
New Insights into the Regulatory Role of Ferroptosis in Ankylosing Spondylitis via Consensus Clustering of Ferroptosis-Related Genes and Weighted Gene Co-Expression Network Analysis


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
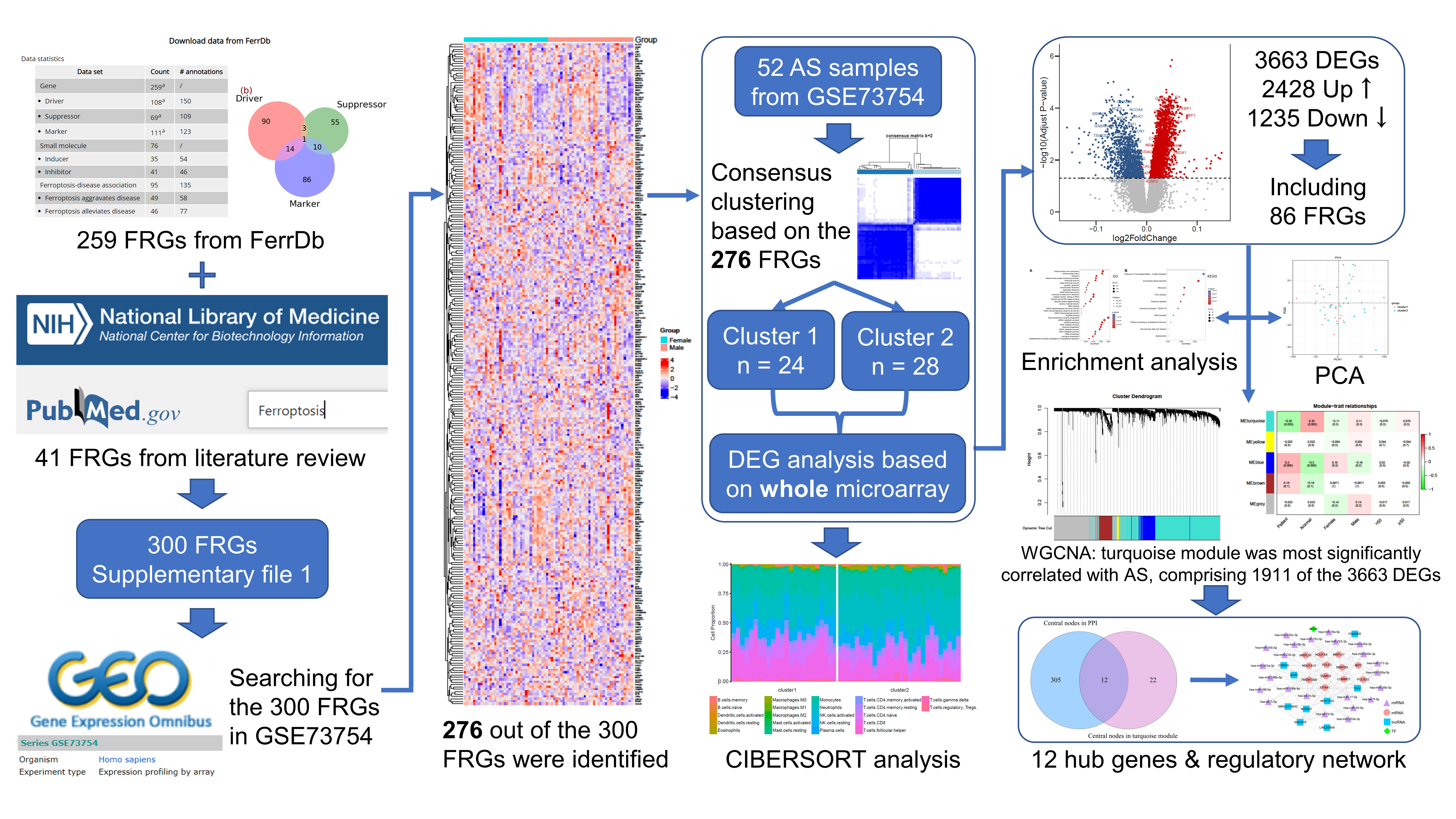
2.1. Data Collection
2.2. Ferroptosis-Related Genes
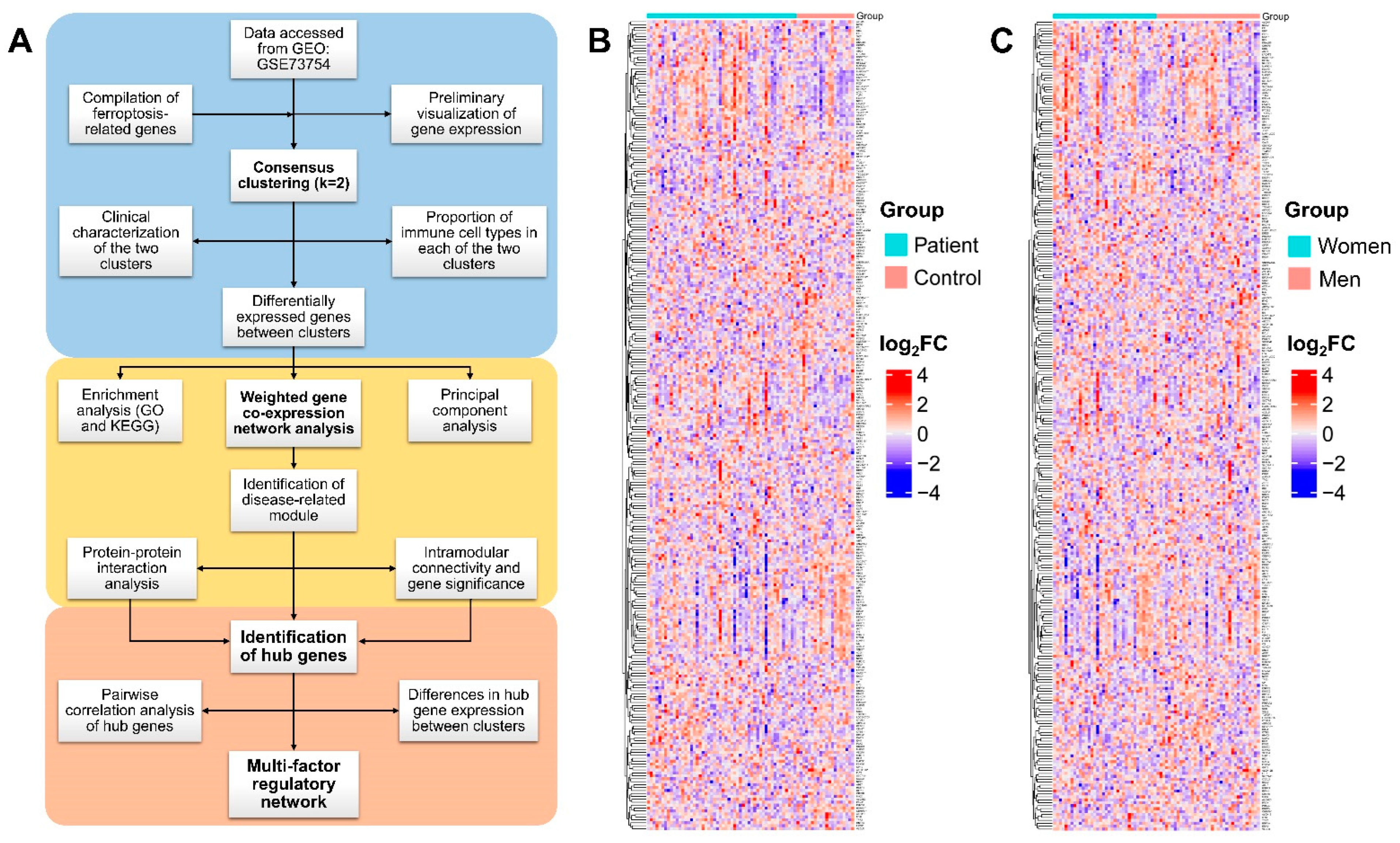
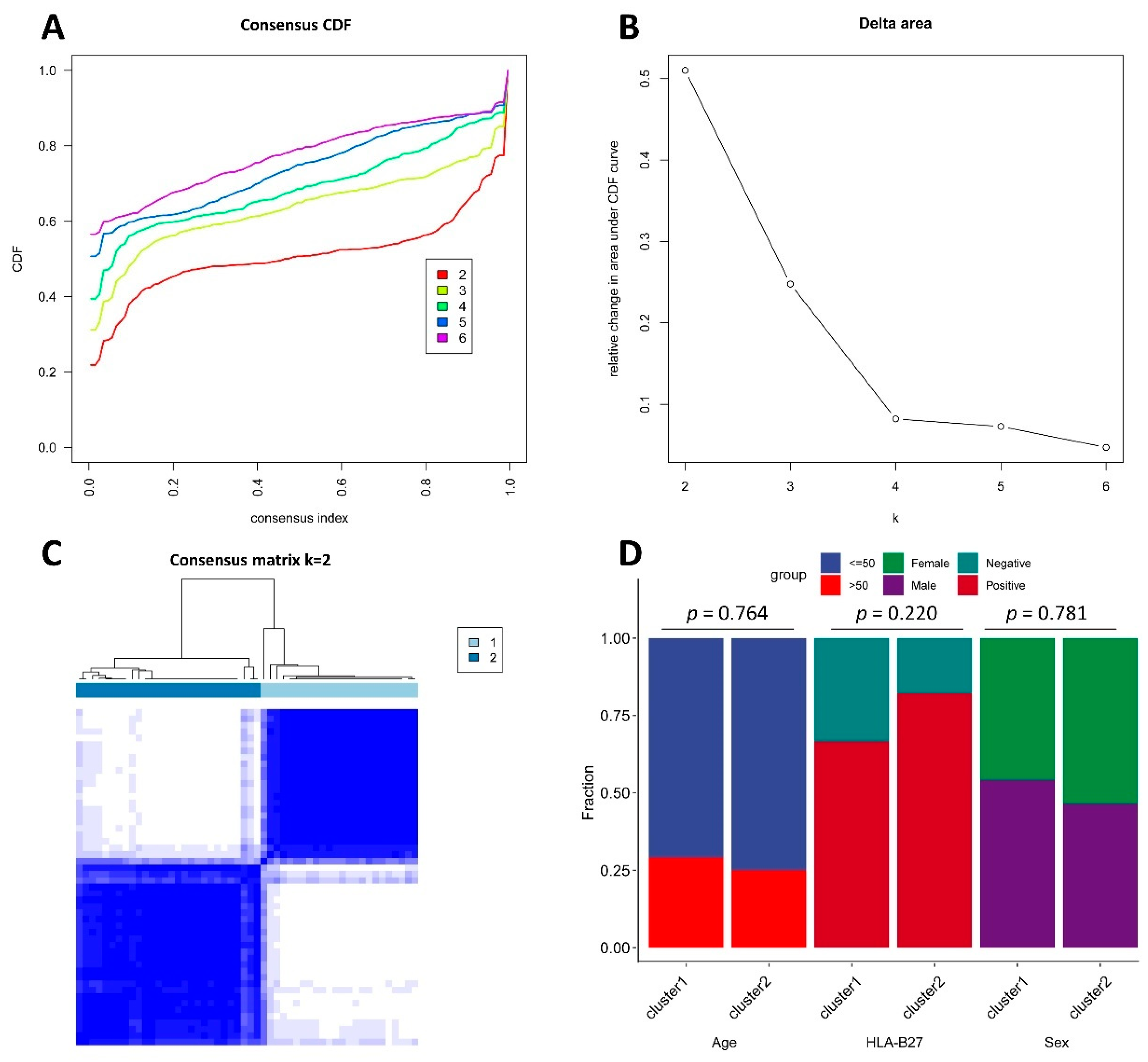
2.3. Consensus Clustering
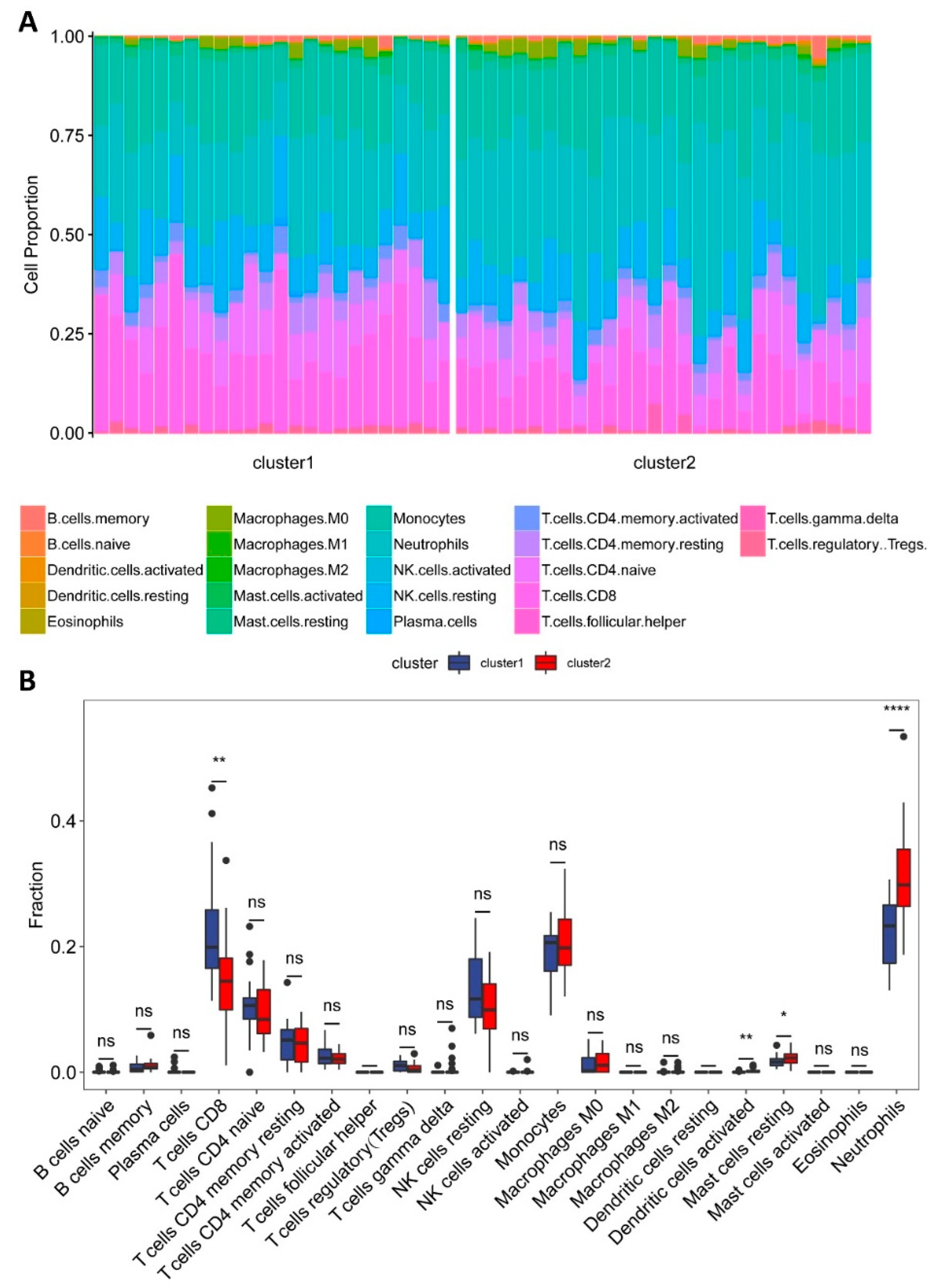
2.4. Estimation of Immune Cell Type Fractions
2.5. Screening for Differentially Expressed Genes
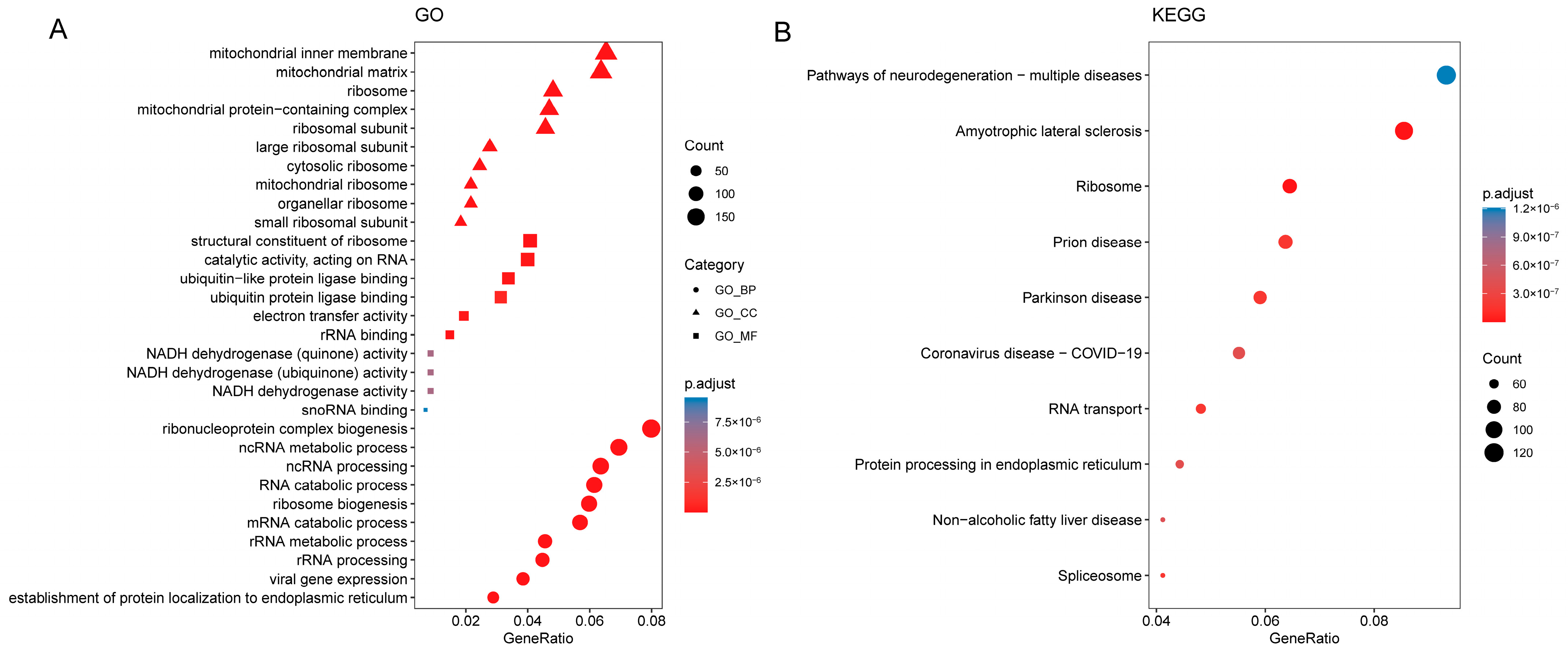
2.6. Gene Ontology and Pathway Enrichment Analysis
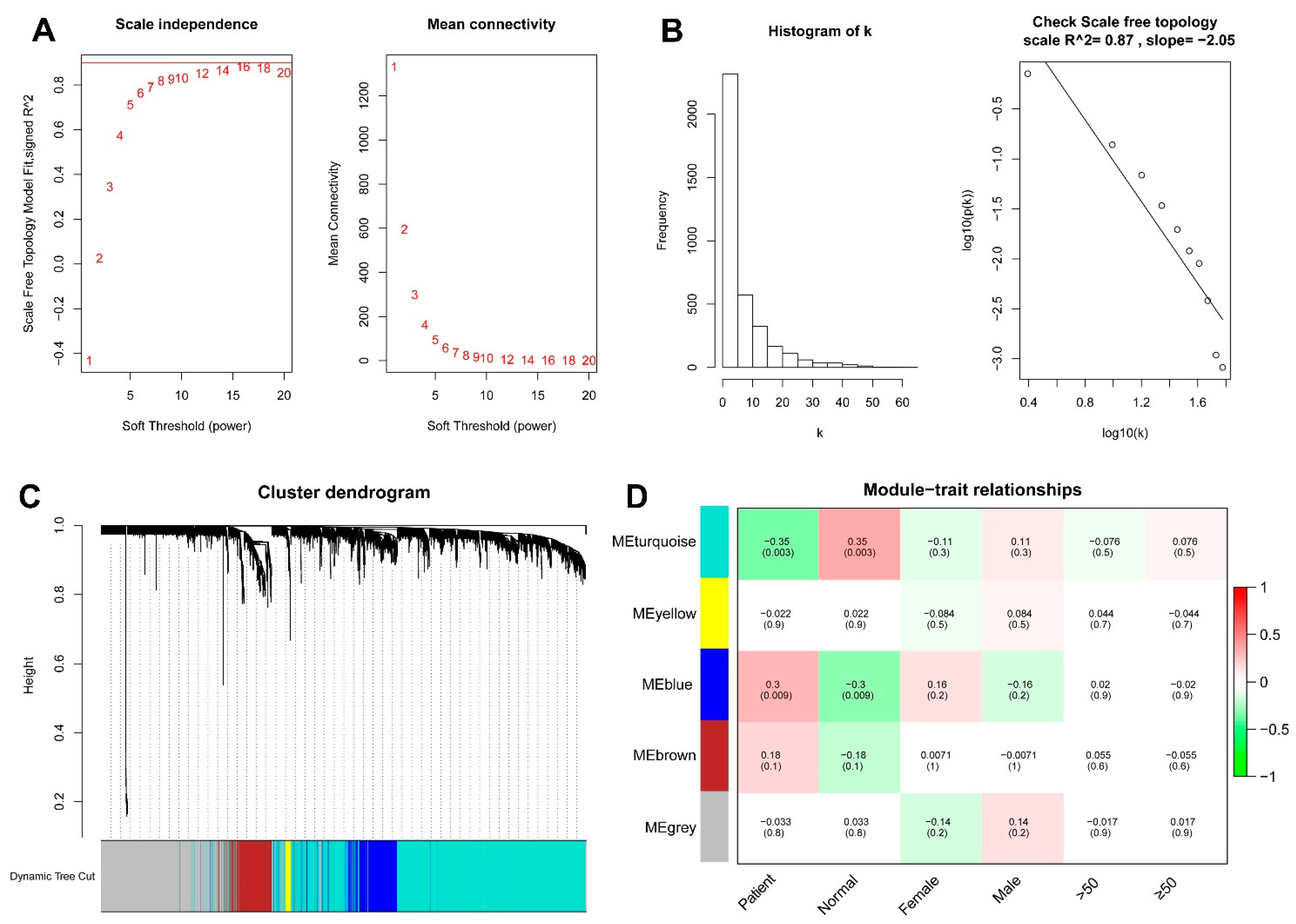
2.7. Weighted Gene Co-Expression Network Analysis
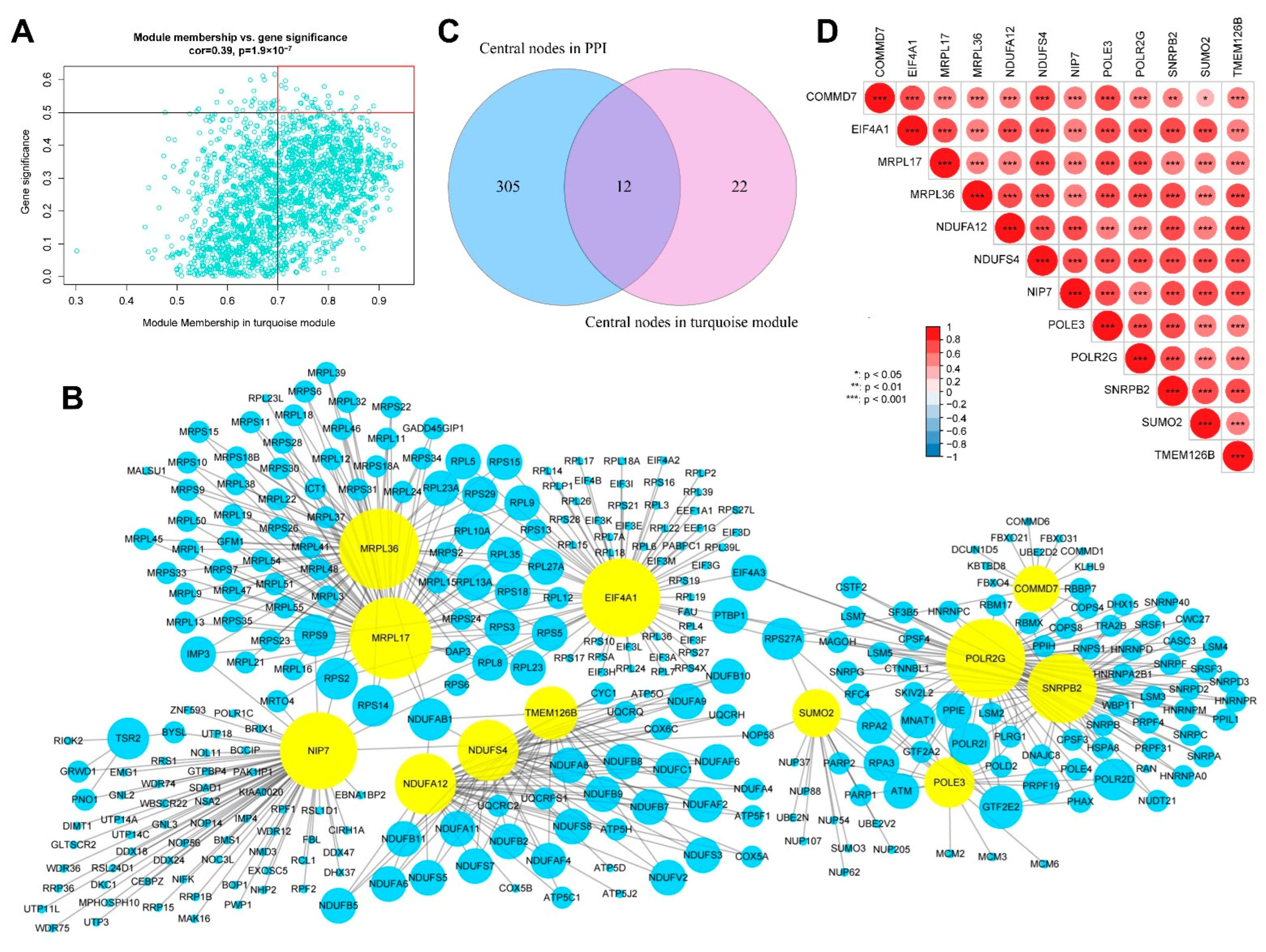
2.8. Identification of Hub Genes in AS
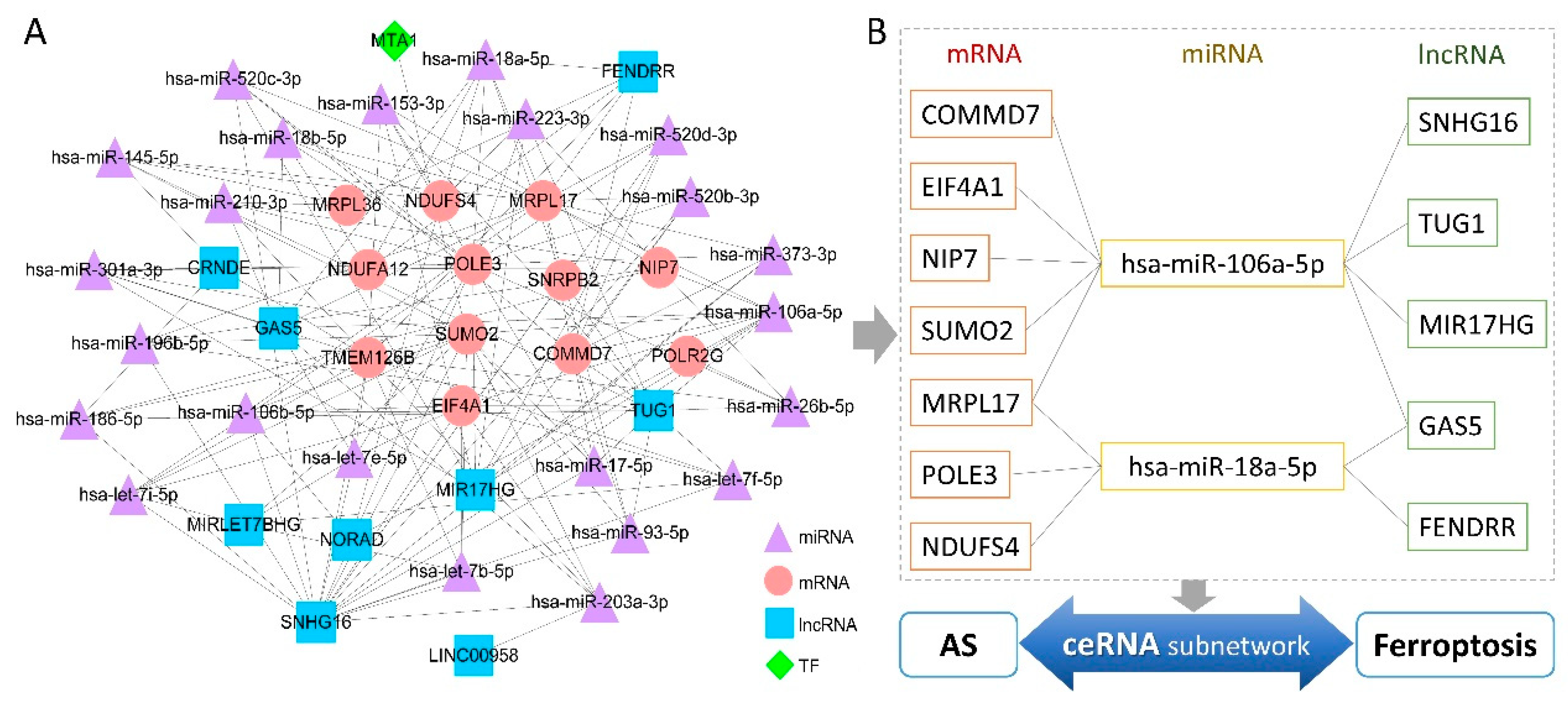
2.9. Construction of Regulatory Network Based on Hub Genes
3. Results
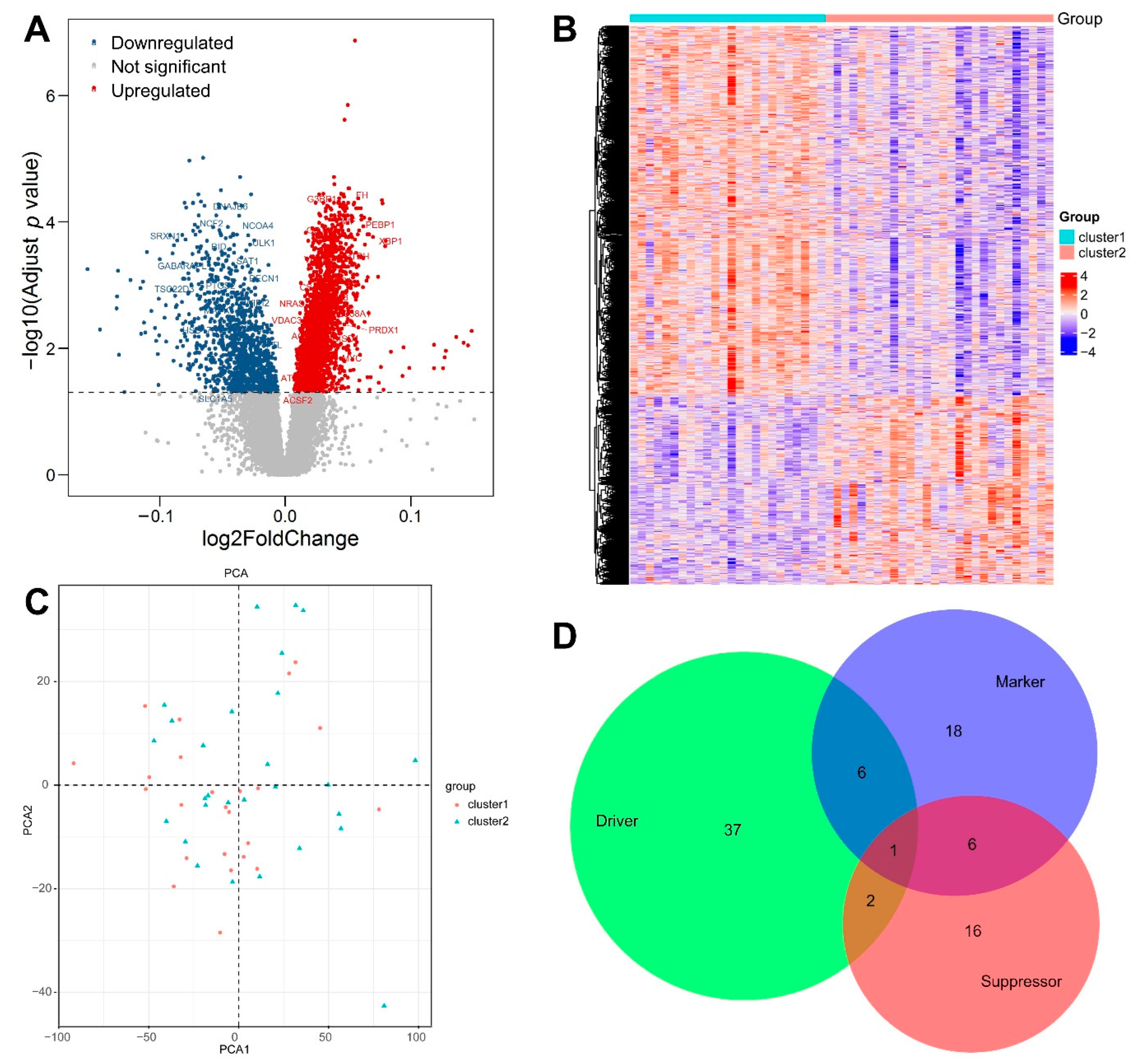
3.1. Consensus Clustering Based on FRGs
3.2. Composition of Immune Cells
3.3. Identification of DEGs between the Two Clusters
3.4. Functional Annotation of DEGs
3.5. WGCNA and the Key Module
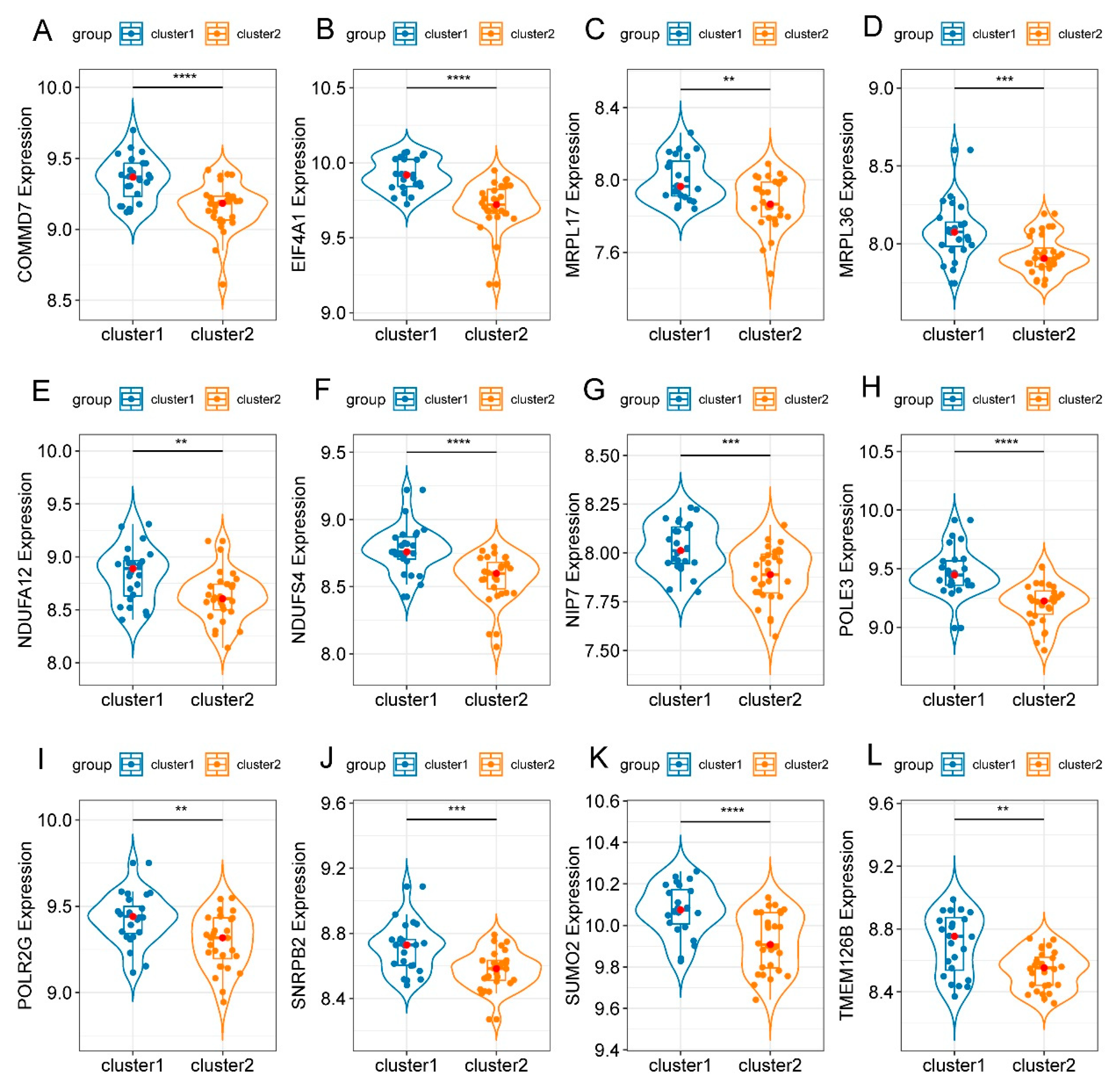
3.6. Identification of Hub Genes
3.7. Multifactor Regulatory Network in AS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sieper, J.; Poddubnyy, D. Axial spondyloarthritis. Lancet 2017, 390, 73–84. [Google Scholar] [CrossRef]
- Nash, P.; Mease, P.J.; Braun, J.; van der Heijde, D. Seronegative spondyloarthropathies: To lump or split? Ann. Rheum. Dis. 2005, 64 (Suppl. S2), ii9–ii13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, L.E.; Jones, G.T.; MacDonald, A.G.; Downham, C.; Sturrock, R.D.; Macfarlane, G.J. Global prevalence of ankylosing spondylitis. Rheumatology 2014, 53, 650–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taurog, J.D.; Chhabra, A.; Colbert, R.A. Ankylosing Spondylitis and Axial Spondyloarthritis. N. Engl. J. Med. 2016, 374, 2563–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, J.; Sieper, J. Ankylosing spondylitis. Lancet 2007, 369, 1379–1390. [Google Scholar] [CrossRef]
- Brown, M.A.; Kenna, T.; Wordsworth, B.P. Genetics of ankylosing spondylitis—Insights into pathogenesis. Nat. Rev. Rheumatol. 2016, 12, 81–91. [Google Scholar] [CrossRef]
- Taams, L.S.; Steel, K.J.A.; Srenathan, U.; Burns, L.A.; Kirkham, B.W. IL-17 in the immunopathogenesis of spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Schett, G.; Lories, R.J.; D’Agostino, M.A.; Elewaut, D.; Kirkham, B.; Soriano, E.R.; McGonagle, D. Enthesitis: From pathophysiology to treatment. Nat. Rev. Rheumatol. 2017, 13, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M.; Schett, G. Effects of the IL-23-IL-17 pathway on bone in spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 631–640. [Google Scholar] [CrossRef]
- Tang, M.; Lu, L.; Yu, X. Interleukin-17A Interweaves the Skeletal and Immune Systems. Front. Immunol. 2020, 11, 625034. [Google Scholar] [CrossRef] [PubMed]
- Maksymowych, W.P. Disease modification in ankylosing spondylitis. Nat. Rev. Rheumatol. 2010, 6, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Lorenz, S.M.; Proneth, B. Targeting Ferroptosis: New Hope for As-Yet-Incurable Diseases. Trends Mol. Med. 2021, 27, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Jiang, M.; Li, K.; Li, H.; Zhou, Y.; Xiao, X.; Xu, Y.; Krishfield, S.; Lipsky, P.E.; Tsokos, G.C.; et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat. Immunol. 2021, 22, 1107–1117. [Google Scholar] [CrossRef]
- Baker, J.F.; Ghio, A.J. Iron homoeostasis in rheumatic disease. Rheumatology 2009, 48, 1339–1344. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.; Schett, G. Anaemia in inflammatory rheumatic diseases. Nat. Rev. Rheumatol. 2013, 9, 205–215. [Google Scholar] [CrossRef]
- Feltelius, N.; Lindh, U.; Venge, P.; Hallgren, R. Ankylosing spondylitis: A chronic inflammatory disease with iron overload in granulocytes and platelets. Ann. Rheum. Dis. 1986, 45, 827–831. [Google Scholar] [CrossRef] [Green Version]
- Dogru, A.; Balkarli, A.; Cetin, G.Y.; Neselioglu, S.; Erel, O.; Tunc, S.E.; Sahin, M. Thiol/disulfide homeostasis in patients with ankylosing spondylitis. Bosn. J. Basic Med. Sci. 2016, 16, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Naziroglu, M.; Akkus, S.; Celik, H. Levels of lipid peroxidation and antioxidant vitamins in plasma and erythrocytes of patients with ankylosing spondylitis. Clin. Biochem. 2011, 44, 1412–1415. [Google Scholar] [CrossRef]
- Yazici, C.; Kose, K.; Calis, M.; Kuzuguden, S.; Kirnap, M. Protein oxidation status in patients with ankylosing spondylitis. Rheumatology 2004, 43, 1235–1239. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gao, L.; Jin, D.; Wang, P.; Yang, B.; Deng, W.; Xie, Z.; Tang, Y.; Wu, Y.; Shen, H. The Relationship of Bone Mineral Density to Oxidant/Antioxidant Status and Inflammatory and Bone Turnover Markers in a Multicenter Cross-Sectional Study of Young Men with Ankylosing Spondylitis. Calcif. Tissue Int. 2015, 97, 12–22. [Google Scholar] [CrossRef]
- Xu, W.D.; Yang, X.Y.; Li, D.H.; Zheng, K.D.; Qiu, P.C.; Zhang, W.; Li, C.Y.; Lei, K.F.; Yan, G.Q.; Jin, S.W.; et al. Up-regulation of fatty acid oxidation in the ligament as a contributing factor of ankylosing spondylitis: A comparative proteomic study. J. Proteom. 2015, 113, 57–72. [Google Scholar] [CrossRef]
- Bayir, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Gracey, E.; Yao, Y.; Green, B.; Qaiyum, Z.; Baglaenko, Y.; Lin, A.; Anton, A.; Ayearst, R.; Yip, P.; Inman, R.D. Sexual Dimorphism in the Th17 Signature of Ankylosing Spondylitis. Arthritis Rheumatol. 2016, 68, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef]
- Hong, Z.; Tang, P.; Liu, B.; Ran, C.; Yuan, C.; Zhang, Y.; Lu, Y.; Duan, X.; Yang, Y.; Wu, H. Ferroptosis-related Genes for Overall Survival Prediction in Patients with Colorectal Cancer can be Inhibited by Gallic acid. Int. J. Biol. Sci. 2021, 17, 942–956. [Google Scholar] [CrossRef]
- Zhuo, S.; Chen, Z.; Yang, Y.; Zhang, J.; Tang, J.; Yang, K. Clinical and Biological Significances of a Ferroptosis-Related Gene Signature in Glioma. Front. Oncol. 2020, 10, 590861. [Google Scholar] [CrossRef]
- Liang, J.Y.; Wang, D.S.; Lin, H.C.; Chen, X.X.; Yang, H.; Zheng, Y.; Li, Y.H. A Novel Ferroptosis-related Gene Signature for Overall Survival Prediction in Patients with Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2430–2441. [Google Scholar] [CrossRef]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.B.; Ellis, J.J.; Pennisi, D.J.; Song, X.; Batra, J.; Hollis, K.; Bradbury, L.A.; Li, Z.; Kenna, T.J.; Brown, M.A. Transcriptome analysis of ankylosing spondylitis patients before and after TNF-alpha inhibitor therapy reveals the pathways affected. Genes Immun. 2017, 18, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troyanskaya, O.G.; Garber, M.E.; Brown, P.O.; Botstein, D.; Altman, R.B. Nonparametric methods for identifying differentially expressed genes in microarray data. Bioinformatics 2002, 18, 1454–1461. [Google Scholar] [CrossRef]
- Gusnanto, A.; Calza, S.; Pawitan, Y. Identification of differentially expressed genes and false discovery rate in microarray studies. Curr. Opin. Lipidol. 2007, 18, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Tang, Q.; He, J.; Li, L.; Yang, N.; Yu, S.; Wang, M.; Zhang, Y.; Lin, J.; Cui, T.; et al. RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. 2022, 50, D326–D332. [Google Scholar] [CrossRef]
- Sieper, J.; Braun, J.; Dougados, M.; Baeten, D. Axial spondyloarthritis. Nat. Rev. Dis. Primers 2015, 1, 15013. [Google Scholar] [CrossRef]
- Ritchlin, C.; Adamopoulos, I.E. Axial spondyloarthritis: New advances in diagnosis and management. BMJ 2021, 372, m4447. [Google Scholar] [CrossRef]
- Liu, L.; Yuan, Y.; Zhang, S.; Xu, J.; Zou, J. Osteoimmunological insights into the pathogenesis of ankylosing spondylitis. J. Cell Physiol. 2021, 236, 6090–6100. [Google Scholar] [CrossRef]
- Ni, S.; Yuan, Y.; Qian, Z.; Zhong, Z.; Lv, T.; Kuang, Y.; Yu, B. Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radic. Biol. Med. 2021, 169, 271–282. [Google Scholar] [CrossRef]
- Duan, J.Y.; Lin, X.; Xu, F.; Shan, S.K.; Guo, B.; Li, F.X.; Wang, Y.; Zheng, M.H.; Xu, Q.S.; Lei, L.M.; et al. Ferroptosis and Its Potential Role in Metabolic Diseases: A Curse or Revitalization? Front. Cell Dev. Biol. 2021, 9, 701788. [Google Scholar] [CrossRef]
- Simao, M.; Cancela, M.L. Musculoskeletal complications associated with pathological iron toxicity and its molecular mechanisms. Biochem. Soc. Trans. 2021, 49, 747–759. [Google Scholar] [CrossRef]
- Yao, X.; Sun, K.; Yu, S.; Luo, J.; Guo, J.; Lin, J.; Wang, G.; Guo, Z.; Ye, Y.; Guo, F. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J. Orthop. Translat. 2021, 27, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zheng, Y.; Sun, W.; Zhang, Z.; Liu, J.; Yang, W.; Yuan, W.; Yi, Y.; Wang, J.; Liu, J. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2alpha-dependent manner. Cell Prolif. 2021, 54, e13134. [Google Scholar] [CrossRef]
- Lu, S.; Song, Y.; Luo, R.; Li, S.; Li, G.; Wang, K.; Liao, Z.; Wang, B.; Ke, W.; Xiang, Q.; et al. Ferroportin-Dependent Iron Homeostasis Protects against Oxidative Stress-Induced Nucleus Pulposus Cell Ferroptosis and Ameliorates Intervertebral Disc Degeneration In Vivo. Oxid. Med. Cell Longev. 2021, 2021, 6670497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, Z.; Xie, Z.; Chen, Y.; Zheng, Z.; Wei, X.; Huang, B.; Shan, Z.; Liu, J.; Fan, S.; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 2020, 160, 552–565. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, S.; Kong, M.; Tu, Q.; Zhang, L.; Ma, X. Single-cell RNA-seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthr. Cartil. 2021, 29, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, S.; Pan, X.; Dai, X.; Pan, G.; Li, Z.; Mai, X.; Tian, Y.; Zhang, S.; Liu, B.; et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J. Cachexia Sarcopenia Muscle 2021, 12, 746–768. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, B.; Shen, D.; Chen, J.; Yu, Z.; Chen, C. Ferroptosis in a sarcopenia model of senescence accelerated mouse prone 8 (SAMP8). Int. J. Biol. Sci. 2021, 17, 151–162. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Zhang, W.; Li, H.; Zhao, W.; Sun, J.; Yang, M. Melatonin Suppresses Ferroptosis Induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Oxid. Med. Cell Longev. 2020, 2020, 9067610. [Google Scholar] [CrossRef]
- Zhang, D.; Li, B.; Guo, R.; Wu, J.; Yang, C.; Jiang, X.; Zhang, C.; Yan, H.; Zhao, Q.; Wang, Z.; et al. RAB5C, SYNJ1, and RNF19B promote male ankylosing spondylitis by regulating immune cell infiltration. Ann. Transl. Med. 2021, 9, 1011. [Google Scholar] [CrossRef]
- Meng, F.; Du, N.; Xu, D.; Kuai, L.; Liu, L.; Xiu, M. Bioinformatics Analysis of the Molecular Mechanism and Potential Treatment Target of Ankylosing Spondylitis. Comput Math. Methods Med. 2021, 2021, 7471291. [Google Scholar] [CrossRef]
- Bowness, P. Hla-B27. Annu. Rev. Immunol. 2015, 33, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.T.; Maragno, M.; Butler, R.; Dupuis, M.L.; Sorrentino, R. CD8(+) T-cell autoreactivity to an HLA-B27-restricted self-epitope correlates with ankylosing spondylitis. J. Clin. Investig. 2000, 106, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracey, E.; Yao, Y.; Qaiyum, Z.; Lim, M.; Tang, M.; Inman, R.D. Altered Cytotoxicity Profile of CD8+ T Cells in Ankylosing Spondylitis. Arthritis Rheumatol. 2020, 72, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Drijvers, J.M.; Gillis, J.E.; Muijlwijk, T.; Nguyen, T.H.; Gaudiano, E.F.; Harris, I.S.; LaFleur, M.W.; Ringel, A.E.; Yao, C.H.; Kurmi, K.; et al. Pharmacologic Screening Identifies Metabolic Vulnerabilities of CD8(+) T Cells. Cancer Immunol. Res. 2021, 9, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Mauro, D.; Thomas, R.; Guggino, G.; Lories, R.; Brown, M.A.; Ciccia, F. Ankylosing spondylitis: An autoimmune or autoinflammatory disease? Nat. Rev. Rheumatol. 2021, 17, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ma, Y.; Wu, M.; Zhang, X.; Yang, J.; Deng, J.; Guan, S.; Gao, X.; Xu, S.; Shuai, Z.; et al. Neutrophil lymphocyte ratio in patients with ankylosing spondylitis: A systematic review and meta-analysis. Mod. Rheumatol. 2020, 30, 141–148. [Google Scholar] [CrossRef]
- Appel, H.; Maier, R.; Wu, P.; Scheer, R.; Hempfing, A.; Kayser, R.; Thiel, A.; Radbruch, A.; Loddenkemper, C.; Sieper, J. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res. Ther. 2011, 13, R95. [Google Scholar] [CrossRef] [Green Version]
- Papagoras, C.; Chrysanthopoulou, A.; Mitsios, A.; Ntinopoulou, M.; Tsironidou, V.; Batsali, A.K.; Papadaki, H.A.; Skendros, P.; Ritis, K. IL-17A expressed on neutrophil extracellular traps promotes mesenchymal stem cell differentiation toward bone-forming cells in ankylosing spondylitis. Eur. J. Immunol. 2021, 51, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Yotsumoto, S.; Muroi, Y.; Chiba, T.; Ohmura, R.; Yoneyama, M.; Magarisawa, M.; Dodo, K.; Terayama, N.; Sodeoka, M.; Aoyagi, R.; et al. Hyperoxidation of ether-linked phospholipids accelerates neutrophil extracellular trap formation. Sci. Rep. 2017, 7, 16026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Xiao, M.; Huang, Y.; Tu, L.; Chen, Z.; Cao, S.; Wei, Q.; Gu, J. Serum Metabolomics Signatures Associated With Ankylosing Spondylitis and TNF Inhibitor Therapy. Front. Immunol. 2021, 12, 630791. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Xie, Z.; Zeng, H.; Wang, P.; Li, J.; Zheng, G.; Wang, S.; Cao, Q.; Li, M.; Liu, W.; et al. Oxidative stress-mediated mitochondrial dysfunction facilitates mesenchymal stem cell senescence in ankylosing spondylitis. Cell Death Dis. 2020, 11, 775. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.C.; Chen, H.C.; Chou, Y.C.; Lin, C.L.; Kao, C.H.; Lo, H.Y.; Liu, F.C.; Yang, T.Y. Positive association of Parkinson’s disease with ankylosing spondylitis: A nationwide population-based study. J. Transl. Med. 2020, 18, 455. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Heo, S.J.; Kim, Y.W.; Yang, S.N.; Moon, H.I. Ankylosing Spondylitis: A Risk Factor for Parkinsonism-A Nationwide Population-Based Study. J. Parkinsons Dis. 2022, 12, 353–360. [Google Scholar] [CrossRef]
- Lari, A.; Gholami Pourbadie, H.; Sharifi-Zarchi, A.; Aslani, S.; Nejatbakhsh Samimi, L.; Jamshidi, A.; Mahmoudi, M. Evaluation of the Ankylosing Spondylitis Transcriptome for Oxidative Phosphorylation Pathway: The Shared Pathway with Neurodegenerative Diseases. Iran. J. Allergy Asthma Immunol. 2021, 20, 563–573. [Google Scholar] [CrossRef]
- Akdis, C.A. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat. Rev. Immunol. 2021, 21, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Lin, P.; Feng, Z.; Lu, H.; Han, Q.; Chen, J.; Zhang, Y.; He, Q.; Nan, G.; Luo, X.; et al. TNFAIP3-DEPTOR complex regulates inflammasome secretion through autophagy in ankylosing spondylitis monocytes. Autophagy 2018, 14, 1629–1643. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Yang, W.; Cai, Z.; Wang, P.; Li, H.; Mi, R.; Jiang, Y.; Xie, Z.; Sui, P.; Wu, Y.; et al. SMAD-specific E3 ubiquitin ligase 2 promotes angiogenesis by facilitating PTX3 degradation in MSCs from patients with ankylosing spondylitis. Stem Cells 2021, 39, 581–599. [Google Scholar] [CrossRef]
- Chang, H.M.; Yeh, E.T.H. SUMO: From Bench to Bedside. Physiol Rev. 2020, 100, 1599–1619. [Google Scholar] [CrossRef]
- Liu, K.; Guo, C.; Lao, Y.; Yang, J.; Chen, F.; Zhao, Y.; Yang, Y.; Yang, J.; Yi, J. A fine-tuning mechanism underlying self-control for autophagy: DeSUMOylation of BECN1 by SENP3. Autophagy 2020, 16, 975–990. [Google Scholar] [CrossRef]
- Ma, R.; Ma, L.; Weng, W.; Wang, Y.; Liu, H.; Guo, R.; Gao, Y.; Tu, J.; Xu, T.L.; Cheng, J.; et al. DUSP6 SUMOylation protects cells from oxidative damage via direct regulation of Drp1 dephosphorylation. Sci. Adv. 2020, 6, eaaz0361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, K.; Yang, J.; Lao, Y.; Deng, L.; Deng, G.; Yi, J.; Sun, X.; Wang, Q. SENP3 Suppresses Osteoclastogenesis by De-conjugating SUMO2/3 from IRF8 in Bone Marrow-Derived Monocytes. Cell Rep. 2020, 30, 1951–1963.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Jiang, C. Identification of potential target genes for ankylosing spondylitis treatment. Medicine 2018, 97, e9760. [Google Scholar] [CrossRef]
- Jin, Z.; Wei, W.; Yang, M.; Du, Y.; Wan, Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014, 20, 483–498. [Google Scholar] [CrossRef] [Green Version]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite. In Practical Guide to Life Science Databases; Abugessaisa, I., Kasukawa, T., Eds.; Springer: Singapore, 2021; pp. 27–56. [Google Scholar]
- Chen, C.; Rong, T.; Li, Z.; Shen, J. Noncoding RNAs Involved in the Pathogenesis of Ankylosing Spondylitis. Biomed. Res. Int. 2019, 2019, 6920281. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, E.S.; Schmitter, D.; Vegter, E.L.; Ter Maaten, J.M.; Valente, M.A.; Liu, L.C.; van der Harst, P.; Pinto, Y.M.; de Boer, R.A.; Meyer, S.; et al. Signature of circulating microRNAs in patients with acute heart failure. Eur. J. Heart Fail. 2016, 18, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Song, L.; Hu, Y.; He, W.; Hou, F.; Yan, P.; Xu, J.; Wang, K.; Tao, Y.; Li, D.; et al. Novel Role of miR-18a-5p and Galanin in Rat Lung Ischemia Reperfusion-Mediated Response. Oxid. Med. Cell Longev. 2021, 2021, 6621921. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, L.; Gan, L.; Hu, Y.; Xiang, P.; Xing, Y.; Zhu, J.; Ye, S. Berberine Acts on C/EBPbeta/lncRNA Gas5/miR-18a-5p Loop to Decrease the Mitochondrial ROS Generation in HK-2 Cells. Front. Endocrinol. 2021, 12, 675834. [Google Scholar] [CrossRef]
- Wang, G.; Yang, Y.; Ma, H.; Shi, L.; Jia, W.; Hao, X.; Liu, W. LncRNA FENDRR Inhibits ox-LDL Induced Mitochondrial Energy Metabolism Disorder in Aortic Endothelial Cells via miR-18a-5p/PGC-1alpha Signaling Pathway. Front. Endocrinol. 2021, 12, 622665. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ge, Z.; Wang, Z.; Gao, Y.; Wang, Y.; Qu, X. Circular RNA RHOT1 promotes progression and inhibits ferroptosis via mir-106a-5p/STAT3 axis in breast cancer. Aging 2021, 13, 8115–8126. [Google Scholar] [CrossRef]
- Huang, C.C.; Law, Y.Y.; Liu, S.C.; Hu, S.L.; Lin, J.A.; Chen, C.J.; Wang, S.W.; Tang, C.H. Adiponectin Promotes VEGF Expression in Rheumatoid Arthritis Synovial Fibroblasts and Induces Endothelial Progenitor Cell Angiogenesis by Inhibiting miR-106a-5p. Cells 2021, 10, 2627. [Google Scholar] [CrossRef]
- Hui, S.; Yang, Y.; Li, J.; Li, N.; Xu, P.; Li, H.; Zhang, Y.; Wang, S.; Lin, G.; Li, S.; et al. Differential miRNAs profile and bioinformatics analyses in bone marrow mesenchymal stem cells from adolescent idiopathic scoliosis patients. Spine J. 2019, 19, 1584–1596. [Google Scholar] [CrossRef]
- Liu, W.; Li, Z.; Cai, Z.; Xie, Z.; Li, J.; Li, M.; Cen, S.; Tang, S.; Zheng, G.; Ye, G.; et al. LncRNA-mRNA expression profiles and functional networks in osteoclast differentiation. J. Cell Mol. Med. 2020, 24, 9786–9797. [Google Scholar] [CrossRef]
- Lan, X.; Ma, H.; Zhang, Z.; Ye, D.; Min, J.; Cai, F.; Luo, J. Downregulation of lncRNA TUG1 is involved in ankylosing spondylitis and is related to disease activity and course of treatment. Biosci. Trends. 2018, 12, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Li, X.; Cui, Y.; Meng, P.; Zeng, G.; Wang, Y.; Wang, Q. Bioinformatics Analysis Identifies Potential Ferroptosis Key Genes in the Pathogenesis of Intracerebral Hemorrhage. Front. Neurosci. 2021, 15, 661663. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Zhang, Q.B.; Liu, T.H.; Yang, Y.Y.; Zheng, J.X.; Zhou, W.J.; Xiong, Q.; Qing, Y.F. Autophagy dysfunction may be involved in the pathogenesis of ankylosing spondylitis. Exp. Ther. Med. 2020, 20, 3578–3586. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Chen, M.; Cao, S.; Yao, Q.; Zhang, X.; Du, S.; Qu, H.; Cheng, Y.; Ma, S.; Zhang, M.; et al. Identification of a ferroptosis-related gene pair biomarker with immune infiltration landscapes in ischemic stroke: A bioinformatics-based comprehensive study. BMC Genom. 2022, 23, 59. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rong, T.; Jia, N.; Wu, B.; Sang, D.; Liu, B. New Insights into the Regulatory Role of Ferroptosis in Ankylosing Spondylitis via Consensus Clustering of Ferroptosis-Related Genes and Weighted Gene Co-Expression Network Analysis. Genes 2022, 13, 1373. https://doi.org/10.3390/genes13081373
Rong T, Jia N, Wu B, Sang D, Liu B. New Insights into the Regulatory Role of Ferroptosis in Ankylosing Spondylitis via Consensus Clustering of Ferroptosis-Related Genes and Weighted Gene Co-Expression Network Analysis. Genes. 2022; 13(8):1373. https://doi.org/10.3390/genes13081373
Chicago/Turabian StyleRong, Tianhua, Ningyi Jia, Bingxuan Wu, Dacheng Sang, and Baoge Liu. 2022. "New Insights into the Regulatory Role of Ferroptosis in Ankylosing Spondylitis via Consensus Clustering of Ferroptosis-Related Genes and Weighted Gene Co-Expression Network Analysis" Genes 13, no. 8: 1373. https://doi.org/10.3390/genes13081373