
Nrf2/HO-1 Signaling Stimulation through Acetyl-11-Keto-Beta-Boswellic Acid (AKBA) Provides Neuroprotection in Ethidium Bromide-Induced Experimental Model of Multiple Sclerosis

 ,
,  , , , and
, , , and
Abstract
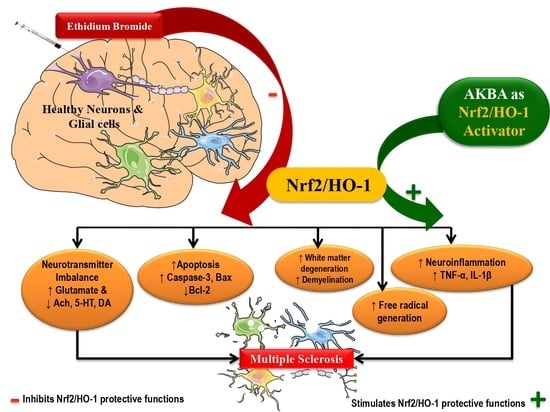
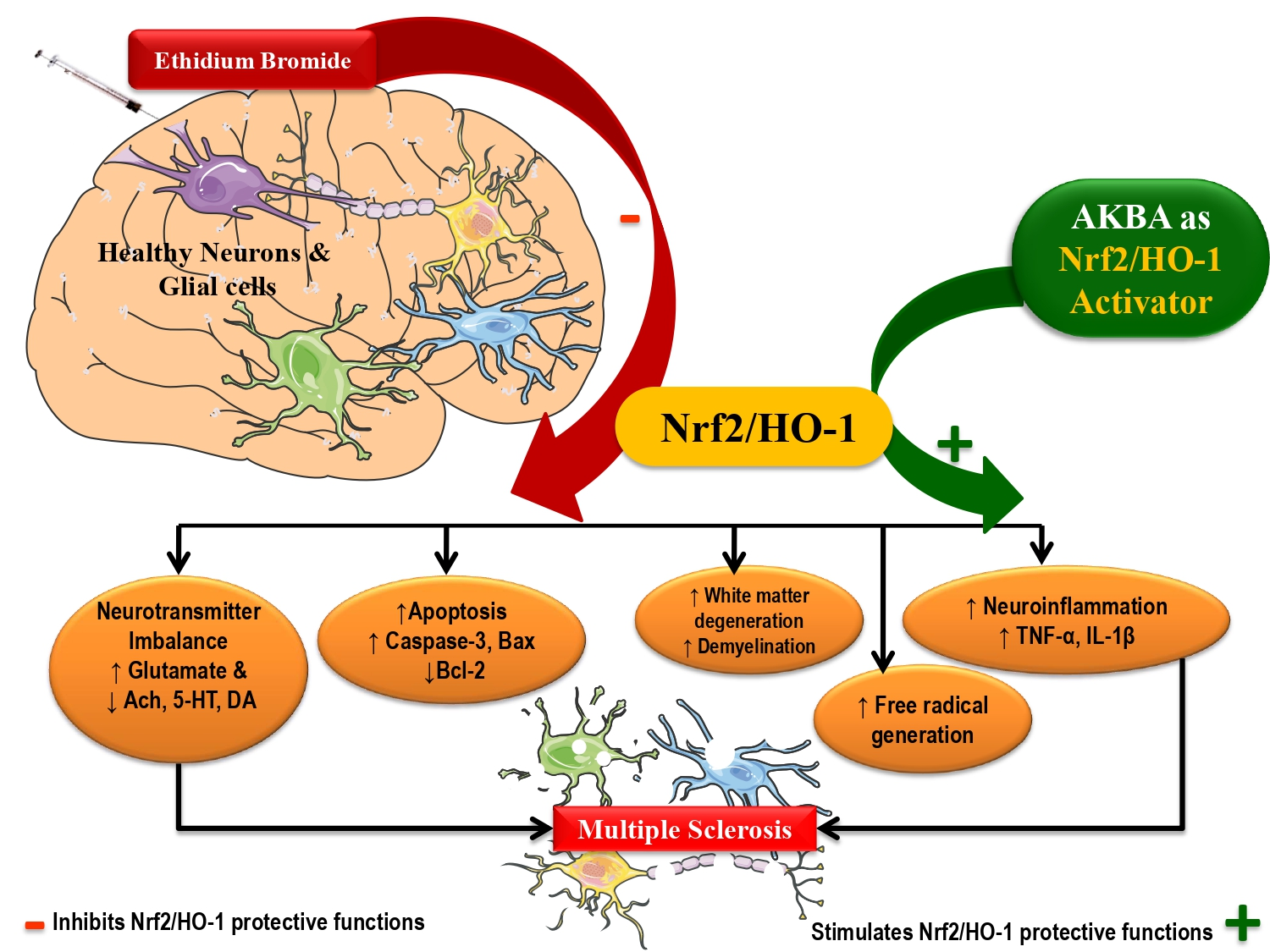
:
{kind=link}
{kind=link}
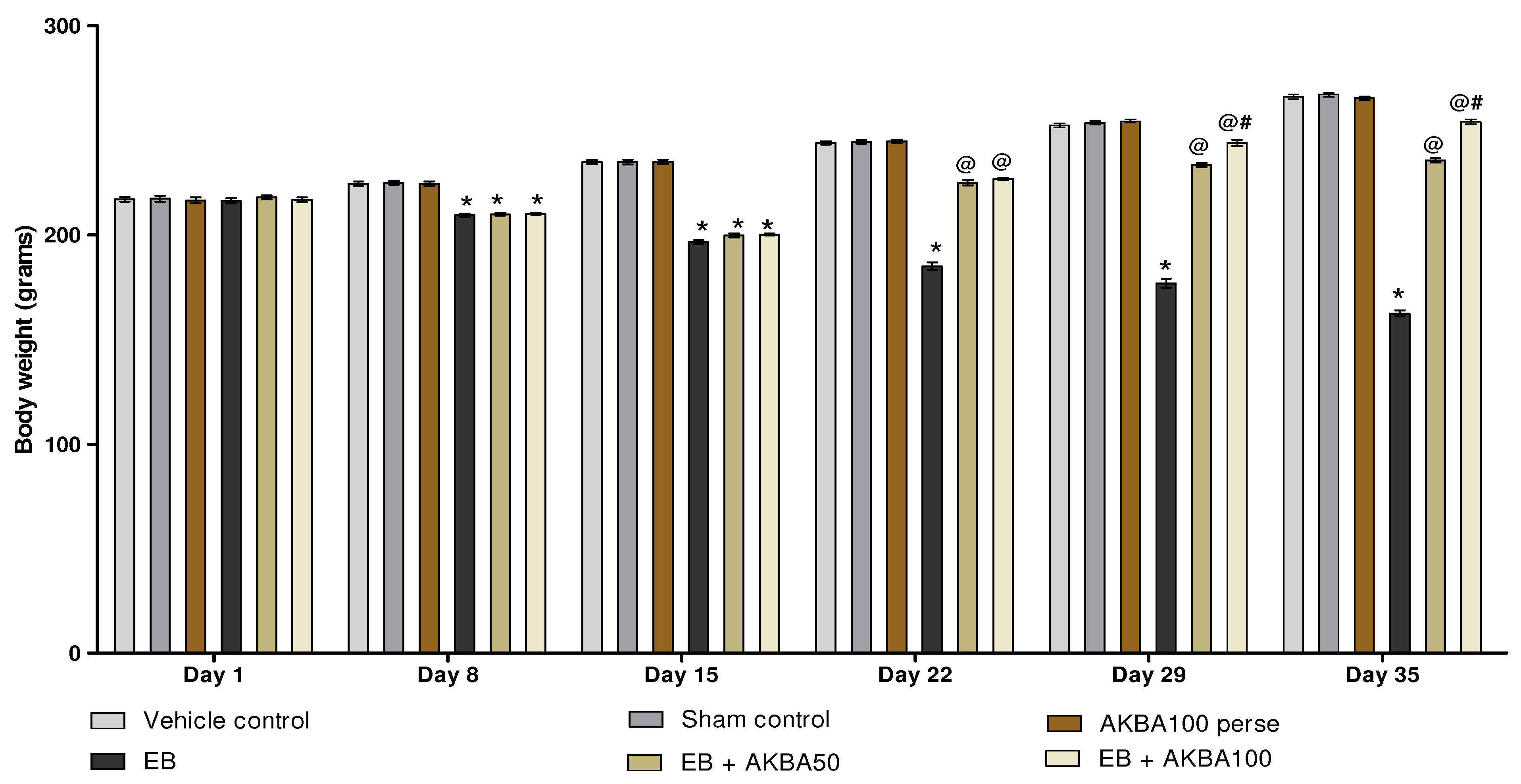{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Drugs and Chemicals
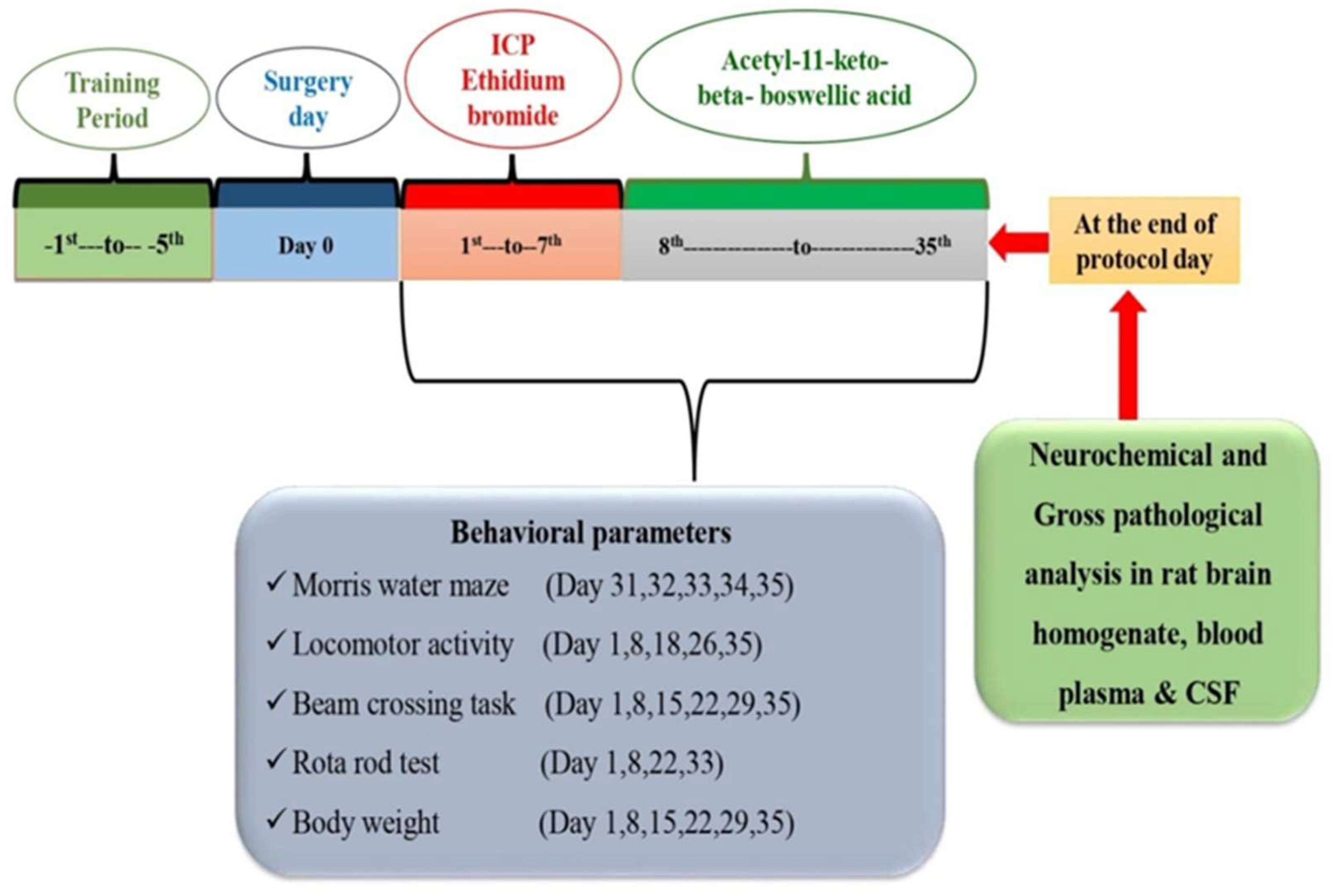
2.3. Protocol Schedule of Animal Experimentation
2.4. Experimental Animal Model of ICP-EB-Induced Experimental MS Rats
2.5. Parameters
2.5.1. Measurement of Weight Variations
2.5.2. Measurement of Relative Brain-Body Weight Ratio
2.6. Assessment of Behaviour Parameters
2.6.1. Morris Water Maze Task
2.6.2. Locomotor Activity
2.6.3. Beam Crossing Task
2.6.4. Rotarod Test
2.7. Neurochemical Parameters
2.7.1. Collection and Preparation of Biological Samples
2.7.2. Assessment of Cellular and Molecular Markers
2.7.3. Assessment of Apoptotic Markers
2.7.4. Assessment of Neurotransmitter Levels
2.7.5. Measurement of TNF- α and IL-1β Levels
2.7.6. Evaluation of Oxidative Stress Markers
2.8. Protein Estimation
2.9. Gross Pathological Examination of Rat Brains
2.10. Assessment of Histopathological Changes
2.10.1. Assessment of Demyelination by Luxol Fast Blue
2.10.2. Statistical Analysis
3. Results
3.1. AKBA Mediated Neuroprotective Function on the Restoration of Body Weight in Experimental Model of Multiple Sclerosis
3.1.1. Improvement in Body Weight after Long-Term Treatment with AKBA
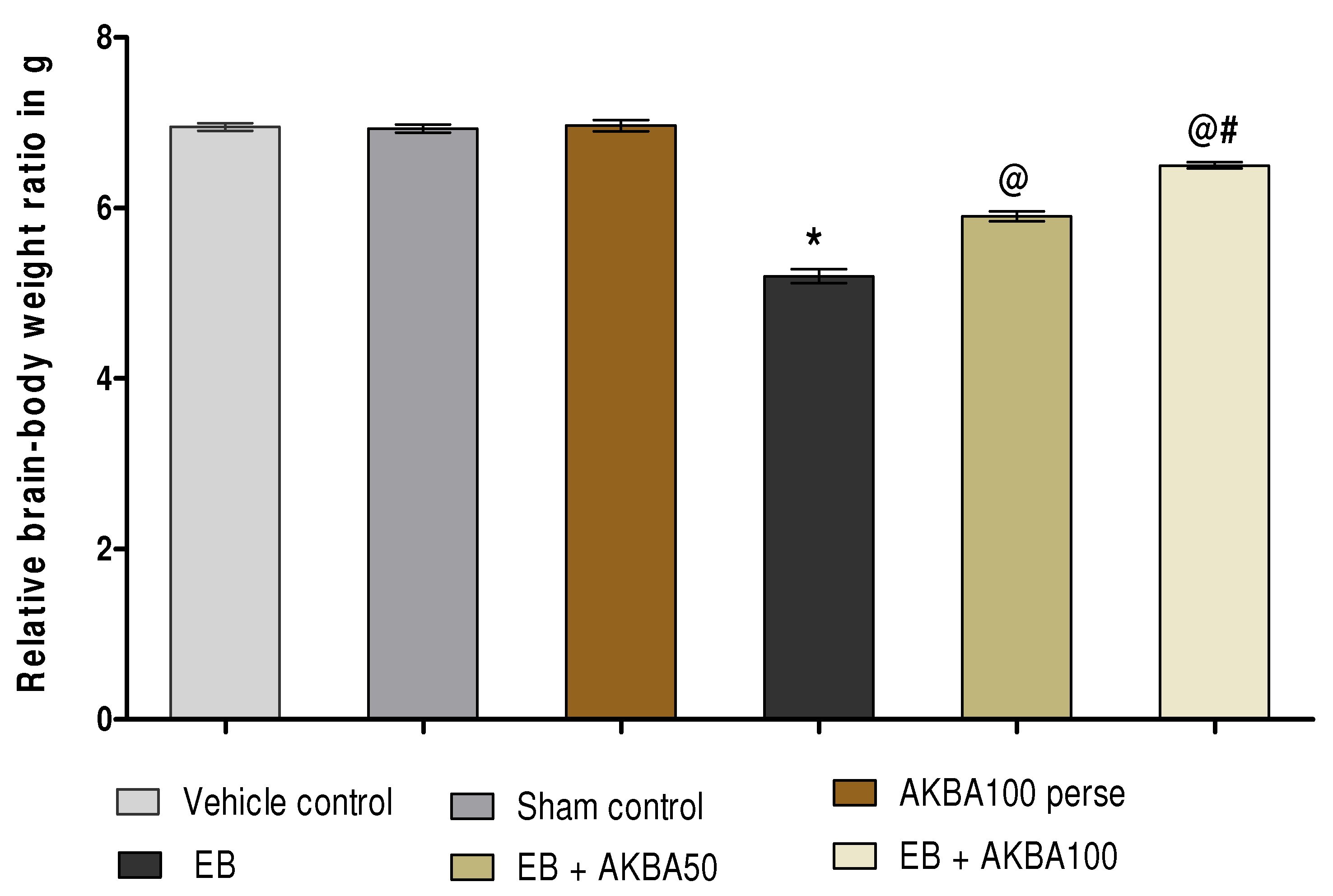
3.1.2. AKBA-Mediated Restored Effect on the Relative Brain-Body Weight Ratio
3.2. AKBA-Mediated Neuroprotective Function on Behavioural Abnormalities in Experimental Model of Multiple Sclerosis
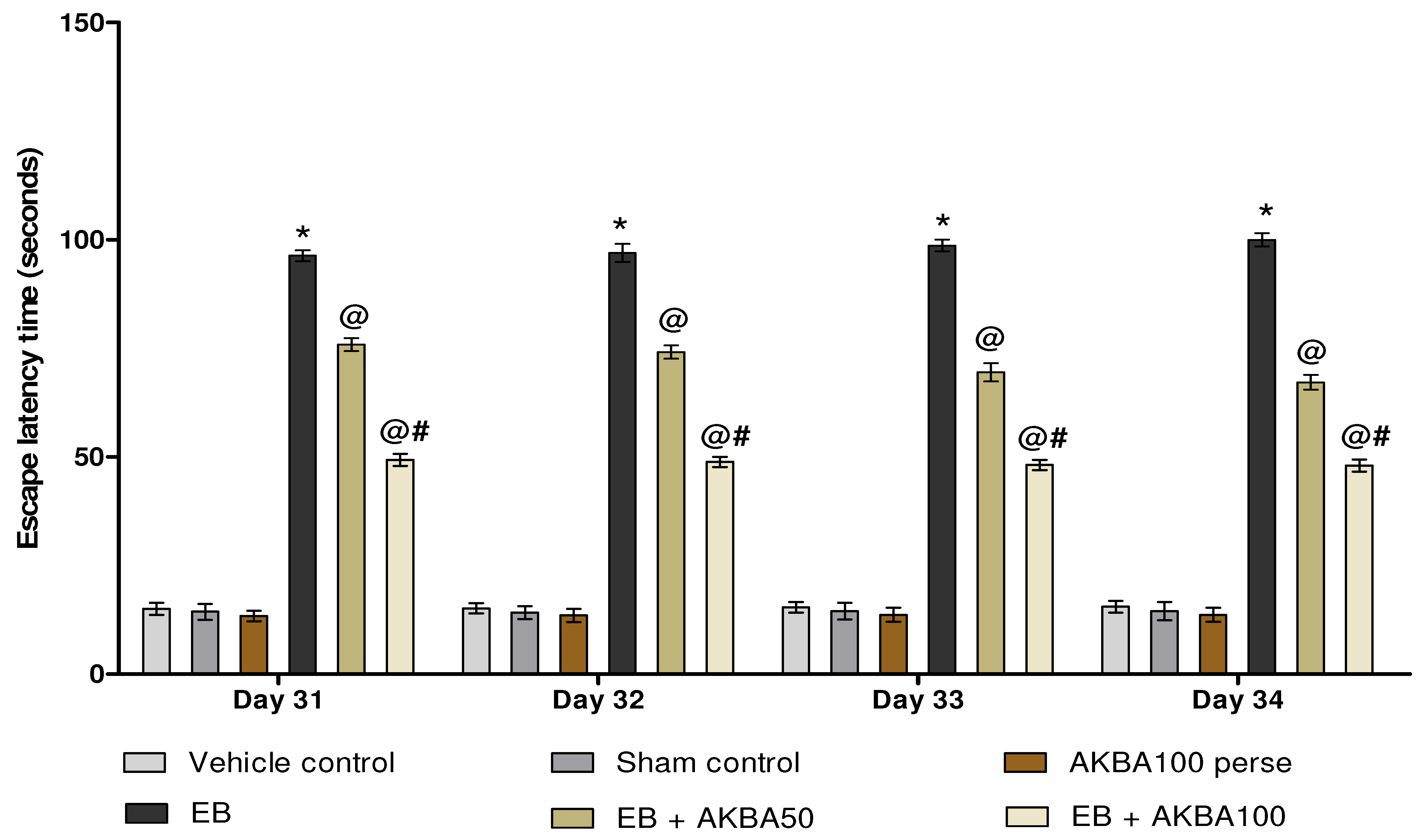
3.2.1. Cognitive Improvement and Memory Consolidation after Long-Term Treatment of AKBA
3.2.2. Improvement of Locomotor Activity after Long-Term Treatment with AKBA
3.2.3. Recovery of Gait Abnormalities after Long-Term Treatment of AKBA
3.2.4. Improvement in Motor Coordination after Long-Term Treatment with AKBA
3.3. AKBA-Mediated Neuroprotective Function on Neurochemical Alterations in Experimental Model of Multiple Sclerosis
3.3.1. Increased Level of Nrf2 and HO-1 after Long-Term Treatment with AKBA
3.3.2. Restoration of Myelin Basic Protein Level after Long-Term Treatment of AKBA
3.3.3. Decreased Caspase-3, Bax, and Increased Bcl-2 Levels after Long-Term Treatment with AKBA
3.3.4. Restoration of Neurotransmitter Levels after Long-Term Treatment of AKBA
3.3.5. Reduction in Inflammatory Cytokines Levels after Long-Term Treatment with AKBA
3.3.6. Amelioration of Oxidative Stress Marker Levels after Long-Term Treatment with AKBA
3.4. AKBA-Mediated Neuroprotective Effect in the Restoration of Gross Pathological Alterations in Experimental Model of Multiple Sclerosis
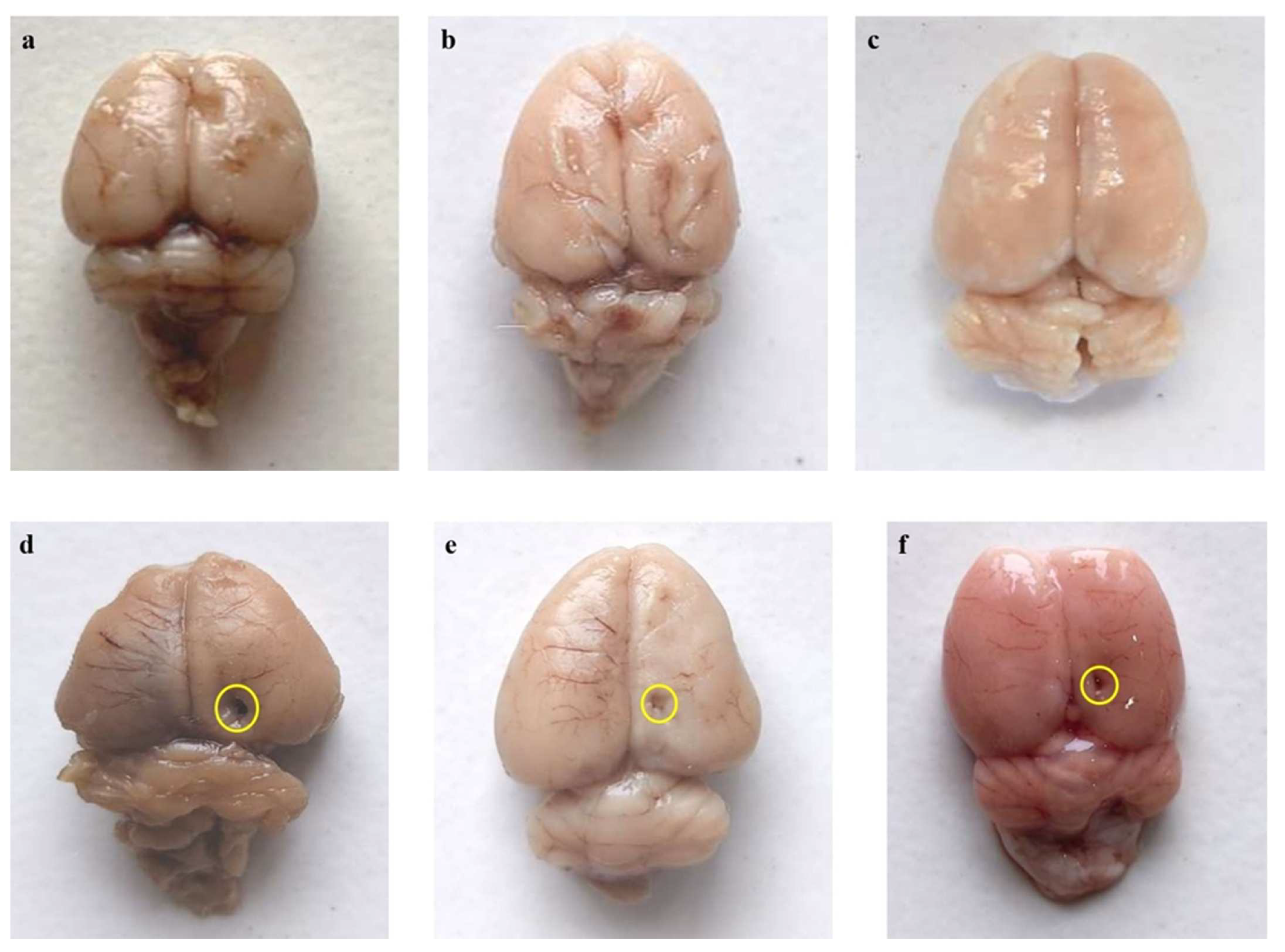
3.4.1. Restoration of Whole-Brain Alterations after Long-Term Treatment with AKBA
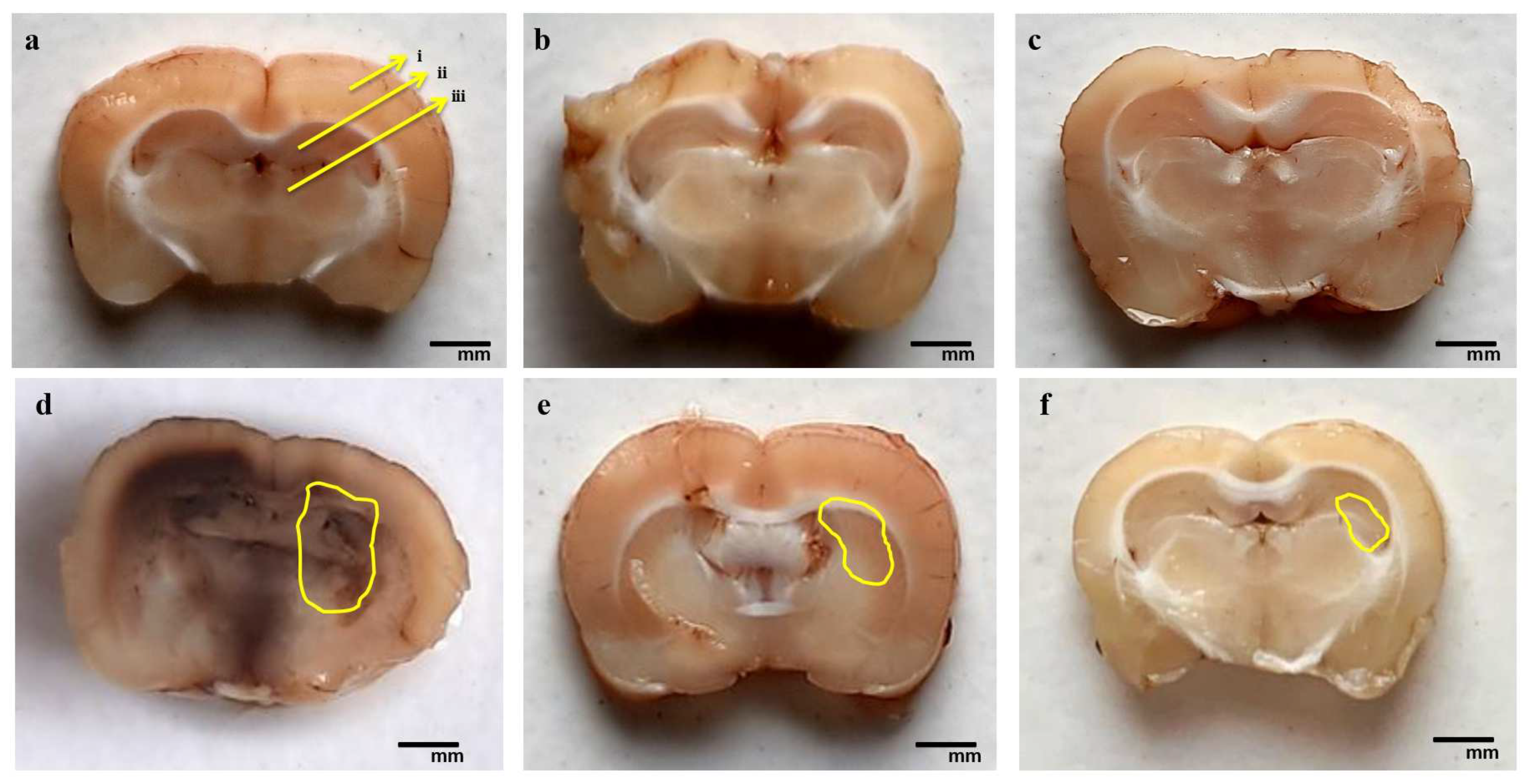
3.4.2. Reduction in Pathological Changes in Brain Sections after Long-Term Treatment with AKBA
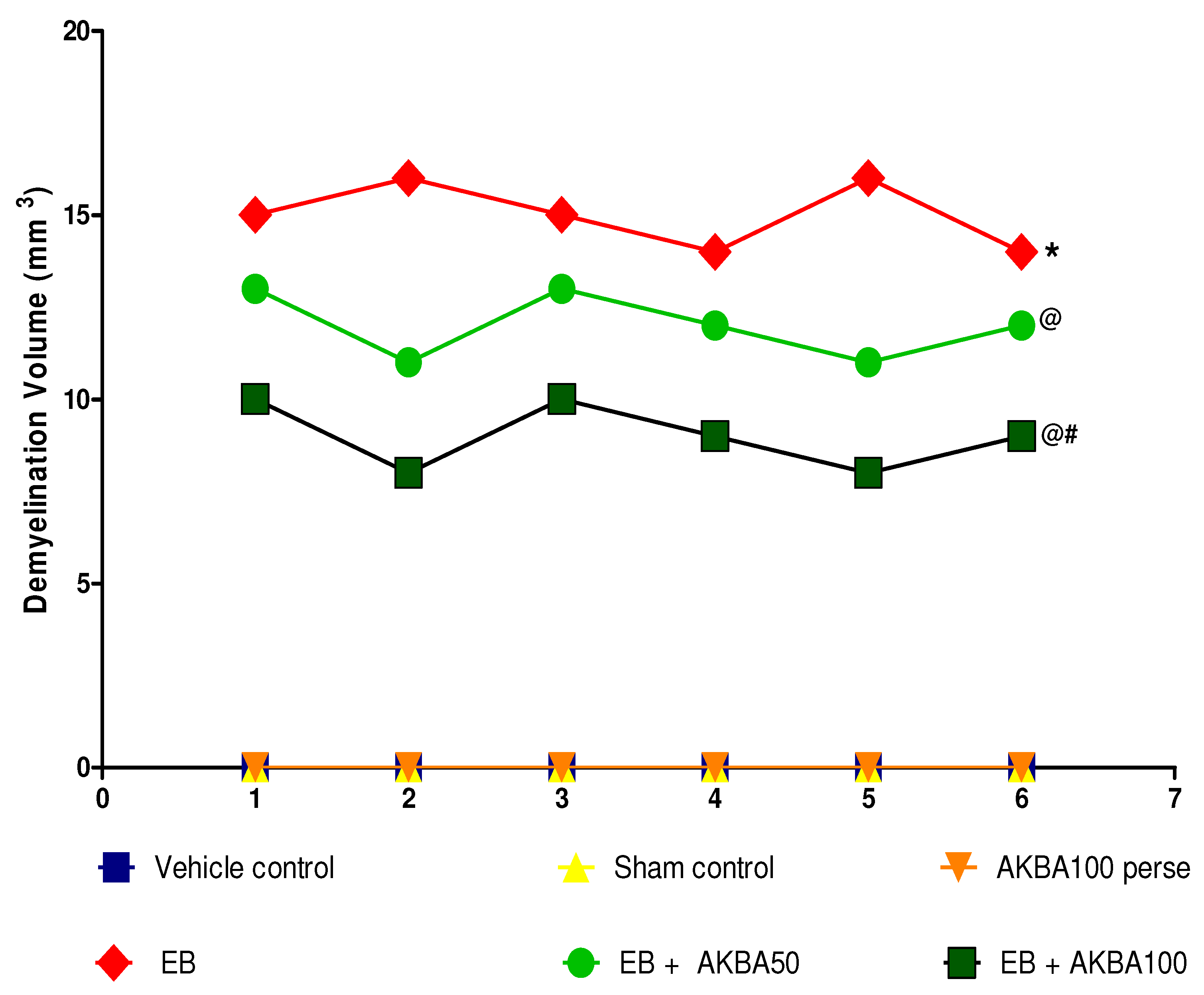
3.4.3. Reduction in Demyelination Volume after Long-Term Treatment with AKBA
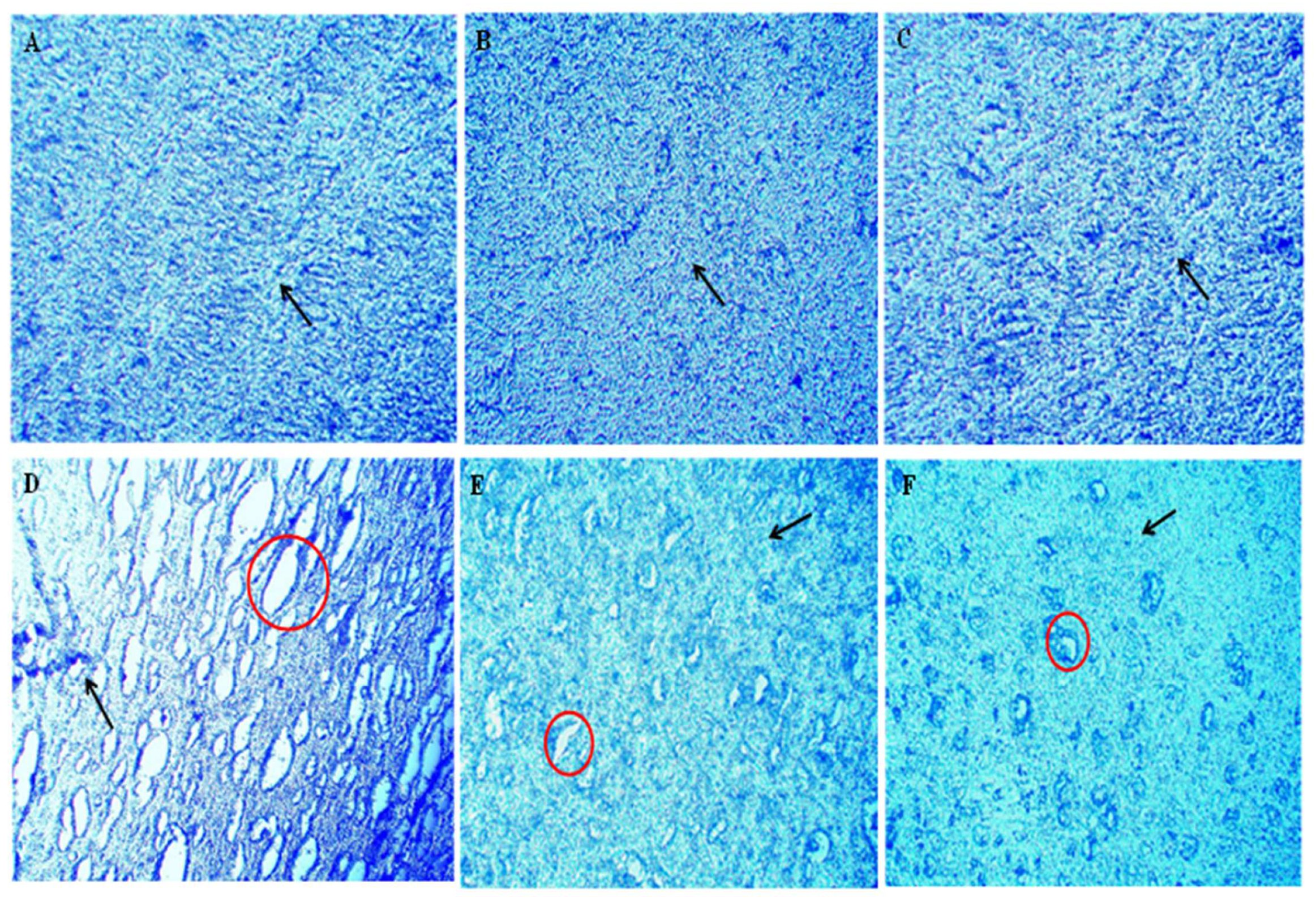
3.5. AKBA-Mediated Neuroprotective Effect in Ethidium Bromide-Induced Histopathological Changes
3.6. AKBA-Mediated Neuroprotective Effect in Ethidium Bromide-Induced Demyelination
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ach | Acetylcholine |
| AchE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| AKBA | Acetyl-11-keto-β-boswellic acid |
| ALS | Amyotrophic lateral sclerosis |
| ARE | Antioxidant response element |
| BCT | Beam crossing task |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| EB | Ethidium bromide |
| ELISA | Enzyme-linked immunosorbent assay |
| ELT | Escape latency time |
| GABA | γ-aminobutyric acid |
| GSH | Glutathione |
| HD | Huntington’s disease |
| HO-1 | Heme oxygenase-1 |
| ICH | Intracerebral hemorrhage |
| ICP | Intracerebropeduncle |
| IL-1 β | Interleukin-1 β |
| Keap1 | Kelch-like ECH-associated protein-1 |
| MBP | Myelin basic protein |
| MDA | Malondialdehyde |
| MS | Multiple sclerosis |
| MWM | Morris water maze |
| NO | Nitric oxide |
| Nrf2 | Nuclear factor erythroid-2-related factor-2 |
| PD | Parkinson’s disease |
| ROS | Reactive oxygen species |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SOD | Superoxide dismutase |
| TNF-α | Tumor necrosis factor-α |
| TSTQ | Time spent in the target quadrant |
References
- Kumar, N.; Sharma, N.; Khera, R.; Gupta, R.; Mehan, S. Guggulsterone ameliorates ethidium bromide-induced experimental model of multiple sclerosis via restoration of behavioral, molecular, neurochemical and morphological alterations in rat brain. Metab. Brain Dis. 2021, 36, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Oveland, E.; Ahmad, I.; Lereim, R.R.; Kroksveen, A.C.; Barsnes, H.; Guldbrandsen, A.; Myhr, K.-M.; Bø, L.; Berven, F.S.; Wergeland, S. Cuprizone and EAE mouse frontal cortex proteomics revealed proteins altered in multiple sclerosis. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mehan, S.; Kapoor, T. Neuroprotective Methodologies in the Treatment of Multiple Sclerosis Current Status of Clinical and Pre-clinical Findings. Curr. Drug Discov. Technol. 2021, 18, 31–46. [Google Scholar] [CrossRef]
- Singh, A.; Upadhayay, S.; Mehan, S. Understanding Abnormal c-JNK/p38MAPK Signaling Overactivation Involved in the Progression of Multiple Sclerosis: Possible Therapeutic Targets and Impact on Neurodegenerative Diseases. Neurotox. Res. 2021, 39, 1630–1650. [Google Scholar] [CrossRef]
- Capriotti, T.; Noel, J.; Brissenden, S. Multiple Sclerosis: An update for home healthcare clinicians. Home Healthc. Now 2018, 36, 169–180. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef]
- Kasper, L.H.; Shoemaker, J. Multiple sclerosis immunology: The healthy immune system vs the MS immune system. Neurology 2009, 74 (Suppl. 1), S2–S8. [Google Scholar] [CrossRef]
- Van Nierop, G.P.; van Luijn, M.M.; Michels, S.S.; Melief, M.J.; Janssen, M.; Langerak, A.W.; Ouwendijk, W.J.; Hintzen, R.Q.; Verjans, G.M. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. 2017, 134, 383–401. [Google Scholar] [CrossRef]
- Bjelobaba, I.; Begovic-Kupresanin, V.; Pekovic, S.; Lavrnja, I. Animal models of multiple sclerosis: Focus on experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2018, 96, 1021–1042. [Google Scholar] [CrossRef]
- Miller, R.H.; Fyffe-Maricich, S.; Caprariello, A.C. Animal models for the study of multiple sclerosis. In Animal Models for the Study of Human Disease; Academic Press: Cambridge, MA, USA, 2017; pp. 967–988. [Google Scholar]
- Youssef, A.E.H.; Dief, A.E.; El Azhary, N.M.; Abdelmonsif, D.A.; El-Fetiany, O.S. LINGO-1 siRNA nanoparticles promote central remyelination in ethidium bromide-induced demyelination in rats. J. Physiol. Biochem. 2019, 75, 89–99. [Google Scholar] [CrossRef]
- Sharma, N.; Shandilya, A.; Kumar, N.; Mehan, S. Dysregulation of SIRT-1 Signaling in Multiple Sclerosis and Neuroimmune Disorders: A Systematic Review of SIRTUIN Activators as Potential Immunomodulators and their Influences on other Dysfunctions. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 1845–1868. [Google Scholar] [CrossRef] [PubMed]
- Goudarzvand, M.; Choopani, S.; Shams, A.; Javan, M.; Khodaii, Z.; Ghamsari, F.; Naghdi, N.; Piryaei, A.; Haghparast, A. Focal Injection of Ethidium Bromide as a Simple Model to Study Cognitive Deficit and Its Improvement. Basic Clin. Neurosci. J. 2016, 7, 63. [Google Scholar]
- Silva, B.A.; Ferrari, C.C. Animal Experimental Models for Understanding and Treating Multiple Sclerosis; SMGE Books: Dover, DE, USA, 2016; pp. 1–16. [Google Scholar]
- Sharma, N.; Upadhayay, S.; Shandilya, A.; Sahu, R.; Singh, A.; Rajkhowa, B.; Mehan, S. Neuroprotection by solanesol against ethidium bromide-induced multiple sclerosis-like neurobehavioral, molecular, and neurochemical alterations in experimental rats. Phytomedicine Plus 2021, 1, 100051. [Google Scholar] [CrossRef]
- Singh, A.; Upadhayay, S.; Mehan, S. Inhibition of c-JNK/p38MAPK signaling pathway by Apigenin prevents neurobehavioral and neurochemical defects in ethidium bromide-induced experimental model of multiple sclerosis in rats: Evidence from CSF, blood plasma and brain samples. Phytomedicine Plus 2021, 1, 100139. [Google Scholar] [CrossRef]
- Huppke, P.; Weissbach, S.; Church, J.A.; Schnur, R.; Krusen, M.; Dreha-Kulaczewski, S.; Kühn-Velten, W.N.; Wolf, A.; Huppke, B.; Millan, F.; et al. Activating de novo mutations in NFE2L2 encoding NRF2 cause a multisystem disorder. Nat. Commun. 2017, 8, 818. [Google Scholar] [CrossRef]
- Michaličková, D.; Hrnčíř, T.; Canová, N.K.; Slanař, O. Targeting Keap1/Nrf2/ARE signaling pathway in multiple sclerosis. Eur. J. Pharmacol. 2020, 873, 172973. [Google Scholar] [CrossRef]
- Funes, S.; Rios, M.; Fernández-Fierro, A.; Covián, C.; Bueno, S.M.; Riedel, C.A.; Mackern-Oberti, J.P.; Kalergis, A.M. Naturally Derived Heme-Oxygenase 1 Inducers and Their Therapeutic Application to Immune-Mediated Diseases. Front. Immunol. 2020, 11, 1467. [Google Scholar] [CrossRef]
- Upadhayay, S.; Mehan, S. Targeting Nrf2/HO-1 anti-oxidant signaling pathway in the progression of multiple sclerosis and influences on neurological dysfunctions. Brain Disord. 2021, 3, 100019. [Google Scholar] [CrossRef]
- Larabee, C.M.; Desai, S.; Agasing, A.; Georgescu, C.; Wren, J.D.; Axtell, R.C.; Plafker, S.M. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol. Vis. 2016, 22, 1503. [Google Scholar]
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The Absence of the Pro-antioxidant Transcription Factor Nrf2 Exacerbates Experimental Autoimmune Encephalomyelitis. Toxicol. Sci. 2010, 114, 237–246. [Google Scholar] [CrossRef]
- Minj, E.; Yadav, R.K.; Mehan, S. Targeting abnormal Nrf2/HO-1 signaling in amyotrophic lateral sclerosis: Current Insights on drug targets and influences on neurological disorders. Curr. Mol. Med. 2021, 21, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, S.; Qi, W.; Xu, X.; Liang, Y. Overexpression of miR-153 promotes oxidative stress in MPP+-induced PD model by negatively regulating the Nrf2/HO-1 signaling pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 4179. [Google Scholar] [PubMed]
- Moretti, D.; Tambone, S.; Cerretani, M.; Fezzardi, P.; Missineo, A.; Sherman, L.T.; Munoz-Sajuan, I.; Harper, S.; Dominquez, C.; Pacifici, R.; et al. NRF2 activation by reversible KEAP1 binding induces the antioxidant response in primary neurons and astrocytes of a Huntington’s disease mouse model. Free. Radic. Biol. Med. 2021, 162, 243–254. [Google Scholar] [CrossRef]
- Lv, C.; Maharjan, S.; Wang, Q.; Sun, Y.; Han, X.; Wang, S.; Mao, Z.; Xin, Y.; Zhang, B. α-Lipoic acid promotes neurological recovery after ischemic stroke by activating the Nrf2/HO-1 pathway to attenuate oxidative damage. Cell. Physiol. Biochem. 2017, 43, 1273–1287. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Dash, P.K.; Kan, Y.W.; Grotta, J.C.; Aronowski, J. Transcription Factor Nrf2 Protects the Brain From Damage Produced by Intracerebral Hemorrhage. Stroke 2007, 38, 3280–3286. [Google Scholar] [CrossRef]
- Sova, M.; Saso, L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug Des. Dev. Ther. 2018, 12, 3181. [Google Scholar] [CrossRef]
- Panieri, E.; Buha, A.; Telkoparan-Akillilar, P.; Cevik, D.; Kouretas, D.; Veskoukis, A.; Skaperda, Z.; Tsatsakis, A.; Wallace, D.; Suzen, S.; et al. Potential Applications of NRF2 Modulators in Cancer Therapy. Antioxidants 2020, 9, 193. [Google Scholar] [CrossRef]
- Fang, J.; Yin, H.; Yang, Z.; Tan, M.; Wang, F.; Chen, K.; Zuo, Z.; Shu, G.; Cui, H.; Ouyang, P.; et al. Vitamin E protects against cadmium-induced sub-chronic liver injury associated with the inhibition of oxidative stress and activation of Nrf2 pathway. Ecotoxicol. Environ. Saf. 2020, 208, 111610. [Google Scholar] [CrossRef]
- Su, X.; Wang, S.; Zhang, H.; Yang, G.; Bai, Y.; Liu, P.; Meng, L.; Jiang, X.; Xin, Y. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 through epigenetic modification. J. Cell. Mol. Med. 2021, 25, 4408–4419. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, Y.; Liu, Z.; Lu, Y.; Zhu, X.; Lan, B.; Mi, Z.; Dang, L.; Li, N.; Zhan, W.; et al. Activation of NRF2 ameliorates oxidative stress and cystogenesis in autosomal dominant polycystic kidney disease. Sci. Transl. Med. 2020, 12, eaba3613. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; de la Vega, M.R.; Wen, Q.; Bharara, M.; Jiang, T.; Zhang, R.; Zhou, S.; Wong, P.K.; Wondrak, G.T.; Zheng, H.; et al. An Essential Role of NRF2 in Diabetic Wound Healing. Diabetes 2016, 65, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the Phase 3 DEFINE and CONFIRM studies. Mult. Scler. J. 2017, 23, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Minj, E.; Upadhayay, S.; Mehan, S. Nrf2/HO-1 Signaling Activator Acetyl-11-keto-beta Boswellic Acid (AKBA)-Mediated Neuroprotection in Methyl Mercury-Induced Experimental Model of ALS. Neurochem. Res. 2021, 46, 2867–2884. [Google Scholar] [CrossRef]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Ageing Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; García-Yagüe, Á.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef]
- Xi, Z.; Chen, X.; Xu, C.; Wang, B.; Zhong, Z.; Sun, Q.; Sun, Y.; Bian, L. Protocatechuic acid attenuates brain edema and blood-brain barrier disruption after intracerebral hemorrhage in mice by promoting Nrf2/HO-1 pathway. NeuroReport 2020, 31, 1274–1282. [Google Scholar] [CrossRef]
- Rajabian, A.; Sadeghnia, H.R.; Hosseini, A.; Mousavi, S.H.; Boroushaki, M.T. 3-Acetyl-11-keto-β-boswellic acid attenuated oxidative glutamate toxicity in neuron-like cell lines by apoptosis inhibition. J Cell Biochem. 2020, 121, 1778–1789. [Google Scholar] [CrossRef]
- Al-Harrasi, A.; Rehman, N.U.; Khan, A.L.; Al-Broumi, M.; Al-Amri, I.; Hussain, J.; Hussain, H.; Csuk, R. Chemical, molecular and structural studies of Boswellia species: β-Boswellic Aldehyde and 3-epi-11β-Dihydroxy BA as precursors in biosynthesis of boswellic acids. PLoS ONE 2018, 13, e0198666. [Google Scholar] [CrossRef]
- Sayed, A.S.; Gomaa, I.E.O.; Bader, M.; Sayed, N.S.E.D.E. Role of 3-Acetyl-11-Keto-Beta-Boswellic Acid in Counteracting LPS-Induced Neuroinflammation via Modulation of miRNA-155. Mol. Neurobiol. 2018, 55, 5798–5808. [Google Scholar] [CrossRef] [PubMed]
- Conti, S.; Vexler, A.; Edry-Botzer, L.; Kalich-Philosoph, L.; Corn, B.W.; Shtraus, N.; Meir, Y.; Hagoel, L.; Shtabsky, A.; Marmor, S.; et al. Combined acetyl-11-keto-β-boswellic acid and radiation treatment inhibited glioblastoma tumor cells. PLoS ONE 2018, 13, e0198627. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhou, B.; Xu, W.; Xue, F.; Nisar, M.F.; Bian, C.; Huang, X.; Yang, L.; Zhang, Y.; Bartsch, J.W.; et al. Nrf2- and Bach1 May Play a Role in the Modulation of Ultraviolet A-Induced Oxidative Stress by Acetyl-11-Keto-β-Boswellic Acid in Skin Keratinocytes. Ski. Pharmacol. Physiol. 2017, 30, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, M.; Yang, Q.; Wang, M.; Wang, Z.; Zhu, Y.; Zhang, Y.; Wang, C.; Jia, Y.; Li, Y.; et al. Antioxidant effects of hydroxysafflor yellow A and acetyl-11-keto-β-boswellic acid in combination on isoproterenol-induced myocardial injury in rats. Int. J. Mol. Med. 2016, 37, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Ichikawa, H.; Badmaev, V.; Aggarwal, B.B. Acetyl-11-keto-β-boswellic acid potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis by suppressing NF-κB and NF-κB-regulated gene expression. J. Immunol. 2006, 176, 3127–3140. [Google Scholar] [CrossRef]
- Wei, C.; Fan, J.; Sun, X.; Yao, J.; Guo, Y.; Zhou, B.; Shang, Y. Acetyl-11-keto-β-boswellic acid ameliorates cognitive deficits and reduces amyloid-β levels in APPswe/PS1dE9 mice through antioxidant and anti-inflammatory pathways. Free Radic. Biol. Med. 2020, 150, 96–108. [Google Scholar] [CrossRef]
- Doaee, P.; Rajaei, Z.; Roghani, M.; Alaei, H.; Kamalinejad, M. Effects of Boswellia serrata resin extract on motor dysfunction and brain oxidative stress in an experimental model of Parkinson’s disease. Avicenna J. Phytomed. 2019, 9, 281–290. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, M.; Wang, M.; Li, Y.; Wen, A. Post treatment with 11-keto-β-boswellic acid ameliorates cerebral ischemia–reperfusion injury: Nrf2/HO-1 pathway as a potential mechanism. Mol. Neurobiol. 2015, 52, 1430–1439. [Google Scholar] [CrossRef]
- Marefati, N.; Beheshti, F.; Memarpour, S.; Bayat, R.; Shafei, M.N.; Sadeghnia, H.R.; Ghazavi, H.; Hosseini, M. The effects of acetyl-11-keto-β-boswellic acid on brain cytokines and memory impairment induced by lipopolysaccharide in rats. Cytokine 2020, 131, 155107. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, Y.; Zhang, B.; Fei, X.; Guo, X.; Jia, Y.; Yu, W. Acetyl-11-keto-β-boswellic acid regulates the repair of rat sciatic nerve injury by promoting the proliferation of Schwann cells. Life Sci. 2020, 254, 116887. [Google Scholar] [CrossRef]
- Mohtashami, L.; Shakeri, A.; Javadi, B. Neuroprotective natural products against experimental autoimmune encephalomyelitis: A review. Neurochem. Int. 2019, 129, 104516. [Google Scholar] [CrossRef] [PubMed]
- Stürner, K.H.; Verse, N.; Yousef, S.; Martin, R.; Sospedra, M. Boswellic acids reduce T h17 differentiation via blockade of IL-1β-mediated IRAK 1 signaling. Eur. J. Immunol. 2014, 44, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, M.; Wang, M.; Wang, M.; Zhang, T.; Park, J.; Zhu, Y.; Guo, C.; Jia, Y.; Li, Y.; et al. Neuroprotection by acetyl-11-keto-β-boswellic acid, in ischemic brain injury involves the Nrf2/HO-1 defense pathway. Sci. Rep. 2014, 4, 7002. [Google Scholar] [CrossRef]
- McMurran, C.E.; Zhao, C.; Franklin, R.J.M. Toxin-based models to investigate demyelination and remyelination Oligodendrocytes. Methods Mol. Biol. 2019, 1936, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, E.M.; Mehan, S.; Upadhayay, S.; Khan, A.; Halawi, M.; Halawi, A.A.; Alsaffar, R.M. Neuroprotective efficacy of 4-Hydroxyisoleucine in experimentally induced intracerebral hemorrhage. Saudi J. Biol. Sci. 2021, 28, 6417–6431. [Google Scholar] [CrossRef]
- Sahu, R.; Mehan, S.; Kumar, S.; Prajapati, A.; Alshammari, A.; Alharbi, M.; Assiri, M.A.; Narula, A.S. Effect of alpha-mangostin in the prevention of behavioural and neurochemical defects in methylmercury-induced neurotoxicity in experimental rats. Toxicol. Rep. 2022, 9, 977–998. [Google Scholar] [CrossRef]
- Duggal, P.; Jadaun, K.S.; Siqqiqui, E.M.; Mehan, S. Investigation of Low Dose Cabazitaxel Potential as Microtubule Stabilizer in Experimental Model of Alzheimer’s Disease: Restoring Neuronal Cytoskeleton. Curr. Alzheimer Res. 2020, 17, 601–615. [Google Scholar] [CrossRef]
- Mehan, S.; Verma, A.; Bedi, K.; Sehgal, V.; Gupta, A.; Meena, H.; Sharma, D. Effect of Mitogen Activated Protein Kinase Inhibitor in Animal Model of Alzheimer’s Diseases. Int. J. Pharma Prof. Res. 2011, 2, 212–223. [Google Scholar]
- Khera, H.; Mehan, S.; Dudi, R. Neuroprotective potential of Mangosteen in 3-nitropropionic acid induced Huntington’s disease like behavioral and biochemical alterations in rats. Pharmaspire 2018, 10, 121–131. [Google Scholar]
- Alam, M.M.; Minj, E.; Yadav, K.R.; Mehan, S. Neuroprotective Potential of Adenyl Cyclase/cAMP/CREB and Mitochondrial CoQ10 Activator in Amyotrophic Lateral Sclerosis Rats. Curr. Bioa. Com. 2021, 17. [Google Scholar] [CrossRef]
- Kumar, M.; Dandapat, S.; Sinha, M.P.; Kumar, A.; Raipat, B.S. Different blood collection methods from rats: A review. Balneo Res. J. 2017, 8, 46–50. [Google Scholar] [CrossRef]
- Kozler, P.; Sobek, O.; Pokorný, J. Signs of Myelin Impairment in Cerebrospinal Fluid After Osmotic Opening of the Blood-Brain Barrier in Rats. Physiol. Res. 2015, 64 (Suppl. 5), S603–S608. [Google Scholar] [CrossRef] [PubMed]
- Rosenling, T.; Stoop, M.P.; Attali, A.; van Aken, H.; Suidgeest, E.; Christin, C.; Stingl, C.; Suits, F.; Horvatovich, P.; Hintzen, R.Q.; et al. Profiling and Identification of Cerebrospinal Fluid Proteins in a Rat EAE Model of Multiple Sclerosis. J. Proteome Res. 2012, 11, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, D.A.; Smith, L.M.; Coffey, M.P.; Aasly, J.O.; LeWitt, P.A. CSF Nrf2 and HSPA8 in Parkinson’s disease patients with and without LRRK2 gene mutations. J. Neural Transm. 2015, 123, 179–187. [Google Scholar] [CrossRef]
- Schipper, H.M.; Chertkow, H.; Mehindate, K.; Frankel, D.; Melmed, C.; Bergman, H. Evaluation of heme oxygenase-1 as a systemic biological marker of sporadic AD. Neurology 2000, 54, 1297–1304. [Google Scholar] [CrossRef]
- Rahi, S.; Gupta, R.; Sharma, A.; Mehan, S. Smo-Shh signaling activator purmorphamine ameliorates neurobehavioral, molecular, and morphological alterations in an intracerebroventricular propionic acid-induced experimental model of autism. Hum. Exp. Toxicol. 2021, 40, 1880–1898. [Google Scholar] [CrossRef]
- Song, J.; Jiao, Y.; Zhang, T.; Zhang, Y.; Huang, X.; Li, H.; Wu, H. Longitudinal Changes in Plasma Caspase-1 and Caspase-3 during the First 2 Years of HIV-1 Infection in CD4Low and CD4High Patient Groups. PLoS ONE 2015, 10, e0121011. [Google Scholar] [CrossRef]
- Tiwari, A.; Khera, R.; Rahi, S.; Mehan, S.; Makeen, H.; Khormi, Y.; Rehman, M.; Khan, A. Neuroprotective Effect of α-Mangostin in Ameliorating Propionic Acid-Induced Experimental Model of Autism in Wistar Rats. Brain Sci. 2021, 11, 288. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, J.; Xiao, J. Correlation of the expression of inflammatory factors with expression of apoptosis-related genes Bax and Bcl-2, in burned rats. Exp. Ther. Med. 2019, 17, 1790–1796. [Google Scholar] [CrossRef]
- Sharma, R.; Rahi, S.; Mehan, S. Neuroprotective potential of solanesol in intracerebroventricular propionic acid induced experimental model of autism: Insights from behavioral and biochemical evidence. Toxicol. Rep. 2019, 6, 1164–1175. [Google Scholar] [CrossRef]
- Khera, R.; Mehan, S.; Bhalla, S.; Kumar, S.; Alshammari, A.; Alharbi, M.; Sadhu, S.S. Guggulsterone Mediated JAK/STAT and PPAR-Gamma Modulation Prevents Neurobehavioral and Neurochemical Abnormalities in Propionic Acid-Induced Experimental Model of Autism. Molecules 2022, 27, 889. [Google Scholar] [CrossRef] [PubMed]
- Rajdev, K.; Siddiqui, E.M.; Jadaun, K.S.; Mehan, S. Neuroprotective potential of solanesol in a combined model of intracerebral and intraventricular hemorrhage in rats [IBRO rep]. IBRO Rep. 2020, 8, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Mehan, S.; Sahu, R.; Kumar, S.; Khan, A.; Makeen, H.A.; Al Bratty, M. Protective effects of apigenin on methylmercury-induced behavioral/neurochemical abnormalities and neurotoxicity in rats. Hum. Exp. Toxicol. 2022, 41, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Mehan, S.; Monga, V.; Rani, M.; Dudi, R.; Ghimire, K. Neuroprotective effect of solanesol against 3-nitropropionic acid-induced Huntington’s disease-like behavioral, biochemical, and cellular alterations: Restoration of coenzyme-Q10-mediated mitochondrial dysfunction. Indian J. Pharmacol. 2018, 50, 309–319. [Google Scholar] [CrossRef]
- Sharma, A.; Bhalla, S.; Mehan, S. PI3K/AKT/mTOR signalling inhibitor chrysophanol ameliorates neurobehavioural and neurochemical defects in propionic acid-induced experimental model of autism in adult rats. Metab. Brain Dis. 2022, 37, 1909–1929. [Google Scholar] [CrossRef]
- Fu, K.; Chen, M.; Zheng, H.; Li, C.; Yang, F.; Niu, Q. Pelargonidin ameliorates MCAO-induced cerebral ischemia/reperfusion injury in rats by the action on the Nrf2/HO-1 pathway. Transl. Neurosci. 2021, 12, 20–31. [Google Scholar] [CrossRef]
- Gupta, R.; Mehan, S.; Sethi, P.; Prajapati, A.; Alshammari, A.; Alharbi, M.; Al-Mazroua, H.A.; Narula, A.S. Smo-Shh Agonist Purmorphamine Prevents Neurobehavioral and Neurochemical Defects in 8-OH-DPAT-Induced Experimental Model of Obsessive-Compulsive Disorder. Brain Sci. 2022, 12, 342. [Google Scholar] [CrossRef]
- Mehan, S.; Kaur, R.; Khanna, D.; Kalra, S.; Parveen, S. Precautionary Ellagic Acid Treatment Ameliorates Chronically Administered Scopolamine Induced Alzheimer’s Type Memory and Cognitive Dysfunctions in Rats. Pharmacologia 2015, 6, 192–212. [Google Scholar] [CrossRef]
- Yu, N.; Hu, S.; Hao, Z. Benificial effect of stachydrine on the traumatic brain injury induced neurodegeneration by attenuating the expressions of Akt/mTOR/PI3K and TLR4/NFκ-B pathway. Transl. Neurosci. 2018, 9, 175–182. [Google Scholar] [CrossRef]
- Deshmukh, R.; Sharma, V.; Mehan, S.; Sharma, N.; Bedi, K. Amelioration of intracerebroventricular streptozotocin induced cognitive dysfunction and oxidative stress by vinpocetine—A PDE1 inhibitor. Eur. J. Pharmacol. 2009, 620, 49–56. [Google Scholar] [CrossRef]
- Mehan, S.; Parveen, S.; Kalra, S. Adenyl cyclase activator forskolin protects against Huntington’s disease-like neurodegenerative disorders. Neural Regen. Res. 2017, 12, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Mehan, S.; Rahi, S.; Tiwari, A.; Kapoor, T.; Rajdev, K.; Sharma, R.; Khera, H.; Kosey, S.; Kukkar, U.; Dudi, R. Adenylate cyclase activator forskolin alleviates intracerebroventricular propionic acid-induced mitochondrial dysfunction of autistic rats. Neural Regen. Res. 2020, 15, 1140–1149. [Google Scholar] [CrossRef]
- Dudi, R.; Mehan, S. Neuroprotection of brain permeable Forskolin ameliorates behavioral, biochemical and histopathological alterations in rat model of intracerebral hemorrhage. Pharmaspire 2018, 10, 68–86. [Google Scholar]
- Bala, R.; Khanna, D.; Mehan, S.; Kalra, S. Experimental evidence for the potential of lycopene in the management of scopolamine induced amnesia. RSC Adv. 2015, 5, 72881–72892. [Google Scholar] [CrossRef]
- Beckmann, N.; Giorgetti, E.; Neuhaus, A.; Zurbruegg, S.; Accart, N.; Smith, P.; Perdoux, J.; Perrot, L.; Nash, M.; Desrayaud, S.; et al. Brain region-specific enhancement of remyelination and prevention of demyelination by the CSF1R kinase inhibitor BLZ945. Acta Neuropathol. Commun. 2018, 6, 9. [Google Scholar] [CrossRef]
- Trapp, B.D.; Vignos, M.; Dudman, J.; Chang, A.; Fisher, E.; Staugaitis, S.M.; Battapady, H.; Mork, S.; Ontaneda, D.; Jones, S.E.; et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: A retrospective study. Lancet Neurol. 2018, 17, 870–884. [Google Scholar] [CrossRef]
- Carassiti, D.; Altmann, D.R.; Petrova, N.; Pakkenberg, B.; Scaravilli, F.; Schmierer, K. Neuronal loss, demyelination and volume change in the multiple sclerosis neocortex. Neuropathol. Appl. Neurobiol. 2017, 44, 377–390. [Google Scholar] [CrossRef]
- Shandilya, A.; Mehan, S.; Kumar, S.; Sethi, P.; Narula, A.S.; Alshammari, A.; Alharbi, M.; Alasmari, A.F. Activation of IGF-1/GLP-1 Signalling via 4-Hydroxyisoleucine Prevents Motor Neuron Impairments in Experimental ALS-Rats Exposed to Methylmercury-Induced Neurotoxicity. Molecules 2022, 27, 3878. [Google Scholar] [CrossRef]
- Carleton, H.M.; Drury, R.A.; Wallington, E.A. Carleton’s Histological Technique; Oxford University Press: Oxford, UK, 1980. [Google Scholar]
- Fathimoghadam, H.; Farbod, Y.; Ghadiri, A.; Fatemi, R. Moderating effects of crocin on some stress oxidative markers in rat brain following demyelination with ethidium bromide. Heliyon 2019, 5, e01213. [Google Scholar] [CrossRef]
- Mazzanti, C.M.; Spanevello, R.; Ahmed, M.; Schmatz, R.; Mazzanti, A.; Salbego, F.Z.; Graça, D.L.; Sallis, E.S.; Morsch, V.M.; Schetinger, M.R.C. Cyclosporine A inhibits acetylcholinesterase activity in rats experimentally demyelinated with ethidium bromide. Int. J. Dev. Neurosci. 2007, 25, 259–264. [Google Scholar] [CrossRef]
- Beckmann, D.V.; Carvalho, F.; Mazzanti, C.M.; dos Santos, R.P.; Andrades, A.O.; Aiello, G.; Rippilinger, A.; Graça, D.L.; Abdalla, F.H.; Oliveira, L.S.; et al. Neuroprotective role of quercetin in locomotor activities and cholinergic neurotransmission in rats experimentally demyelinated with ethidium bromide. Life Sci. 2014, 103, 79–87. [Google Scholar] [CrossRef]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE signaling pathway: Key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef]
- Hammer, A.; Waschbisch, A.; Kuhbandner, K.; Bayas, A.; Lee, D.-H.; Duscha, A.; Haghikia, A.; Gold, R.; Linker, R.A. The NRF2 pathway as potential biomarker for dimethyl fumarate treatment in multiple sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 668–676. [Google Scholar] [CrossRef]
- Kooi, E.-J.; Van Horssen, J.; Witte, M.E.; Amor, S.; Bø, L.; Dijkstra, C.D.; Van Der Valk, P.; Geurts, J.J.G. Abundant extracellular myelin in the meninges of patients with multiple sclerosis. Neuropathol. Appl. Neurobiol. 2009, 35, 283–295. [Google Scholar] [CrossRef]
- Kim, J.K.; Mastronardi, F.G.; Wood, D.D.; Lubman, D.M.; Zand, R.; Moscarello, M.A. Multiple Sclerosis: An important role for post-translational modifications of myelin basic protein in pathogenesis. Mol. Cell. Proteom. 2003, 2, 453–462. [Google Scholar] [CrossRef]
- Salem, N.A.; Assaf, N.; Ismail, M.F.; Khadrawy, Y.A.; Samy, M. Ozone Therapy in Ethidium Bromide-Induced Demyelination in Rats: Possible Protective Effect. Cell. Mol. Neurobiol. 2016, 36, 943–954. [Google Scholar] [CrossRef]
- Lamers, K.J.; de Reus, H.P.; Jongen, P.J. Myelin basic protein in CSF as indicator of disease activity in multiple sclerosis. Mult. Scler. 1998, 4, 124–126. [Google Scholar] [CrossRef]
- Cohen, S.R.; Brooks, B.R.; Herndon, R.M.; McKhann, G.M. A diagnostic index of active demyelination: Myelin basic protein in cerebrospinal fluid. Ann. Neurol. 1980, 8, 25–31. [Google Scholar] [CrossRef]
- Weil, M.-T.; Möbius, W.; Winkler, A.; Ruhwedel, T.; Wrzos, C.; Romanelli, E.; Bennett, J.L.; Enz, L.; Goebels, N.; Nave, K.-A.; et al. Loss of Myelin Basic Protein Function Triggers Myelin Breakdown in Models of Demyelinating Diseases. Cell Rep. 2016, 16, 314–322. [Google Scholar] [CrossRef]
- Yang, L.; Tan, D.; Piao, H. Myelin Basic Protein Citrullination in Multiple Sclerosis: A Potential Therapeutic Target for the Pathology. Neurochem. Res. 2016, 41, 1845–1856. [Google Scholar] [CrossRef]
- Tan, M.; Ouyang, Y.; Jin, M.; Chen, M.; Liu, P.; Chao, X.; Chen, Z.; Chen, X.; Ramassamy, C.; Gao, Y.; et al. Downregulation of Nrf2/HO-1 pathway and activation of JNK/c-Jun pathway are involved in homocysteic acid-induced cytotoxicity in HT-22 cells. Toxicol Lett. 2013, 223, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Call, C.L.; De Biase, L.M.; Bergles, D.E. Neuron–glial interactions and neurotransmitter signaling to cells of the oligodendrocyte lineage. Patterning Cell Type Specif. Dev. CNS PNS 2020, 891–918. [Google Scholar]
- Butt, A.M.; Fern, R.; Matute, C. Neurotransmitter signaling in white matter. Glia 2014, 62, 1762–1779. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; Van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef]
- Schmierer, K.; Parkes, H.G.; So, P.-W.; An, S.F.; Brandner, S.; Ordidge, R.; Yousry, T.A.; Miller, D.H. High field (9.4 Tesla) magnetic resonance imaging of cortical grey matter lesions in multiple sclerosis. Brain 2010, 133, 858–867. [Google Scholar] [CrossRef]
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Kovacs, G.G.; Kutzelnigg, A.; Lassmann, H.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef]




















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upadhayay, S.; Mehan, S.; Prajapati, A.; Sethi, P.; Suri, M.; Zawawi, A.; Almashjary, M.N.; Tabrez, S. Nrf2/HO-1 Signaling Stimulation through Acetyl-11-Keto-Beta-Boswellic Acid (AKBA) Provides Neuroprotection in Ethidium Bromide-Induced Experimental Model of Multiple Sclerosis. Genes 2022, 13, 1324. https://doi.org/10.3390/genes13081324
Upadhayay S, Mehan S, Prajapati A, Sethi P, Suri M, Zawawi A, Almashjary MN, Tabrez S. Nrf2/HO-1 Signaling Stimulation through Acetyl-11-Keto-Beta-Boswellic Acid (AKBA) Provides Neuroprotection in Ethidium Bromide-Induced Experimental Model of Multiple Sclerosis. Genes. 2022; 13(8):1324. https://doi.org/10.3390/genes13081324
Chicago/Turabian StyleUpadhayay, Shubham, Sidharth Mehan, Aradhana Prajapati, Pranshul Sethi, Manisha Suri, Ayat Zawawi, Majed N. Almashjary, and Shams Tabrez. 2022. "Nrf2/HO-1 Signaling Stimulation through Acetyl-11-Keto-Beta-Boswellic Acid (AKBA) Provides Neuroprotection in Ethidium Bromide-Induced Experimental Model of Multiple Sclerosis" Genes 13, no. 8: 1324. https://doi.org/10.3390/genes13081324