De Been, M.; Van Schaik, W.; Cheng, L.; Corander, J.; Willems, R.J. Recent recombination events in the core genome are associated with adaptive evolution in Enterococcus faecium. Genome Biol. Evol. 2013, 5, 1524–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sletvold, H.; Johnsen, P.J.; Wikmark, O.G.; Simonsen, G.S.; Sundsfjord, A.; Nielsen, K.M. Tn1546 is part of a larger plasmid-encoded genetic unit horizontally disseminated among clonal Enterococcus faecium lineages. J. Antimicrob. Chemother. 2010, 65, 1894–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halvorsen, E.M.; Williams, J.J.; Bhimani, A.J.; Billings, E.A.; Hergenrother, P.J. Txe, an endoribonuclease of the enterococcal Axe-Txe toxin-antitoxin system, cleaves mRNA and inhibits protein synthesis. Microbiology 2011, 157, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szakacs, T.A.; Kalan, L.; McConnell, M.J.; Eshaghi, A.; Shahinas, D.; McGeer, A.; Wright, G.D.; Low, D.E.; Patel, S.N. Outbreak of vancomycin-susceptible Enterococcus faecium containing the wild-type vanA gene. J. Clin. Microbiol. 2014, 52, 1682–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sletvold, H.; Johnsen, P.J.; Simonsen, G.S.; Aasnæs, B.; Sundsfjord, A.; Nielsen, K.M. Comparative DNA analysis of two vanA plasmids from Enterococcus faecium strains isolated from poultry and a poultry farmer in Norway. Antimicrob. Agents Chemother. 2007, 51, 736–739. [Google Scholar] [CrossRef] [Green Version]
- Sletvold, H.; Johnsen, P.J.; Hamre, I.; Simonsen, G.S.; Sundsfjord, A.; Nielsen, K.M. Complete sequence of Enterococcus faecium pVEF3 and the detection of an ω-ε-ζ toxin-antitoxin module and an ABC transporter. Plasmid 2008, 60, 75–85. [Google Scholar] [CrossRef]
- Guardabassi, L.; Perichon, B.; Van Heijenoort, J.; Blanot, D.; Courvalin, P. Glycopeptide resistance vanA operons in Paenibacillus strains isolated from soil. Antimicrob. Agents Chemother. 2005, 49, 4227–4233. [Google Scholar] [CrossRef] [Green Version]
- Fraimow, H.; Knob, C.; Herrero, I.A.; Patel, R. Putative VanRS-like two-component regulatory system associated with the inducible glycopeptide resistance cluster of Paenibacillus popilliae. Antimicrob. Agents Chemother. 2005, 49, 2625–2633. [Google Scholar] [CrossRef] [Green Version]
- Ligozzi, M.; Lo Cascio, G.; Fontana, R. vanA gene cluster in a vancomycin-resistant clinical isolate of Bacillus circulans. Antimicrob. Agents Chemother. 1998, 42, 2055–2059. [Google Scholar] [CrossRef]
- Knight, D.R.; Androga, G.O.; Ballard, S.A.; Howden, B.P.; Riley, T.V. A phenotypically silent vanB2 operon carried on a Tn1549-like element in Clostridium difficile. Msphere 2016, 1, e00177-16. [Google Scholar] [CrossRef] [Green Version]
- Kruse, T.; Levisson, M.; de Vos, W.M.; Smidt, H. vanI: A novel d-Ala-d-Lac vancomycin resistance gene cluster found in Desulfitobacterium hafniense. Microb. Biotechnol. 2014, 7, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Sekiguchi, Y. Cultivation of uncultured Chloroflexi subphyla: Significance and ecophysiology of formerly uncultured Chloroflexi “subphylum I” with natural and biotechnological relevance. Microbes Environ. 2009, 24, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.J.; Xia, F.; Overbeek, R.A.; Olsen, G.J. Genomes of the class Erysipelotrichia clarify the firmicute origin of the class Mollicutes. Int. J. Syst. Evol. Microbiol. 2013, 63, 2727–2741. [Google Scholar] [CrossRef]
- Yabe, S.; Sakai, Y.; Abe, K.; Yokota, A. Diversity of Ktedonobacteria with actinomycetes-like morphology in terrestrial environments. Microbes Environ. 2017, 32, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, 327–331. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, L.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Gan, R.; Zhang, F.; Ren, C.; Yu, L.; Si, Y.; Huang, Z. PHISDetector: A tool to detect diverse in silico phage–host interaction signals for virome studies. Genom. Proteom. Bioinform. 2022, in press. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Sitnikova, T.; Rzhetsky, A.; Nei, M. Interior-branch and bootstrap tests of phylogenetic trees. Mol. Biol. Evol. 1995, 12, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Battistuzzi, F.U.; Feijao, A.; Hedges, S.B. A genomic timescale of prokaryote evolution: Insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol. Biol. 2004, 4, 44. [Google Scholar] [CrossRef] [Green Version]
- Schleifer, K.H. Phylum XIII. Firmicutes Gibbons and Murray 1978, 5 (Firmacutes [sic] Gibbons and Murray 1978, 5). In Bergey’s Manual® of Systematic Bacteriology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 3, p. 1450. [Google Scholar]
- Ogata, Y.; Sakamoto, M.; Ohkuma, M.; Hattori, M.; Suda, W. Complete genome sequence of Longicatena caecimuris strain 3BBH23, isolated from healthy Japanese feces. Microbiol. Resour. Announc. 2021, 10, e00282-21. [Google Scholar] [CrossRef]
- Reynolds, P.E.; Ambur, O.H.; Casadewall, B.; Courvalin, P. The VanYD d,d-carboxypeptidase of Enterococcus faecium BM4339 is a penicillin-binding protein. Microbiology 2001, 147, 2571–2578. [Google Scholar] [CrossRef] [Green Version]
- Cavaletti, L.; Monciardini, P.; Bamonte, R.; Schumann, P.; Ronde, M.; Sosio, M.; Donadio, S. New lineage of filamentous, spore-forming, Gram-positive bacteria from soil. Appl. Environ. Microbiol. 2006, 72, 4360–4369. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P.; Gourbeyre, E.; Varani, A.; Ton-Hoang, B.; Chandler, M. Everyman’s guide to bacterial insertion sequences. Microbiol. Spectr. 2015, 3, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Maniloff, J.; Ackermann, H.W. Taxonomy of bacterial viruses: Establishment of tailed virus genera and the order Caudovirales. Arch. Virol. 1998, 143, 2051–2063. [Google Scholar] [CrossRef]
- Fontana, R.; Ligozzi, M.; Pedrotti, C.; Padovani, E.M.; Cornaglia, G. Vancomycin-resistant Bacillus circulans carrying the vanA gene responsible for vancomycin resistance in enterococci. Eur. J. Clin. Microbiol. Infect. Dis. 1997, 16, 473–474. [Google Scholar] [CrossRef]
- Aminov, R.I.; Mackie, R.I. Evolution and ecology of antibiotic resistance genes. FEMS Microbiol. Lett. 2007, 271, 147–161. [Google Scholar] [CrossRef]
- Heaton, M.P.; Discotto, L.F.; Pucci, M.J.; Handwerger, S. Mobilization of vancomycin resistance by transposon-mediated fusion of a VanA plasmid with an Enterococcus faecium sex pheromone-response plasmid. Gene 1996, 171, 9–17. [Google Scholar] [CrossRef]
- Krumholz, L.R.; Bryant, M.P. Clostridium pfennigii sp. nov. uses methoxyl groups of monobenzenoids and produces butyrate. Int. J. Syst. Bacteriol. 1985, 35, 454–456. [Google Scholar] [CrossRef]
- Šul’ák, M.; Sikorová, L.; Jankuvová, J.; Javorský, P.; Pristaš, P. Variability of Actinobacteria, a minor component of rumen microflora. Folia Microbiol. 2012, 57, 351–353. [Google Scholar] [CrossRef]
- Wambui, J.; Stevens, M.J.A.; Cernela, N.; Stephan, R. Unraveling the genotypic and phenotypic diversity of the psychrophilic Clostridium estertheticum complex, a meat spoilage agent. Front. Microbiol. 2022, 13, 856810. [Google Scholar] [CrossRef]
- Kim, Y.B.; Kim, J.Y.; Kim, J.; Song, H.S.; Whon, T.W.; Lee, S.H.; Yoo, S.R.; Myoung, J.; Son, H.S.; Roh, S.W. Aminipila terrae sp. nov., a strictly anaerobic bacterium isolated from river sediment. Arch. Microbiol. 2021, 203, 3163–3169. [Google Scholar] [CrossRef]
- Passari, A.K.; Leo, V.V.; Chandra, P.; Kumar, B.; Nayak, C.; Hashem, A.; Abd Allah, E.F.; Alqarawi, A.A.; Singh, B.P. Bioprospection of actinobacteria derived from freshwater sediments for their potential to produce antimicrobial compounds. Microb. Cell Fact. 2018, 17, 68. [Google Scholar] [CrossRef]
- Nakahara, N.; Nobu, M.K.; Takaki, Y.; Miyazaki, M.; Tasumi, E.; Sakai, S.; Ogawara, M.; Yoshida, N.; Tamaki, H.; Yamanaka, Y.; et al. Aggregatilinea lenta gen. nov., sp. nov., a slow-growing, facultatively anaerobic bacterium isolated from subseafloor sediment, and proposal of the new order Aggregatilineales ord. nov. within the class Anaerolineae of the phylum Chloroflexi. Int. J. Syst. Evol. Microbiol. 2019, 69, 1185–1194. [Google Scholar] [CrossRef]
- Biddle, A.S.; Leschine, S.; Huntemann, M.; Han, J.; Chen, A.; Kyrpides, N.; Markowitz, V.; Palaniappan, K.; Ivanova, N.; Mikhailova, N.; et al. The complete genome sequence of Clostridium indolis DSM 755T. Stand. Genom. Sci. 2015, 9, 1089–1104. [Google Scholar] [CrossRef] [Green Version]
- Mechichi, T.; Labat, M.; Patel, B.K.C.; Woo, T.H.; Thomas, P.; Garcia, J. New aromatic O-demethylating homoacetogen from an olive mill wastewater treatment digester. Int. J. Syst. Bacteriol. 1997, 49, 1201–1209. [Google Scholar] [CrossRef] [Green Version]
- Danylec, N.; Stoll, D.A.; Huch, M. Draft genome sequences of type strains of Gordonibacter faecihominis, Paraeggerthella hongkongensis, Parvibacter caecicola, Slackia equolifaciens, Slackia faecicanis, and Slackia isoflavoniconvertens. Microbiol. Resour. Announc. 2019, 36, e01532-18. [Google Scholar] [CrossRef] [Green Version]
- Clavel, T.; Charrier, C.; Braune, A.; Wenning, M.; Blaut, M.; Haller, D. Isolation of bacteria from the ileal mucosa of TNFdeltaARE mice and description of Enterorhabdus mucosicola gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 1805–1812. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classes: | Genera: | Bacterial Strains Carrying van Genes: |
|---|---|---|
| Anaerolineae | Aggregatilinea (Al.) | Aggregatilinea lenta MO-CFX2 |
| Bacilli | Alicyclobacillus (Alic.) | Alicyclobacillus shizuokensis NBRC 103103 |
| Alkalihalobacillus | Alkalihalobacillus sp. EGI L200015 | |
| Anoxybacillus (Anox.) | Anoxybacillus sediminis PCH 117 | |
| Bacillus | Bacillus sp. JCA | |
| Brevibacillus (Bba.) | Brevibacillus halotolerans J5TS2; Brevibacillus laterosporus ACRRF; Bba. laterosporus G25-128; Bba. laterosporus OSY-I1; Bba. laterosporus SAM19; Bba. laterosporus B9 (plasmid); Brevibacillus sp. MCWH; Brevibacillus sp. NL20B1; Brevibacillus sp. SKDU10; Brevibacillus sp. VP | |
| Cohnella (Coh.) | Cohnella zeiphila CBP-2801 | |
| Neobacillus (Nba.) | Neobacillus jeddahensis MGYG-HGUT-01469; Nba. jeddahensis JCE | |
| Paenibacillus (Pba.) | Paenibacillus apiarius MW-14; Paenibacillus dendritiformis J6TS7; Paenibacillus elgii MER 157; Pba. elgii SWL-W8; Paenibacillus sp. DXL2; Paenibacillus hemerocallicola KCTC 33185; Paenibacillus jilunlii DSM 23019; Paenibacillus macerans 3CT49; Pba. macerans DSM 24746; Pba. macerans HMSSN-036; Pba. macerans FSL R5-0527; Pba. macerans MER 50.2; Paenibacillus mesophilus SYSU K30004; Paenibacillus nasutitermitis CGMCC 1.15178; Paenibacillus oralis KCOM 3021; Paenibacillus physcomitrellae CGMCC 1.15044; Pba. physcomitrellae XB; Paenibacillus piri MS74; Paenibacillus sonchi IIRRBNF1; Pba. sonchi X19-5; Paenibacillus sp. cl123; Paenibacillus sp. SM 69; Paenibacillus sp. UNCCL117; Paenibacillus sp. YIM B00362; Paenibacillus sp. YIM B00624; Paenibacillus thiaminolyticus BO5; Paenibacillus zanthoxyli JH29; Paenibacillus flagellatus DXL2 | |
| Thermoactinomyces | Thermoactinomyces sp. CICC 10735 | |
| Thermobacillus (Tba.) | Thermobacillus xylanilyticus XE | |
| Weizmannia (W.) | Weizmannia ginsengihumi Gsoil 114 | |
| Clostridia | Dubious genus | [Clostridium] indolis DSM 755; [Clostridium] methoxybenzovorans SR3; [Clostridium] scindens MSK.5.24; [Clostridium] symbiosum MSK.7.21 |
| Unidentified genus | Lachnospiraceae bacterium M18-1 | |
| Aminipila (Am.) | Aminipila terrae CBA3637 | |
| Anaerobacterium (An.) | Anaerobacterium chartisolvens DSM 27016 | |
| Bariatricus (Bar.) | Bariatricus massiliensis DFI.1.181 | |
| Beduini (Bed.) | Beduini massiliensis GM1 | |
| Blautia (Bl.) | Blautia producta ER3; Blautia pseudococcoides SCSK; Blautia sp. NBRC 113351 | |
| Candidatus Formimonas | Candidatus Formimonas warabiya DCMF | |
| Clostridium (Cli.) | Clostridium argentinense 113/29; Clostridium sp. M3/9; Clostridium tagluense CS002 | |
| Cuneatibacter | Cuneatibacter sp. NSJ-177 | |
| Extibacter (Ext.) | Extibacter muris DSM 28560 | |
| Lachnotalea | Lachnotalea sp. AF33-28 | |
| Oxobacter (Ox.) | Oxobacter pfennigii DSM 3222 | |
| Ruminococcus (Rcc.) | Ruminococcus gauvreauii DSM 19829; Ruminococcus gauvreauii MGYG-HGUT-01690 | |
| Ruthenibacterium (Rba.) | Ruthenibacterium lactatiformans 668 | |
| Erysipelotrichia | Unidentified genus | Erysipelotrichaceae bacterium 66202529 |
| Longicatena (Lct.) | Longicatena caecimuris 3BBH23 | |
| Ktedonobacteria | Unidentified genus | Ktedonobacteria bacterium brp13 |
| Dictyobacter (Db.) | Dictyobacter alpinus Uno16 | |
| Ktedonobacter (Kb.) | Ktedonobacter racemifer DSM 44963; Ktedonobacter sp. SOSP1-52 | |
| Ktedonosporobacter (Ksb.) | Ktedonosporobacter rubrisoli SCAWS-G2 | |
| Reticulobacter (Rb.) | Reticulibacter mediterranei 150040 | |
| Thermosporothrix | Thermosporothrix sp. COM3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yushchuk, O.; Binda, E.; Fedorenko, V.; Marinelli, F. Occurrence of vanHAX and Related Genes beyond the Actinobacteria Phylum. Genes 2022, 13, 1960. https://doi.org/10.3390/genes13111960
Yushchuk O, Binda E, Fedorenko V, Marinelli F. Occurrence of vanHAX and Related Genes beyond the Actinobacteria Phylum. Genes. 2022; 13(11):1960. https://doi.org/10.3390/genes13111960
Chicago/Turabian StyleYushchuk, Oleksandr, Elisa Binda, Victor Fedorenko, and Flavia Marinelli. 2022. "Occurrence of vanHAX and Related Genes beyond the Actinobacteria Phylum" Genes 13, no. 11: 1960. https://doi.org/10.3390/genes13111960