
TWEAK and TNFα, Both TNF Ligand Family Members and Multiple Sclerosis-Related Cytokines, Induce Distinct Gene Response in Human Brain Microvascular Endothelial Cells
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture Reagents
2.2. Microarray Assay
2.3. Real-Time Quantitative PCR (qPCR)
2.4. Western Blot Analysis
3. Results
3.1. Modulation of Genes Involved in Leukocyte Extravasation at the 24 h Timepoint
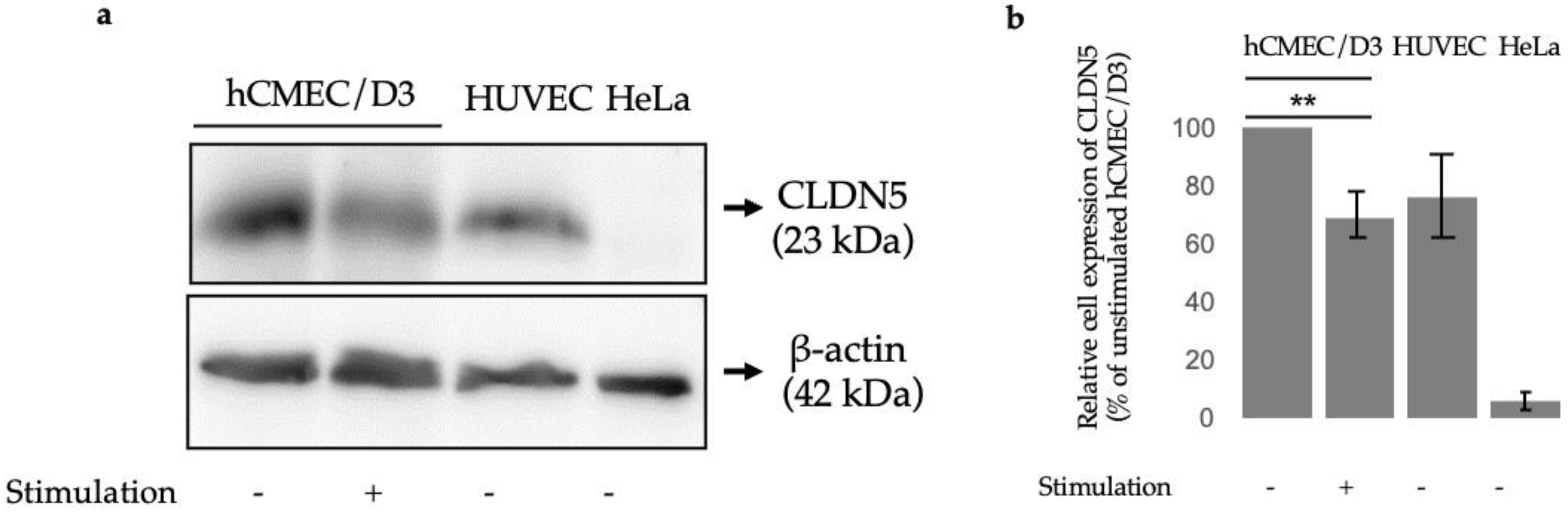
3.2. Claudin-5 Protein Expression after 24 h TWEAK Stimulation of hCMEC/D3 Cells
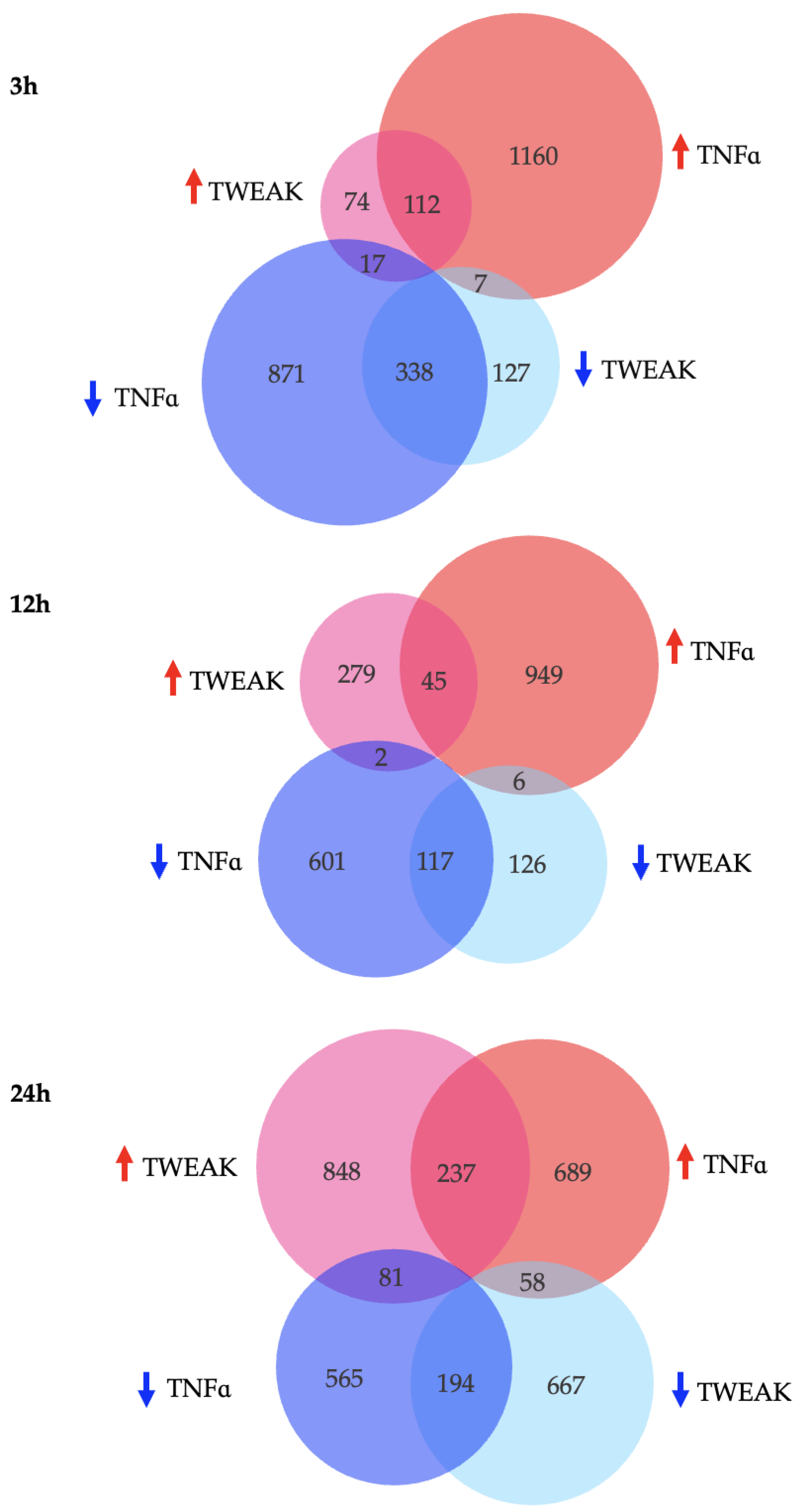
3.3. Overall Transcriptional Profiles of Human Brain Microvascular Endothelial Cells in Response to TWEAK or TNFα at 3, 12, and 24 h
3.4. Canonical Pathway Analysis after 24 h TWEAK or TNFα Incubation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chicheportiche, Y.; Bourdon, P.R.; Xu, H.; Hsu, Y.-M.; Scott, H.; Hession, C.; Garcia, I.; Browning, J.L. TWEAK, a New Secreted Ligand in the Tumor Necrosis Factor Family That Weakly Induces Apoptosis. J. Biol. Chem. 1997, 272, 32401–32410. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, M.; Raju, R.; Radhakrishnan, A.; Nanjappa, V.; Muthusamy, B.; Singh, K.; Kuppusamy, D.; Lingala, B.T.; Pan, A.; Mathur, P.P.; et al. A Bioinformatics Resource for TWEAK-Fn14 Signaling Pathway. J. Signal. Transduct. 2012, 2012, 376470. [Google Scholar] [CrossRef] [PubMed]
- Boulamery, A.; Desplat-Jégo, S. Regulation of Neuroinflammation: What Role for the Tumor Necrosis Factor-Like Weak Inducer of Apoptosis/Fn14 Pathway? Front. Immunol. 2017, 8, 1534. [Google Scholar] [CrossRef]
- Serafini, B.; Magliozzi, R.; Rosicarelli, B.; Reynolds, R.; Zheng, T.S.; Aloisi, F. Expression of TWEAK and Its Receptor Fn14 in the Multiple Sclerosis Brain: Implications for Inflammatory Tissue Injury. J. Neuropathol. Exp. Neurol. 2008, 67, 1137–1148. [Google Scholar] [CrossRef]
- Desplat-Jégo, S.; Varriale, S.; Creidy, R.; Terra, R.; Bernard, D.; Khrestchatisky, M.; Izui, S.; Chicheportiche, Y.; Boucraut, J. TWEAK Is Expressed by Glial Cells, Induces Astrocyte Proliferation and Increases EAE Severity. J. Neuroimmunol. 2002, 133, 116–123. [Google Scholar] [CrossRef]
- Desplat-Jégo, S.; Creidy, R.; Varriale, S.; Allaire, N.; Luo, Y.; Bernard, D.; Hahm, K.; Burkly, L.; Boucraut, J. Anti-TWEAK Monoclonal Antibodies Reduce Immune Cell Infiltration in the Central Nervous System and Severity of Experimental Autoimmune Encephalomyelitis. Clin. Immunol. 2005, 117, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, A.; Stephan, D.; Ranjeva, M.-P.; Ranjeva, J.-P.; Pelletier, J.; Audoin, B.; Khrestchatisky, M.; Desplat-Jégo, S. High Levels of Serum Soluble TWEAK Are Associated with Neuroinflammation during Multiple Sclerosis. J. Transl. Med. 2019, 17, 51. [Google Scholar] [CrossRef]
- Stephan, D.; Sbai, O.; Wen, J.; Couraud, P.-O.; Putterman, C.; Khrestchatisky, M.; Desplat-Jégo, S. TWEAK/Fn14 Pathway Modulates Properties of a Human Microvascular Endothelial Cell Model of Blood Brain Barrier. J. Neuroinflamm. 2013, 10, 781. [Google Scholar] [CrossRef]
- Lynch, C.N.; Wang, Y.C.; Lund, J.K.; Chen, Y.W.; Leal, J.A.; Wiley, S.R. TWEAK Induces Angiogenesis and Proliferation of Endothelial Cells. J. Biol. Chem. 1999, 274, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Saas, P.; Boucraut, J.; Walker, P.R.; Quiquerez, A.-L.; Billot, M.; Desplat-Jego, S.; Chicheportiche, Y.; Dietrich, P.-Y. TWEAK Stimulation of Astrocytes and the Proinflammatory Consequences. Glia 2000, 32, 102–107. [Google Scholar] [CrossRef]
- Weksler, B.B.; Subileau, E.A.; Perrière, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain Barrier-specific Properties of a Human Adult Brain Endothelial Cell Line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Yamaguchi, H.; Katsukura, Y.; Asashima, T.; Terasaki, T. MRNA Expression Levels of Tight Junction Protein Genes in Mouse Brain Capillary Endothelial Cells Highly Purified by Magnetic Cell Sorting. J. Neurochem. 2008, 104, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Blecharz, K.G.; Haghikia, A.; Stasiolek, M.; Kruse, N.; Drenckhahn, D.; Gold, R.; Roewer, N.; Chan, A.; Förster, C.Y. Glucocorticoid Effects on Endothelial Barrier Function in the Murine Brain Endothelial Cell Line CEND Incubated with Sera from Patients with Multiple Sclerosis. Mult. Scler. 2010, 16, 293–302. [Google Scholar] [CrossRef]
- Kluger, M.S.; Clark, P.R.; Tellides, G.; Gerke, V.; Pober, J.S. Claudin-5 Controls Intercellular Barriers of Human Dermal Microvascular but Not Human Umbilical Vein Endothelial Cells. ATVB 2013, 33, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Cunniffe, C.; Ryan, F.; Lambkin, H.; Brankin, B. Expression of Tight and Adherens Junction Proteins in Cervical Neoplasia. Br. J. Biomed. Sci. 2012, 69, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Luissint, A.-C.; Federici, C.; Guillonneau, F.; Chrétien, F.; Camoin, L.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O. Guanine Nucleotide-Binding Protein Gαi2: A New Partner of Claudin-5 That Regulates Tight Junction Integrity in Human Brain Endothelial Cells. J. Cereb. Blood Flow Metab. 2012, 32, 860–873. [Google Scholar] [CrossRef]
- Stewart, R.J.; Kashour, T.S.; Marsden, P.A. Vascular Endothelial Platelet Endothelial Adhesion Molecule-1 (PECAM-1) Expression Is Decreased by TNF-Alpha and IFN-Gamma. Evidence for Cytokine-Induced Destabilization of Messenger Ribonucleic Acid Transcripts in Bovine Endothelial Cells. J. Immunol. 1996, 156, 1221–1228. [Google Scholar]
- Willis, C.L.; Meske, D.S.; Davis, T.P. Protein Kinase C Activation Modulates Reversible Increase in Cortical Blood–Brain Barrier Permeability and Tight Junction Protein Expression during Hypoxia and Posthypoxic Reoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1847–1859. [Google Scholar] [CrossRef]
- Diouf, B.; Cheng, Q.; Krynetskaia, N.F.; Yang, W.; Cheok, M.; Pei, D.; Fan, Y.; Cheng, C.; Krynetskiy, E.Y.; Geng, H.; et al. Somatic Deletions of Genes Regulating MSH2 Protein Stability Cause DNA Mismatch Repair Deficiency and Drug Resistance in Human Leukemia Cells. Nat. Med. 2011, 17, 1298–1303. [Google Scholar] [CrossRef]
- Morey, J.S.; Ryan, J.C.; Van Dolah, F.M. Microarray Validation: Factors Influencing Correlation between Oligonucleotide Microarrays and Real-Time PCR. Biol. Proced. Online 2006, 8, 175–193. [Google Scholar] [CrossRef]
- Etienne, W.; Meyer, M.H.; Peppers, J.; Meyer, R.A. Comparison of MRNA Gene Expression by RT-PCR and DNA Microarray. BioTechniques 2004, 36, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-Selective Loosening of the Blood-Brain Barrier in Claudin-5–Deficient Mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Campbell, M. The Dynamic Blood-Brain Barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Saint-Laurent, O.; Godschalk, A.; Terouz, S.; Briels, C.; Larouche, S.; Bourbonnière, L.; Larochelle, C.; Prat, A. Focal Disturbances in the Blood–Brain Barrier Are Associated with Formation of Neuroinflammatory Lesions. Neurobiol. Dis. 2015, 74, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Basivireddy, J.; Kollar, A.; Biron, K.E.; Reickmann, P.; Jefferies, W.A.; McQuaid, S. Blood–Brain Barrier Disruption and Enhanced Vascular Permeability in the Multiple Sclerosis Model EAE. J. Neuroimmunol. 2010, 229, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Sirivichayakul, S.; Kanchanatawan, B.; Vodjani, A. Breakdown of the Paracellular Tight and Adherens Junctions in the Gut and Blood Brain Barrier and Damage to the Vascular Barrier in Patients with Deficit Schizophrenia. Neurotox. Res. 2019, 36, 306–322. [Google Scholar] [CrossRef]
- Kiliç, F.; Işik, Ü.; Usta, A.; Demirdaş, A. Serum Tumor Necrosis Factor-like Weak Inducer of Apoptosis Levels Are Elevated in Schizophrenia. Braz. J. Psychiatry 2021, 43, 242–246. [Google Scholar] [CrossRef]
- Usta, A.; Kılıç, F.; Demirdaş, A.; Işık, Ü.; Doğuç, D.K.; Bozkurt, M. Serum Zonulin and Claudin-5 Levels in Patients with Schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 767–773. [Google Scholar] [CrossRef]
- Mueller, A.M.; Pedré, X.; Kleiter, I.; Hornberg, M.; Steinbrecher, A.; Giegerich, G. Targeting Fibroblast Growth Factor-Inducible-14 Signaling Protects from Chronic Relapsing Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2005, 159, 55–65. [Google Scholar] [CrossRef]
- Maecker, H.; Varfolomeev, E.; Kischkel, F.; Lawrence, D.; LeBlanc, H.; Lee, W.; Hurst, S.; Danilenko, D.; Li, J.; Filvaroff, E.; et al. TWEAK Attenuates the Transition from Innate to Adaptive Immunity. Cell 2005, 123, 931–944. [Google Scholar] [CrossRef]
- Zhang, X.W.; Schramm, R.; Liu, Q.; Ekberg, H.; Jeppsson, B.; Thorlacius, H. Important Role of CD18 in TNF-a-Induced Leukocyte Adhesion in Muscle and Skin Venules in Vivo. Inflamm. Res. 2000, 49, 529–534. [Google Scholar] [CrossRef]
- Zeng, M.; Zhang, H.; Lowell, C.; He, P. Tumor Necrosis Factor-α-Induced Leukocyte Adhesion and Microvessel Permeability. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H2420–H2430. [Google Scholar] [CrossRef] [PubMed]
- Petrache, I.; Verin, A.D.; Crow, M.T.; Birukova, A.; Liu, F.; Garcia, J.G. Differential Effect of MLC Kinase in TNF-Alpha-Induced Endothelial Cell Apoptosis and Barrier Dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1168–L1178. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the Regulation of Endothelial Permeability. Vascul. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- Abu Taha, A.; Schnittler, H.-J. Dynamics between Actin and the VE-Cadherin/Catenin Complex: Novel Aspects of the ARP2/3 Complex in Regulation of Endothelial Junctions. Cell Adh. Migr. 2014, 8, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Szulcek, R.; Beckers, C.M.L.; Hodzic, J.; de Wit, J.; Chen, Z.; Grob, T.; Musters, R.J.P.; Minshall, R.D.; van Hinsbergh, V.W.M.; van Nieuw Amerongen, G.P. Localized RhoA GTPase Activity Regulates Dynamics of Endothelial Monolayer Integrity. Cardiovasc. Res. 2013, 99, 471–482. [Google Scholar] [CrossRef]
- Heemskerk, N.; Schimmel, L.; Oort, C.; van Rijssel, J.; Yin, T.; Ma, B.; van Unen, J.; Pitter, B.; Huveneers, S.; Goedhart, J.; et al. F-Actin-Rich Contractile Endothelial Pores Prevent Vascular Leakage during Leukocyte Diapedesis through Local RhoA Signalling. Nat. Commun 2016, 7, 10493. [Google Scholar] [CrossRef]
- Marcos-Ramiro, B.; García-Weber, D.; Millán, J. TNF-Induced Endothelial Barrier Disruption: Beyond Actin and Rho. Thromb. Haemost 2014, 112, 1088–1102. [Google Scholar] [CrossRef]
- Zhang, Z.; Schittenhelm, J.; Meyermann, R.; Schluesener, H.J. Lesional Accumulation of RhoA + Cells in Brains of Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Neuropathol. Appl. Neurobiol. 2008, 34, 231–240. [Google Scholar] [CrossRef]
- Gavard, J.; Patel, V.; Gutkind, J.S. Angiopoietin-1 Prevents VEGF-Induced Endothelial Permeability by Sequestering Src through MDia. Dev. Cell 2008, 14, 25–36. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-Mediated Disruption of Endothelial CLN-5 Promotes Blood-Brain Barrier Breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karar, J.; Maity, A. PI3K/AKT/MTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef]
- Hamada, K.; Sasaki, T.; Koni, P.A.; Natsui, M.; Kishimoto, H.; Sasaki, J.; Yajima, N.; Horie, Y.; Hasegawa, G.; Naito, M.; et al. The PTEN/PI3K Pathway Governs Normal Vascular Development and Tumor Angiogenesis. Genes Dev. 2005, 19, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wang, Y.; Song, X.; Ning, H.; Zhang, Y.; Teng, Y.; Wang, J.; Yang, X. Brain Endothelial PTEN/AKT/NEDD4-2/MFSD2A Axis Regulates Blood-Brain Barrier Permeability. Cell Rep. 2021, 36, 109327. [Google Scholar] [CrossRef] [PubMed]
- Ikner, A.; Ashkenazi, A. TWEAK Induces Apoptosis through a Death-Signaling Complex Comprising Receptor-Interacting Protein 1 (RIP1), Fas-Associated Death Domain (FADD), and Caspase-8. J. Biol. Chem. 2011, 286, 21546–21554. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Schwenzer, R.; Haas, E.; Mühlenbeck, F.; Schubert, G.; Scheurich, P.; Tschopp, J.; Wajant, H. TWEAK Can Induce Cell Death via Endogenous TNF and TNF Receptor 1. Eur. J. Immunol. 1999, 29, 1785–1792. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
 | Gene | TWEAK | TNFα |
| GNAI2 | 2.913 | ||
| PXN | 2.824 | −1.453 | |
| ACTC1 | 2.447 | ||
| THY1 | 2.400 | −1.650 | |
| RAC2 | 2.217 | −1.045 | |
| CTNND1 | 2.199 | ||
| ACTB | 1.988 | −1.421 | |
| MMP9 | 1.912 | 1.206 | |
| ITGA3 | 1.658 | ||
| CLDN11 | 1.646 | −1.329 | |
| CXCL12 | 1.571 | 3.635 | |
| PECAM1 | 2.791 | ||
| CD44 | −2.058 | ||
| CLDN5 | −1.771 | −1.153 | |
| JAM1 | −1.988 | −1.332 | |
| CLDN23 | −2.214 | 1.952 | |
| PIK3C2B | −2.549 | −1.093 | |
| PRKCZ | −2.889 |
 | Gene | TWEAK | TNFα | ||
| Microarray | qPCR | Microarray | qPCR | ||
| THY1 | 2.40 | −1.11 | −1.65 | 1.14 | |
| CXCL12 | 1.57 | −1.04 | 3.64 | 2.17 | |
| CLDN5 | −1.77 | −6.08 | −1.15 | −4.99 | |
| JAM1 | −1.99 | −1.47 | −1.33 | 1.11 | |
| TWEAK | TNFα | |||
|---|---|---|---|---|
| Z-Score | p-Value | Z-Score | p-Value | |
| Canonical Pathways | ||||
| Integrin Signaling | 3.570 | *** | 2.449 | ** |
| VEGF Signaling | 3.530 | *** | 1.698 | *** |
| Actin Cytoskeleton Signaling | 3.430 | *** | 2.335 | *** |
| Ephrin Receptor Signaling | 3.380 | *** | 2.887 | |
| Signaling By Rho Family GTPases | 3.240 | *** | 1.134 | *** |
| Regulation Of Actin-base Motility By Rho | 2.887 | ** | 1.732 | ** |
| Amyotrophic Lateral Sclerosis Signaling | 2.830 | *** | 0.577 | |
| IL-8 Signaling | 2.530 | *** | 1.800 | *** |
| RhoA Signaling | 2.357 | ** | 2.183 | ** |
| Rac Signaling | 2.183 | ** | 1.414 | *** |
| NGF Signaling | 2.065 | ** | 0.302 | |
| Neuroinflammation Signaling Pathway | 1.915 | * | 0.174 | ** |
| Leukocyte Extravasation Signaling | 1.877 | ** | −0.218 | ** |
| Death Receptor Signaling | 0.000 | ** | 2.138 | ** |
| Sirtuin Signaling Pathway | −0.392 | ** | 2.400 | ** |
| RhoGDI Signaling | −2.236 | ** | −0.218 | ** |
| PTEN Signaling | −3.578 | *** | −2.065 | *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stephan, D.; Roger, A.; Aghzadi, J.; Carmona, S.; Picard, C.; Dales, J.-P.; Desplat-Jégo, S. TWEAK and TNFα, Both TNF Ligand Family Members and Multiple Sclerosis-Related Cytokines, Induce Distinct Gene Response in Human Brain Microvascular Endothelial Cells. Genes 2022, 13, 1714. https://doi.org/10.3390/genes13101714
Stephan D, Roger A, Aghzadi J, Carmona S, Picard C, Dales J-P, Desplat-Jégo S. TWEAK and TNFα, Both TNF Ligand Family Members and Multiple Sclerosis-Related Cytokines, Induce Distinct Gene Response in Human Brain Microvascular Endothelial Cells. Genes. 2022; 13(10):1714. https://doi.org/10.3390/genes13101714
Chicago/Turabian StyleStephan, Delphine, Anais Roger, Jehanne Aghzadi, Sylvie Carmona, Christophe Picard, Jean-Philippe Dales, and Sophie Desplat-Jégo. 2022. "TWEAK and TNFα, Both TNF Ligand Family Members and Multiple Sclerosis-Related Cytokines, Induce Distinct Gene Response in Human Brain Microvascular Endothelial Cells" Genes 13, no. 10: 1714. https://doi.org/10.3390/genes13101714