
Gene–microRNA Network Analysis Identified Seven Hub Genes in Association with Progression and Prognosis in Non-Small Cell Lung Cancer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquirement
2.2. Identification of DEG
2.3. Gene Ontology and Pathway Enrichment Analysis
2.4. Genomic Specificity Analysis
2.5. Regulatory Network Analysis
2.6. Identification of Differentially Expressed miRNA
2.7. Target Gene Analysis of miRNA
2.8. miRNA Gene Regulatory Network Analysis
2.9. Verification Analysis by Independent Dataset
3. Results
3.1. Differentially Expressed Genes in NSCLC
3.2. Functional Enrichment Analysis of DEG
3.3. Genomic Specificity Analysis by ShinyGO
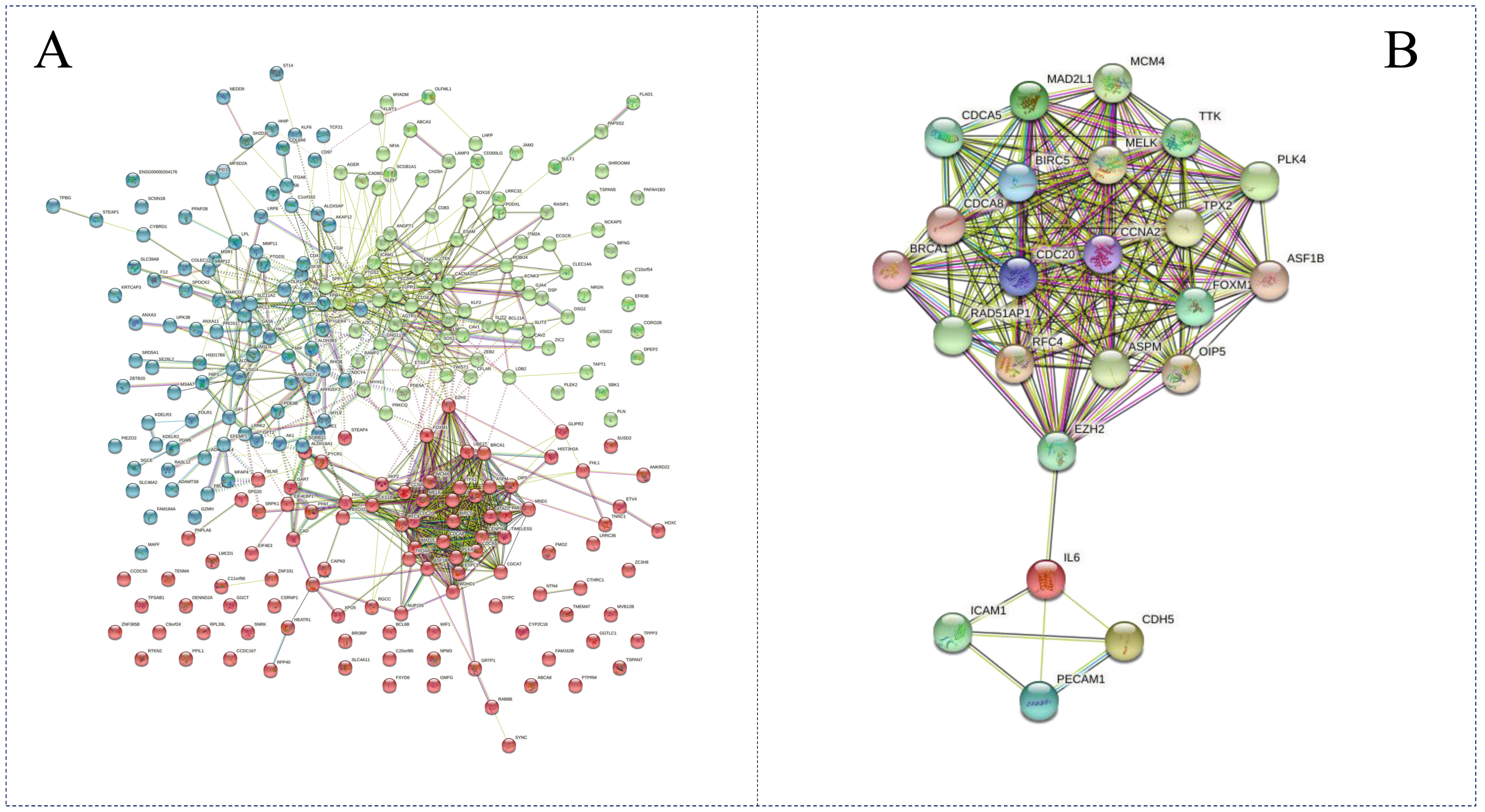
3.4. PPI Visualization of DEGs
3.5. Identification of DEM
3.6. DEG–DEM Interaction Prediction
3.7. DEM Target Enrichment Analysis
3.8. Network Analysis of the DEG-DEM Interaction
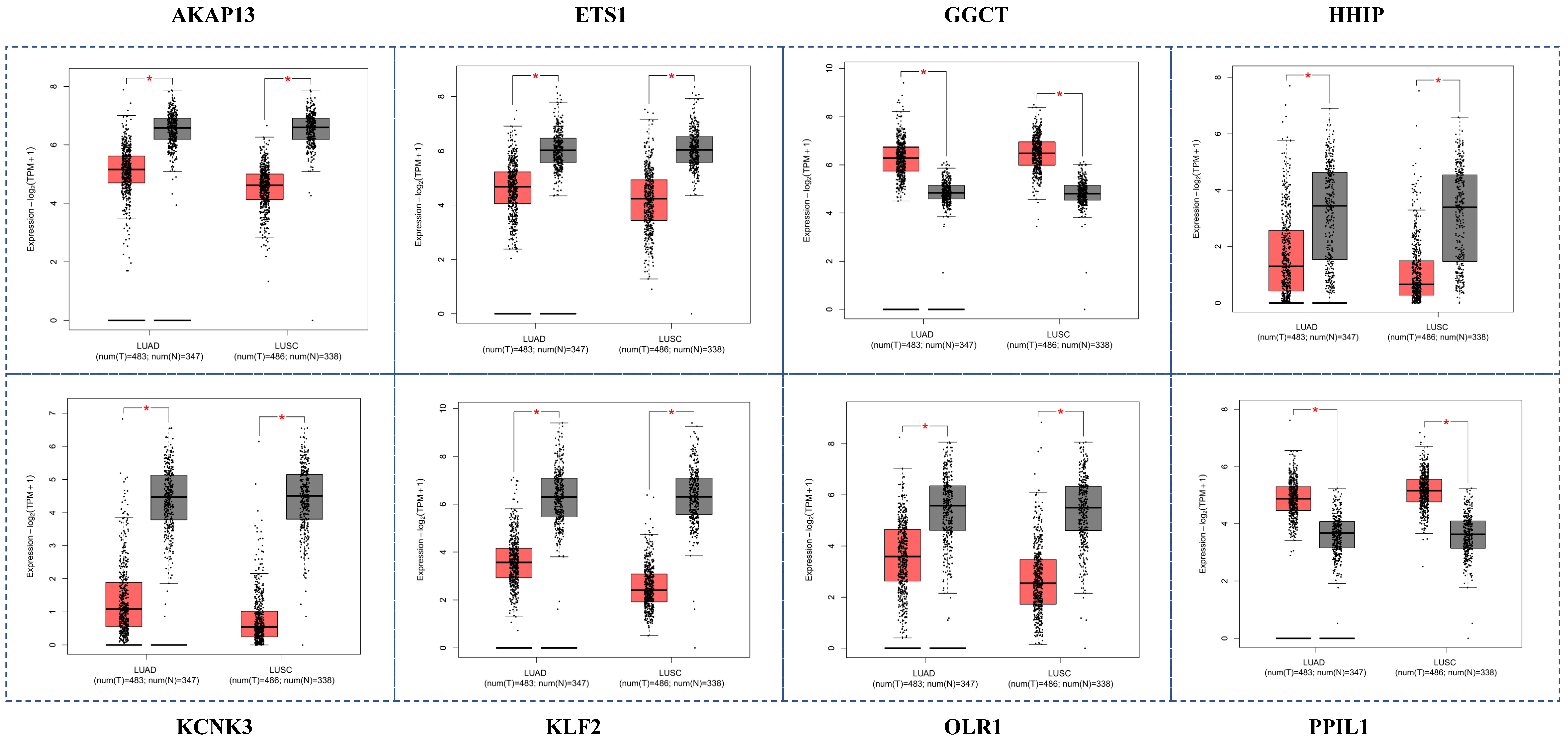
3.9. Validation of Hub Genes Using GEPIA
3.10. Survival Analysis of Hub Genes Using KMplot
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, S.Y.; Pan, Y.; Mao, Y.Y.; Chen, Y.; He, Y.Y. Current progress and mechanisms of bone metastasis in lung cancer: A narrative review. Transl. Lung Cancer R 2021, 10, 439–451. [Google Scholar] [CrossRef]
- Hanna, N.H.; Robinson, A.G.; Temin, S.; Baker, S., Jr.; Brahmer, J.R.; Ellis, P.M.; Gaspar, L.E.; Haddad, R.Y.; Hesketh, P.J.; Jain, D. Therapy for stage IV non–small-cell lung cancer with driver alterations: ASCO and OH (CCO) joint guideline update. J. Clin. Oncol. 2021, 39, 1040–1091. [Google Scholar] [CrossRef] [PubMed]
- Chhatre, S.; Vachani, A.; Allison, R.R.; Jayadevappa, R. Survival Outcomes with Photodynamic Therapy, Chemotherapy and Radiation in Patients with Stage III or Stage IV Non-Small Cell Lung Cancer. Cancers 2021, 13, 803. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, M.; Zeng, X.; Wan, A.T.-Y.; Tsui, S.K.-W. In silico analysis of proteins and microRNAs related to human African trypanosomiasis in tsetse fly. Comput. Biol. Chem. 2020, 88, 107347. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wan, A.T.Y.; Liu, X.Y.; Xiong, Q.; Liu, Z.G.; Tsui, S.K.W. Identification, functional annotation and stability analysis of miRNA in Dermatophagoides pteronyssinus. Allergy 2019, 75, 1237–1240. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.G.; Zhao, Y.W.; Wang, F.L.; Chen, Y.; Fei, X.B. miR-224 enhances invasion and metastasis by targeting HOXD10 in non-small cell lung cancer cells. Oncol. Lett. 2018, 15, 7069–7075. [Google Scholar] [CrossRef]
- Gao, Z.J.; Yuan, W.D.; Yuan, J.Q.; Yuan, K.; Wang, Y. miR-486-5p functions as an oncogene by targeting PTEN in non-small cell lung cancer. Pathol. Res. Pract. 2018, 214, 700–705. [Google Scholar] [CrossRef]
- Xiong, R.; Sun, X.X.; Wu, H.R.; Xu, G.W.; Wang, G.X.; Sun, X.H.; Xu, M.Q.; Xie, M.R. Mechanism research of miR-34a regulates Axl in non-small-cell lung cancer with gefitinib-acquired resistance. Thorac. Cancer 2020, 11, 156–165. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.L.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W.Z. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.M.; Yao, R.A. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets (vol 49, pg D605, 2021). Nucleic Acids Res. 2021, 49, 10800. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.D.; Cui, S.D.; Huang, Y.X.; Tang, Y.; Xu, J.T.; Bao, J.Y.; Li, Y.L.; Wen, J.; Zuo, H.L.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Kehl, T.; Kern, F.; Backes, C.; Fehlmann, T.; Stockel, D.; Meese, E.; Lenhof, H.P.; Keller, A. miRPathDB 2.0: A novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020, 48, D142–D147. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Li, C.W.; Tang, Z.F.; Zhang, W.J.; Ye, Z.C.; Liu, F.L. GEPIA2021: Integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res. 2021, 49, W242–W246. [Google Scholar] [CrossRef]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet. Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Lei, X.; Wang, F.; Wu, F.-X.; Zhang, A.; Pedrycz, W. Protein complex identification through Markov clustering with firefly algorithm on dynamic protein–protein interaction networks. Inf. Sci. 2016, 329, 303–316. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Zhao, B.; Han, H.; Chen, J.; Zhang, Z.; Li, S.; Fang, F.; Zheng, Q.; Ma, Y.; Zhang, J.; Wu, N.; et al. MicroRNA let-7c inhibits migration and invasion of human non-small cell lung cancer by targeting ITGB3 and MAP4K3. Cancer Lett. 2014, 342, 43–51. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, L.; Hu, Q.; Xia, J.; Sun, J.; Wang, X.; Xiong, H.; Gurbani, D.; Li, L.; Liu, Y. MicroRNA-218 functions as a tumor suppressor in lung cancer by targeting IL-6/STAT3 and negatively correlates with poor prognosis. Mol. Cancer 2017, 16, 141. [Google Scholar] [CrossRef]
- Huang, H.; Huang, J.Y.; Yao, J.; Li, N.; Yang, Z.Z. miR-125a regulates HAS1 and inhibits the proliferation, invasion and metastasis by targeting STAT3 in non-small cell lung cancer cells. J. Cell Biochem. 2020, 121, 3197–3207. [Google Scholar] [CrossRef]
- Ba, Z.; Zhou, Y.; Yang, Z.; Xu, J.; Zhang, X. miR-324-5p upregulation potentiates resistance to cisplatin by targeting FBXO11 signalling in non-small cell lung cancer cells. J. Biochem. 2019, 166, 517–527. [Google Scholar] [CrossRef]
- Wang, H.L.; Li, K.Z.; Li, J.L.; Hu, B.L. Prognostic value of AKAP13 methylation and expression in lung squamous cell carcinoma. Biomark. Med. 2020, 14, 503–512. [Google Scholar] [CrossRef]
- Leithner, K.; Hirschmugl, B.; Li, Y.J.; Tang, B.; Papp, R.; Nagaraj, C.; Stacher, E.; Stiegler, P.; Lindenmann, J.; Olschewski, A.; et al. TASK-1 Regulates Apoptosis and Proliferation in a Subset of Non-Small Cell Lung Cancers. PLoS ONE 2016, 11, e0157453. [Google Scholar] [CrossRef]
- Jiang, W.B.; Xu, X.Q.; Deng, S.L.; Luo, J.; Xu, H.; Wang, C.; Sun, T.T.; Lei, G.Q.; Zhang, F.L.; Yang, C.; et al. Methylation of kruppel-like factor 2 (KLF2) associates with its expression and non-small cell lung cancer progression. Am. J. Trans. Res. 2017, 9, 2024–2037. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | Gene Number | Count | Percent (%) | p-Value |
|---|---|---|---|---|---|
| GAD_DISEASE_CLASS | CANCER |  | 86 | 31.2 | 1.90 × 10−5 |
| ---Lung Cancer | 22 | 8 | 2.00 × 10−3 | ||
| ---Cervical cancer | 7 | 2.5 | 4.50 × 10−3 | ||
| ---Breast Cancer | 21 | 7.6 | 4.90 × 10−3 | ||
| ---Prostate cancer | 18 | 6.5 | 9.50 × 10−3 | ||
| GAD_DISEASE_CLASS | RENAL |  | 46 | 16.7 | 7.00 × 10−5 |
| GAD_DISEASE_CLASS | UNKNOWN |  | 47 | 17 | 6.80 × 10−4 |
| GAD_DISEASE_CLASS | CARDIOVASCULAR |  | 100 | 36.2 | 7.40 × 10−4 |
| GAD_DISEASE_CLASS | REPRODUCTION |  | 31 | 11.2 | 1.70 × 10−3 |
| GAD_DISEASE_CLASS | IMMUNE |  | 72 | 26.1 | 3.50 × 10−3 |
| GAD_DISEASE_CLASS | DEVELOPMENTAL |  | 41 | 14.9 | 5.50 × 10−3 |
| GAD_DISEASE_CLASS | OTHER |  | 42 | 15.2 | 8.90 × 10−3 |
| No. | DEM (miRNA) | DEG (Gene) | Protein ID | Binding Energy |
|---|---|---|---|---|
| (kcal/mol) | ||||
| 1 | hsa-miR-484 | KDELR2 | NM_006854 | −33.4 |
| 2 | hsa-miR-574-5p | STEAP4 | NM_001205316 | −33 |
| 3 | hsa-miR-331-3p | LMCD1 | NM_001278233 | −32.6 |
| 4 | hsa-miR-324-5p | CFLAR | NM_001308043 | −31.8 |
| 5 | hsa-miR-423-3p | LRRC32 | NM_001128922 | −31.7 |
| 6 | hsa-miR-423-5p | GPI | NM_001289789 | −31.2 |
| 7 | hsa-miR-125a-5p | COLEC12 | NM_130386 | −31 |
| 8 | hsa-miR-296-5p | GRK5 | NM_005308 | −30.6 |
| 9 | hsa-miR-361-5p | ARHGEF3 | NM_001289698 | −30.5 |
| 10 | hsa-miR-574-3p | GLIPR2 | NM_001287014 | −29.5 |
| 11 | hsa-miR-107 | CCDC50 | NM_174908 | −27.7 |
| 12 | hsa-miR-342-3p | SLC11A1 | NM_000578 | −27.4 |
| 13 | hsa-miR-361-3p | CXCL12 | NM_000609 | −27.1 |
| 14 | hsa-miR-199b-5p | MAFF | NM_012323 | −26.4 |
| 15 | hsa-miR-199a-5p | MAFF | NM_012323 | −26.3 |
| 16 | hsa-miR-146b-5p | TIMELESS | NM_001330295 | −25.7 |
| 17 | hsa-miR-532-5p | CSRNP1 | NM_001320560 | −25.6 |
| miRNA | Database | Pathway | Targeted Gene Hits | Expected Hits | p-Value |
|---|---|---|---|---|---|
| hsa-miR-125a-5p | WikiPathways | Non-small cell lung cancer | 9 | 0.997 | 1.06 × 10−5 |
| hsa-miR-331-3p | WikiPathways | Non-small cell lung cancer | 3 | 0.156 | 0.002 |
| hsa-miR-199a-5p | WikiPathways | Non-small cell lung cancer | 4 | 0.705 | 0.014 |
| hsa-miR-423-5p | WikiPathways | Non-small cell lung cancer | 41 | 28.09 | 0.023 |
| hsa-miR-324-5p | KEGG | Non-small cell lung cancer | 23 | 13.86 | 0.046 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Wang, H.; Zhao, Z.; Jin, Y.; Zhang, Z.; Tan, J.; Hu, F. Gene–microRNA Network Analysis Identified Seven Hub Genes in Association with Progression and Prognosis in Non-Small Cell Lung Cancer. Genes 2022, 13, 1480. https://doi.org/10.3390/genes13081480
Yang Z, Wang H, Zhao Z, Jin Y, Zhang Z, Tan J, Hu F. Gene–microRNA Network Analysis Identified Seven Hub Genes in Association with Progression and Prognosis in Non-Small Cell Lung Cancer. Genes. 2022; 13(8):1480. https://doi.org/10.3390/genes13081480
Chicago/Turabian StyleYang, Zhiyuan, Hongqi Wang, Zixin Zhao, Yunlong Jin, Zhengnan Zhang, Jiayi Tan, and Fuyan Hu. 2022. "Gene–microRNA Network Analysis Identified Seven Hub Genes in Association with Progression and Prognosis in Non-Small Cell Lung Cancer" Genes 13, no. 8: 1480. https://doi.org/10.3390/genes13081480