
The Role of Cytotoxic T-Lymphocyte Antigen 4 in the Pathogenesis of Multiple Sclerosis


Abstract
:
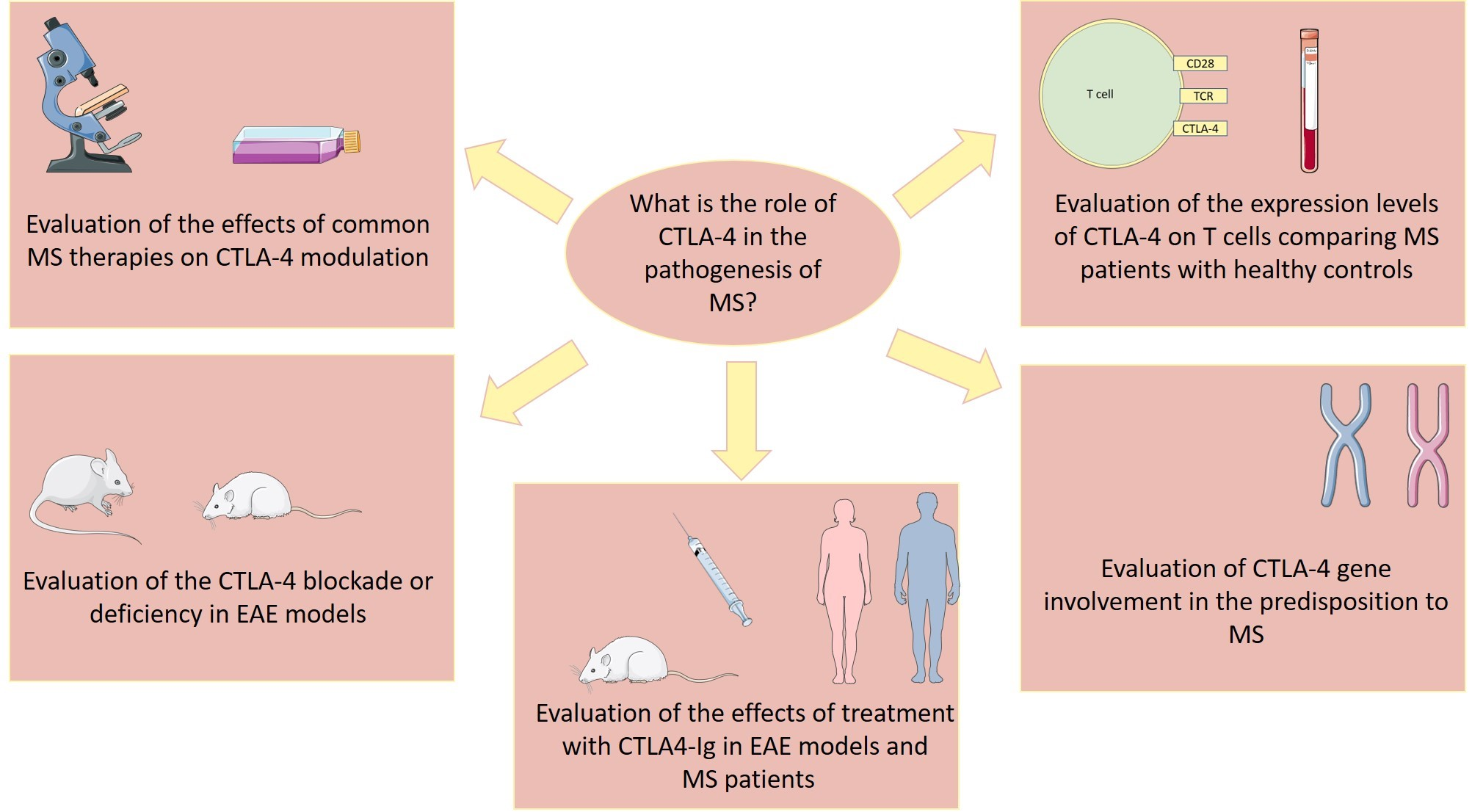
1. Introduction
2. Preclinical Studies
2.1. In Silico, In Vitro, and Ex Vivo Studies
2.2. In Vivo Studies
3. Clinical studies
3.1. Genetic Studies
3.2. Other Clinical Studies
3.2.1. Clinical Studies Investigating the Effects of Common MS Therapies on CTLA-4
3.2.2. Case Reports
3.3. Clinical Trials
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D.; Author, C. Diagnosis and Treatment of Multiple Sclerosis A Review Clinical Review & Education JAMA|Review. Number 2021, 325, 765. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, A.R.; Robertson, N.; la Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Prim. 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Schiess, N.; Calabresi, P.A. Multiple Sclerosis. Semin. Neurol. 2016, 36, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Vandercappellen, J.; King, M.; Brichetto, G. Symptom Interconnectivity in Multiple Sclerosis: A Narrative Review of Potential Underlying Biological Disease Processes. Neurol. Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue “Crosstalk between Depression, Anxiety, and Dementia: Comorbidity in Behavioral Neurology and Neuropsychiatry”. Biomedicines 2021, 9, 517. [Google Scholar] [CrossRef]
- Calabrese, M.; Agosta, F.; Rinaldi, F.; Mattisi, I.; Grossi, P.; Favaretto, A.; Atzori, M.; Bernardi, V.; Barachino, L.; Rinaldi, L.; et al. Cortical lesions and atrophy associated with cognitive impairment in relapsing-remitting multiple sclerosis. Arch. Neurol. 2009, 66, 1144–1150. [Google Scholar] [CrossRef] [Green Version]
- Benedict, R.H.B.; Amato, M.P.; DeLuca, J.; Geurts, J.J.G. Cognitive impairment in multiple sclerosis: Clinical management, MRI, and therapeutic avenues. Lancet. Neurol. 2020, 19, 860–871. [Google Scholar] [CrossRef]
- Battaglia, S.; Serio, G.; Scarpazza, C.; D’Ausilio, A.; Borgomaneri, S. Frozen in (e)motion: How reactive motor inhibition is influenced by the emotional content of stimuli in healthy and psychiatric populations. Behav. Res. Ther. 2021, 146, 103963. [Google Scholar] [CrossRef]
- Battaglia, S.; Fabius, J.H.; Moravkova, K.; Fracasso, A.; Borgomaneri, S. The Neurobiological Correlates of Gaze Perception in Healthy Individuals and Neurologic Patients. Biomedicines 2022, 10, 627. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, S.; Thayer, J.F. Functional interplay between central and autonomic nervous systems in human fear conditioning. Trends Neurosci. 2022, 45, 504–506. [Google Scholar] [CrossRef]
- Battaglia, S.; Orsolini, S.; Borgomaneri, S.; Barbieri, R.; Diciotti, S.; di Pellegrino, G. Characterizing cardiac autonomic dynamics of fear learning in humans. Psychophysiology 2022, e14122. [Google Scholar] [CrossRef]
- Komatsu, H.; Watanabe, E.; Fukuchi, M. Psychiatric neural networks and precision therapeutics by machine learning. Biomedicines 2021, 9, 403. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, G.; Gambino, C.M.; Sasso, B.L.; Scazzone, C.; Giglio, R.V.; Agnello, L.; Ciaccio, M. Serum Vitamin D as a Biomarker in Autoimmune, Psychiatric and Neurodegenerative Diseases. Diagnostics 2022, 12, 130. [Google Scholar] [CrossRef]
- Zheng, C.; He, L.; Liu, L.; Zhu, J.; Jin, T. The efficacy of vitamin D in multiple sclerosis: A meta-analysis. Mult. Scler. Relat. Disord. 2018, 23, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Doosti-Irani, A.; Tamtaji, O.R.; Mansournia, M.A.; Ghayour- Mobarhan, M.; Ferns, G.; Daneshvar Kakhaki, R.; Rezaei Shahmirzadi, A.; Asemi, Z. The effects of vitamin D supplementation on expanded disability status scale in people with multiple sclerosis: A critical, systematic review and metaanalysis of randomized controlled trials. Clin. Neurol. Neurosurg. 2019, 187, 105564. [Google Scholar] [CrossRef] [PubMed]
- Quirant-Sánchez, B.; Mansilla, M.J.; Navarro-Barriuso, J.; Presas-Rodríguez, S.; Teniente-Serra, A.; Fondelli, F.; Ramo-Tello, C.; Martínez-Cáceres, E. Combined therapy of vitamin d3-tolerogenic dendritic cells and interferon-β in a preclinical model of multiple sclerosis. Biomedicines 2021, 9, 1758. [Google Scholar] [CrossRef]
- Majlath, Z.; Annus, A.; Vecsei, L. Kynurenine System and Multiple Sclerosis, Pathomechanism and Drug Targets with An Emphasis on Laquinimod. Curr. Drug Targets 2018, 19, 805–814. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Monitoring the kynurenine system: Concentrations, ratios or what else? Adv. Clin. Exp. Med. 2021, 30, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the etiological links behind neurodegenerative diseases: Inflammatory cytokines and bioactive kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Song, F.; Fernandez-Escobar, A.; Luo, G.; Wang, J.-H.; Sun, Y. The Properties of Cytokines in Multiple Sclerosis: Pros and Cons. Am. J. Med. Sci. 2018, 356, 552–560. [Google Scholar] [CrossRef]
- Cavalli, E.; Mazzon, E.; Basile, M.S.; Mammana, S.; Pennisi, M.; Fagone, P.; Kalfin, R.; Martinovic, V.; Ivanovic, J.; Andabaka, M.; et al. In Silico and In Vivo Analysis of IL37 in Multiple Sclerosis Reveals Its Probable Homeostatic Role on the Clinical Activity, Disability, and Treatment with Fingolimod. Molecules 2019, 25, 20. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, E.; Mazzon, E.; Basile, M.S.; Mangano, K.; Di Marco, R.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Upregulated Expression of Macrophage Migration Inhibitory Factor, Its Analogue D-Dopachrome Tautomerase, and the CD44 Receptor in Peripheral CD4 T Cells from Clinically Isolated Syndrome Patients with Rapid Conversion to Clinical Defined Multiple Sclerosis. Medicina 2019, 55, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammana, S.; Bramanti, P.; Mazzon, E.; Cavalli, E.; Basile, M.S.; Fagone, P.; Petralia, M.C.; McCubrey, J.A.; Nicoletti, F.; Mangano, K. Preclinical evaluation of the PI3K/Akt/mTOR pathway in animal models of multiple sclerosis. Oncotarget 2018, 9, 8263–8277. [Google Scholar] [CrossRef] [Green Version]
- Dello Russo, C.; Lisi, L.; Feinstein, D.L.; Navarra, P. mTOR kinase, a key player in the regulation of glial functions: Relevance for the therapy of multiple sclerosis. Glia 2013, 61, 301–311. [Google Scholar] [CrossRef]
- Cotsapas, C.; Mitrovic, M.; Hafler, D. Multiple sclerosis. Handb. Clin. Neurol. 2018, 148, 723–730. [Google Scholar] [CrossRef]
- Immovilli, P.; Morelli, N.; Terracciano, C.; Rota, E.; Marchesi, E.; Vollaro, S.; De Mitri, P.; Zaino, D.; Bazzurri, V.; Guidetti, D. Multiple Sclerosis Treatment in the COVID-19 Era: A Risk-Benefit Approach. Neurol. Int. 2022, 14, 368–377. [Google Scholar] [CrossRef]
- Avan, R.; Sahebnasagh, A.; Hashemi, J.; Monajati, M.; Faramarzi, F.; Henney, N.C.; Montecucco, F.; Jamialahmadi, T.; Sahebkar, A. Update on Statin Treatment in Patients with Neuropsychiatric Disorders. Life 2021, 11, 1365. [Google Scholar] [CrossRef]
- Abdalla, M.A.; Zakhary, C.M.; Rushdi, H.; Hamdan, J.A.; Youssef, K.N.; Khan, A.; Khan, S. The Effectiveness of Statins as Potential Therapy for Multiple Sclerosis: A Systematic Review of Randomized Controlled trials. Cureus 2021, 13, e18092. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, D.; Montalban, X. Reaching an evidence-based prognosis for personalized treatment of multiple sclerosis. Nat. Rev. Neurol. 2019, 15, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Vandenbroeck, K. Pharmacogenomics and multiple sclerosis: Moving toward individualized medicine. Curr. Neurol. Neurosci. Rep. 2011, 11, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Aguilar, L.; Pérez-Ramírez, C.; Maldonado-Montoro, M.d.M.; Carrasco-Campos, M.I.; Membrive-Jiménez, C.; Martínez-Martínez, F.; García-Collado, C.; Calleja-Hernández, M.Á.; Ramírez-Tortosa, M.C.; Jiménez-Morales, A. Effect of genetic polymorphisms on therapeutic response in multiple sclerosis relapsing-remitting patients treated with interferon-beta. Mutat. Res.-Rev. Mutat. Res. 2020, 785, 108322. [Google Scholar] [CrossRef]
- Zarzuelo-Romero, M.J.; Pérez-Ramírez, C.; Cura, Y.; Carrasco-Campos, M.I.; Marangoni-Iglecias, L.M.; Ramírez-Tortosa, M.C.; Jiménez-Morales, A. Influence of genetic polymorphisms on clinical outcomes of glatiramer acetate in multiple sclerosis patients. J. Pers. Med. 2021, 11, 1032. [Google Scholar] [CrossRef]
- Zarzuelo Romero, M.J.; Pérez Ramírez, C.; Carrasco Campos, M.I.; Sánchez Martín, A.; Calleja Hernández, M.Á.; Ramírez Tortosa, M.C.; Jiménez Morales, A. Therapeutic value of single nucleotide polymorphisms on the efficacy of new therapies in patients with multiple sclerosis. J. Pers. Med. 2021, 11, 335. [Google Scholar] [CrossRef]
- Fagone, P.; Mazzon, E.; Mammana, S.; Di Marco, R.; Spinasanta, F.; Basile, M.; Petralia, M.; Bramanti, P.; Nicoletti, F.; Mangano, K. Identification of CD4+ T cell biomarkers for predicting the response of patients with relapsing-remitting multiple sclerosis to natalizumab treatment. Mol. Med. Rep. 2019, 20, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Ziemssen, T.; Akgün, K.; Brück, W. Molecular biomarkers in multiple sclerosis. J. Neuroinflam. 2019, 16, 272. [Google Scholar] [CrossRef] [Green Version]
- Basile, M.S.; Mazzon, E.; Mangano, K.; Pennisi, M.; Petralia, M.C.; Lombardo, S.D.; Nicoletti, F.; Fagone, P.; Cavalli, E. Impaired expression of tetraspanin 32 (TSPAN32) in memory T cells of patients with multiple sclerosis. Brain Sci. 2020, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Matute-Blanch, C.; Río, J.; Villar, L.M.; Midaglia, L.; Malhotra, S.; Álvarez-Cermeño, J.C.; Vidal-Jordana, A.; Montalban, X.; Comabella, M. Chitinase 3-like 1 is associated with the response to interferon-beta treatment in multiple sclerosis. J. Neuroimmunol. 2017, 303, 62–65. [Google Scholar] [CrossRef]
- Novakova, L.; Zetterberg, H.; Sundström, P.; Axelsson, M.; Khademi, M.; Gunnarsson, M.; Malmeström, C.; Svenningsson, A.; Olsson, T.; Piehl, F.; et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology 2017, 89, 2230–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Vécsei, L. Monitoring the redox status in multiple sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Afshar, B.; Khalifehzadeh-Esfahani, Z.; Seyfizadeh, N.; Rezaei Danbaran, G.; Hemmatzadeh, M.; Mohammadi, H. The role of immune regulatory molecules in multiple sclerosis. J. Neuroimmunol. 2019, 337, 577061. [Google Scholar] [CrossRef] [PubMed]
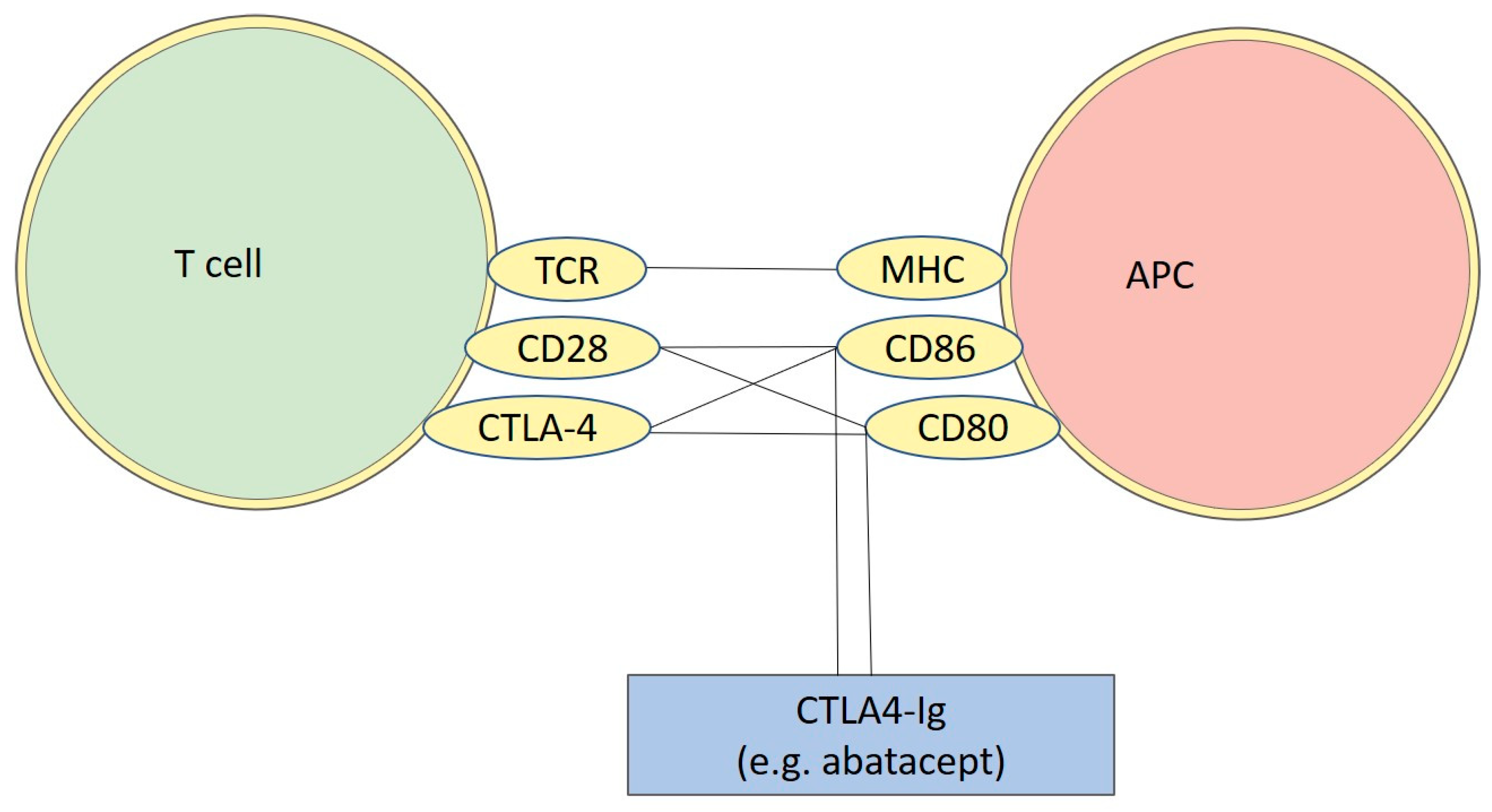
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhu, H.X.; Yao, Y.; Bian, Z.H.; Zheng, Y.J.; Li, L.; Moutsopoulos, H.M.; Gershwin, M.E.; Lian, Z.X. Immune checkpoint molecules. Possible future therapeutic implications in autoimmune diseases. J. Autoimmun. 2019, 104, 102333. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Edner, N.M.; Carlesso, G.; Rush, J.S.; Walker, L.S.K. Targeting co-stimulatory molecules in autoimmune disease. Nat. Rev. Drug Discov. 2020, 19, 860–883. [Google Scholar] [CrossRef]
- Watanabe, N.; Nakajima, H. Coinhibitory molecules in autoimmune diseases. Clin. Dev. Immunol. 2012, 2012, 269756. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, C.R.; Jayswal, R.; Adams, V.; Anthony, L.B.; Villano, J.L. Multiple Sclerosis Outcomes after Cancer Immunotherapy. Clin. Transl. Oncol. 2019, 21, 1336. [Google Scholar] [CrossRef] [PubMed]
- Gettings, E.J.; Hackett, C.T.; Scott, T.F. Severe relapse in a multiple sclerosis patient associated with ipilimumab treatment of melanoma. Mult. Scler. J. 2015, 21, 670. [Google Scholar] [CrossRef]
- Cao, Y.; Nylander, A.; Ramanan, S.; Goods, B.A.; Ponath, G.; Zabad, R.; Chiang, V.L.S.; Vortmeyer, A.O.; Hafler, D.A.; Pitt, D. CNS demyelination and enhanced myelin-reactive responses after ipilimumab treatment. Neurology 2016, 86, 1553–1556. [Google Scholar] [CrossRef] [Green Version]
- Cuzzubbo, S.; Javeri, F.; Tissier, M.; Roumi, A.; Barlog, C.; Doridam, J.; Lebbe, C.; Belin, C.; Ursu, R.; Carpentier, A.F. Neurological adverse events associated with immune checkpoint inhibitors: Review of the literature. Eur. J. Cancer 2017, 73, 1–8. [Google Scholar] [CrossRef]
- Fellner, A.; Makranz, C.; Lotem, M.; Bokstein, F.; Taliansky, A.; Rosenberg, S.; Blumenthal, D.T.; Mandel, J.; Fichman, S.; Kogan, E.; et al. Neurologic complications of immune checkpoint inhibitors. J. Neurooncol. 2018, 137, 601–609. [Google Scholar] [CrossRef]
- Dalakas, M.C. Neurological complications of immune checkpoint inhibitors: What happens when you ‘take the brakes off’ the immune system. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418799864. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, L.A.; Held, K.; Beltrán, E.; Berking, C.; Prinz, J.C.; Junker, A.; Tietze, J.K.; Ertl-Wagner, B.; Straube, A.; Kümpfel, T.; et al. CTLA4 as Immunological Checkpoint in the Development of Multiple Sclerosis. Ann. Neurol. 2016, 80, 294. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; Rosenzweig, N.; Tsitsou-Kampeli, A.; Sharif, A.M.; Matcovitch-Natan, O.; Kertser, A.; David, E.; Amit, I.; Schwartz, M. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat. Med. 2016, 22, 135–137. [Google Scholar] [CrossRef]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2019, 141, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Maerz, M.D.; Bahnson, H.T.; Somasundaram, A.; McCarthy, L.H.; Speake, C.; Buckner, J.H. Fewer LAG-3 + T Cells in Relapsing-Remitting Multiple Sclerosis and Type 1 Diabetes. J. Immunol. 2022, 208, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Choudhury, S.; Banerjee, R.; Basu, P.; Kumar, H. Soluble LAG-3 and Toll-interacting protein: Novel upstream neuro-inflammatory markers in Parkinson’s disease. Parkinsonism Relat. Disord. 2021, 91, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, O.P.; Larsen, Z.M.; Pociot, F. CTLA-4 in autoimmune diseases--a general susceptibility gene to autoimmunity? Genes Immun. 2000, 1, 170–184. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Yuan, W.; Wu, X. Association of CTLA-4 polymorphisms with increased risks of myasthenia gravis. Ann. Hum. Genet. 2018, 82, 358–369. [Google Scholar] [CrossRef]
- Fang, T.K.; Yan, C.J.; Du, J. CTLA-4 methylation regulates the pathogenesis of myasthenia gravis and the expression of related cytokines. Medicine 2018, 97, e0620. [Google Scholar] [CrossRef]
- Rosenzweig, N.; Dvir-Szternfeld, R.; Tsitsou-Kampeli, A.; Keren-Shaul, H.; Ben-Yehuda, H.; Weill-Raynal, P.; Cahalon, L.; Kertser, A.; Baruch, K.; Amit, I.; et al. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat. Commun. 2019, 10, 465. [Google Scholar] [CrossRef]
- Latta-Mahieu, M.; Elmer, B.; Bretteville, A.; Wang, Y.; Lopez-Grancha, M.; Goniot, P.; Moindrot, N.; Ferrari, P.; Blanc, V.; Schussler, N.; et al. Systemic immune-checkpoint blockade with anti-PD1 antibodies does not alter cerebral amyloid-β burden in several amyloid transgenic mouse models. Glia 2018, 66, 492–504. [Google Scholar] [CrossRef]
- Rogers, N.K.; Romero, C.; Sanmartín, C.D.; Ponce, D.P.; Salech, F.; López, M.N.; Gleisner, A.; Tempio, F.; Behrens, M.I. Inverse Relationship Between Alzheimer’s Disease and Cancer: How Immune Checkpoints Might Explain the Mechanisms Underlying Age-Related Diseases. J. Alzheimers. Dis. 2020, 73, 443–454. [Google Scholar] [CrossRef]
- Ceeraz, S.; Nowak, E.C.; Burns, C.M.; Noelle, R.J. Immune checkpoint receptors in regulating immune reactivity in rheumatic disease. Arthritis Res. Ther. 2014, 16, 469. [Google Scholar] [CrossRef] [Green Version]
- Basile, M.S.; Mazzon, E.; Fagone, P.; Longo, A.; Russo, A.; Fallico, M.; Bonfiglio, V.; Nicoletti, F.; Avitabile, T.; Reibaldi, M. Immunobiology of Uveal Melanoma: State of the Art and Therapeutic Targets. Front. Oncol. 2019, 9, 1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Vignali, D.A.A. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The yin and yang of co-inhibitory receptors: Toward anti-tumor immunity without autoimmunity. Cell Res. 2020, 30, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joller, N. Immune checkpoint in CNS Autoimmunity. Immunol. Rev. 2013, 248, 122–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, Y.; Han, J.; Zhu, J.; Jin, T. Role of the PD-1/PD-L1 Signaling in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis: Recent Insights and Future Directions. Mol. Neurobiol. 2021, 58, 6249–6271. [Google Scholar] [CrossRef]
- Oliveira, E.M.L.; Bar-Or, A.; Waliszewska, A.I.; Cai, G.; Anderson, D.E.; Krieger, J.I.; Hafler, D.A. CTLA-4 dysregulation in the activation of myelin basic protein reactive T cells may distinguish patients with multiple sclerosis from healthy controls. J. Autoimmun. 2003, 20, 71–81. [Google Scholar] [CrossRef]
- Viglietta, V.; Bourcier, K.; Buckle, G.J.; Healy, B.; Weiner, H.L.; Hafler, D.A.; Egorova, S.; Guttmann, C.R.G.; Rusche, J.R.; Khoury, S.J. CTLA4Ig treatment in patients with multiple sclerosis: An open-label, phase 1 clinical trial. Neurology 2008, 71, 917–924. [Google Scholar] [CrossRef]
- Khoury, S.J.; Rochon, J.; Ding, L.; Byron, M.; Ryker, K.; Tosta, P.; Gao, W.; Freedman, M.S.; Arnold, D.L.; Sayre, P.H.; et al. ACCLAIM: A randomized trial of abatacept (CTLA4-Ig) for relapsing-remitting multiple sclerosis. Mult. Scler. 2017, 23, 686. [Google Scholar] [CrossRef] [Green Version]
- Glatigny, S.; Höllbacher, B.; Motley, S.J.; Tan, C.; Hundhausen, C.; Buckner, J.H.; Smilek, D.; Khoury, S.J.; Ding, L.; Qin, T.; et al. Abatacept Targets T Follicular Helper and Regulatory T Cells, Disrupting Molecular Pathways That Regulate Their Proliferation and Maintenance. J. Immunol. 2019, 202, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Mena, E.; Rohowsky-Kochan, C. Expression of costimulatory molecules on peripheral blood mononuclear cells in multiple sclerosis. Acta Neurol. Scand. 1999, 100, 92–96. [Google Scholar] [CrossRef]
- Lavon, I.; Heli, C.; Brill, L.; Charbit, H.; Vaknin-Dembinsky, A. Blood Levels of Co-inhibitory-Receptors: A Biomarker of Disease Prognosis in Multiple Sclerosis. Front. Immunol. 2019, 10, 835. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, A.; Rad, I.A.; Ahmadi-Salmasi, B. CTLA-4, PD-1 and TIM-3 expression predominantly downregulated in MS patients. J. Neuroimmunol. 2018, 323, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, K.; Zhong, X.; Dai, Y.; Wu, A.; Li, Y.; Hu, X. Plasma sCD28, sCTLA-4 levels in neuromyelitis optica and multiple sclerosis during relapse. J. Neuroimmunol. 2012, 243, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Eschborn, M.; Pawlitzki, M.; Wirth, T.; Nelke, C.; Pfeuffer, S.; Schulte-Mecklenbeck, A.; Lohmann, L.; Rolfes, L.; Pape, K.; Eveslage, M.; et al. Evaluation of Age-Dependent Immune Signatures in Patients With Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2021, 8, e1094. [Google Scholar] [CrossRef] [PubMed]
- Kosmaczewska, A.; Bilinska, M.; Ciszak, L.; Noga, L.; Pawlak, E.; Szteblich, A.; Podemski, R.; Frydecka, I. Different patterns of activation markers expression and CD4+ T-cell responses to ex vivo stimulation in patients with clinically quiescent multiple sclerosis (MS). J. Neuroimmunol. 2007, 189, 137–146. [Google Scholar] [CrossRef] [PubMed]
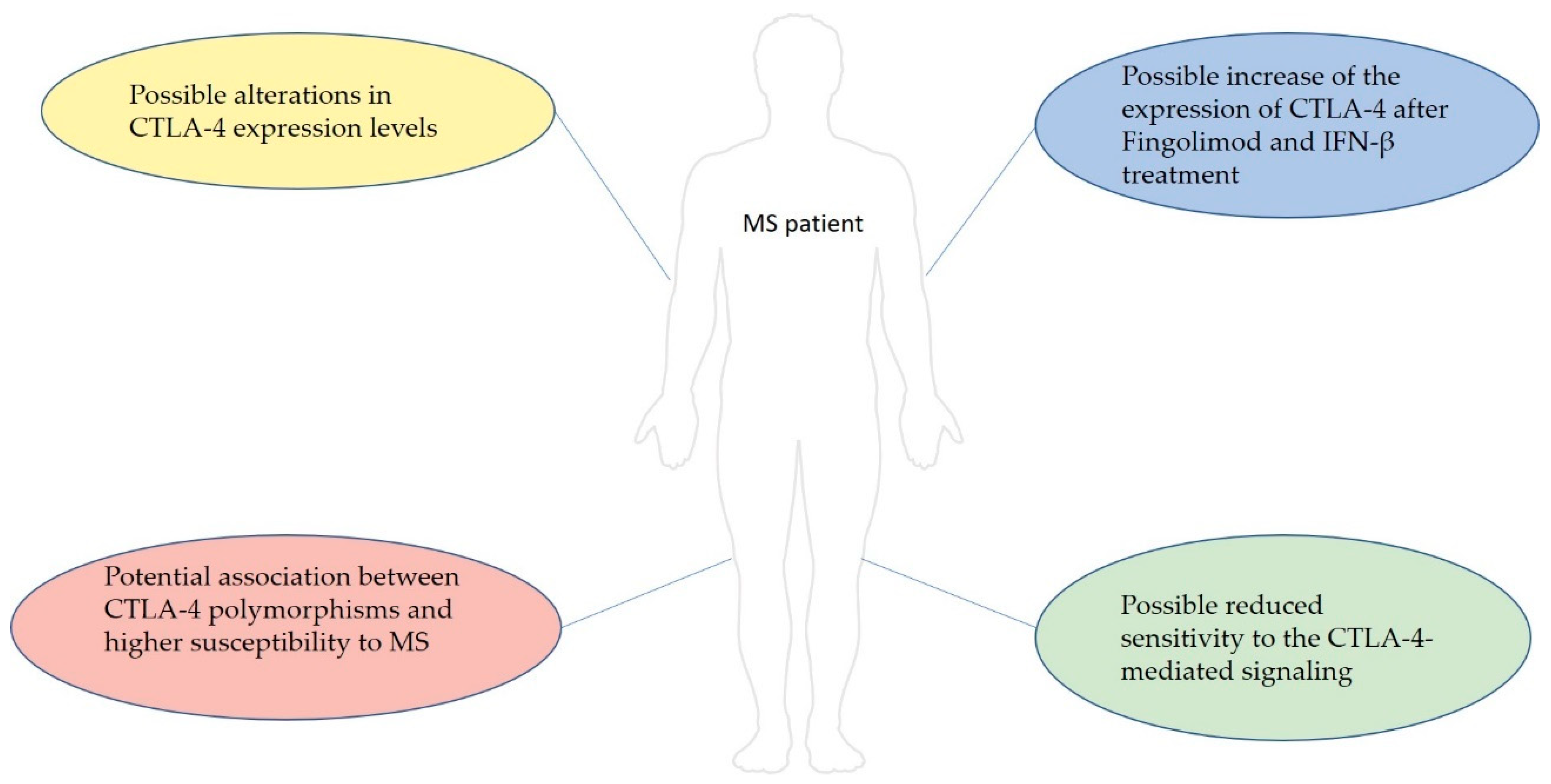
- Derakhshani, A.; Shadbad, M.A.; Asadzadeh, Z.; Safarpour, H.; Heydari, A.; Baradaran, B.; Leone, P.; Racanelli, V. Regulation of CTLA-4 and PD-L1 Expression in Relapsing-Remitting Multiple Sclerosis Patients after Treatment with Fingolimod, IFNβ-1α, Glatiramer Acetate, and Dimethyl Fumarate Drugs. J. Pers. Med. 2021, 11, 721. [Google Scholar] [CrossRef]
- Hallal-Longo, D.E.M.; Mirandola, S.R.; Oliveira, E.C.; Farias, A.S.; Pereira, F.G.; Metze, I.L.; Brandão, C.O.; Ruocco, H.H.; Damasceno, B.P.; Santos, L.M.B. Diminished myelin-specific T cell activation associated with increase in CTLA4 and Fas molecules in multiple sclerosis patients treated with IFN-beta. J. Interferon Cytokine Res. 2007, 27, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Sellebjerg, F.; Krakauer, M.; Khademi, M.; Olsson, T.; Sørensen, P.S. FOXP3, CBLB and ITCH gene expression and cytotoxic T lymphocyte antigen 4 expression on CD4(+) CD25(high) T cells in multiple sclerosis. Clin. Exp. Immunol. 2012, 170, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Pentón-Rol, G.; Cervantes-Llanos, M.; Cabrera-Gómez, J.A.; Alonso-Ramírez, R.; Valenzuela-Silva, C.; Rodríguez-Lara, R.; Montero-Casimiro, E.; Bello-Rivero, I.; López-Saura, P. Treatment with type I interferons induces a regulatory T cell subset in peripheral blood mononuclear cells from multiple sclerosis patients. Int. Immunopharmacol. 2008, 8, 881–886. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.S.; Mamula, M.J. Experimental autoimmune encephalomyelitis in the Wistar rat: Dependence of MBP-specific T cell responsiveness on B7 costimulation. Autoimmunity 2002, 35, 191–199. [Google Scholar] [CrossRef]
- Almolda, B.; González, B.; Castellano, B. Activated microglial cells acquire an immature dendritic cell phenotype and may terminate the immune response in an acute model of EAE. J. Neuroimmunol. 2010, 223, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.H.; Girard, T.J.; Giacoletto, K.S.; Evans, R.J.; Keeling, R.M.; Lin, R.F.; Trotter, J.L.; Karr, R.W. Long-term inhibition of murine experimental autoimmune encephalomyelitis using CTLA-4-Fc supports a key role for CD28 costimulation. J. Clin. Invest. 1995, 95, 2783–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, A.H.; San, M.; Keeling, R.M.; Karr, R.W. CTLA-4-Fc treatment of ongoing EAE improves recovery, but has no effect upon relapse rate. Implications for the mechanisms involved in disease perpetuation. J. Neuroimmunol. 1999, 96, 144–147. [Google Scholar] [CrossRef]
- Perrin, P.J.; Scott, D.; Quigley, L.; Albert, P.S.; Feder, O.; Gray, G.S.; Abe, R.; June, C.H.; Racke, M.K. Role of B7:CD28/CTLA-4 in the induction of chronic relapsing experimental allergic encephalomyelitis. J. Immunol. 1995, 154, 1481–1490. [Google Scholar]
- Croxford, J.L.; O’Neill, J.K.; Ali, R.R.; Browne, K.; Byrnes, A.P.; Dallman, M.J.; Wood, M.J.A.; Feldmann, M.; Baker, D. Local gene therapy with CTLA4-immunoglobulin fusion protein in experimental allergic encephalomyelitis. Eur. J. Immunol. 1998, 28, 3904–3916. [Google Scholar] [CrossRef]
- Khoury, S.; Akalin, E.; Chandraker, A.; Turka, L.; Linsley, P.; Sayegh, M.; Hancock, W. CD28-B7 costimulatory blockade by CTLA4Ig prevents actively induced experimental autoimmune encephalomyelitis and inhibits Th1 but spares Th2 cytokines in the central nervous system. J. Immunol. 1995, 155, 4521–4524. [Google Scholar]
- Vogel, I.; Kasran, A.; Cremer, J.; Kim, Y.J.; Boon, L.; Van Gool, S.W.; Ceuppens, J.L. CD28/CTLA-4/B7 costimulatory pathway blockade affects regulatory T-cell function in autoimmunity. Eur. J. Immunol. 2015, 45, 1832–1841. [Google Scholar] [CrossRef]
- Kuchroo, V.K.; Prabhu Das, M.; Brown, J.A.; Ranger, A.M.; Zamvil, S.S.; Sobel, R.A.; Weiner, H.L.; Nabavi, N.; Glimcher, L.H. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: Application to autoimmune disease therapy. Cell 1995, 80, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, A.A.; Sullivan, T.J.; Sobel, R.A.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 (CTLA-4) limits the expansion of encephalitogenic T cells in experimental autoimmune encephalomyelitis (EAE)-resistant BALB/c mice. Proc. Natl. Acad. Sci. USA 2002, 99, 3013. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, A.A.; Sullivan, T.J.; Krummel, M.F.; Sobel, R.A.; Allison, J.P. Specific blockade of CTLA-4/B7 interactions results in exacerbated clinical and histologic disease in an actively-induced model of experimental allergic encephalomyelitis. J. Neuroimmunol. 1997, 73, 57–62. [Google Scholar] [CrossRef]
- Karandikar, N.J.; Vanderlugt, C.L.; Walunas, T.L.; Miller, S.D.; Bluestone, J.A. CTLA-4: A negative regulator of autoimmune disease. J. Exp. Med. 1996, 184, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Perrin, P.J.; Maldonado, J.H.; Davis, T.A.; June, C.H.; Racke, M.K. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J. Immunol. 1996, 157, 1333–1336. [Google Scholar] [PubMed]
- Karandikar, N.J.; Eagar, T.N.; Vanderlugt, C.L.; Bluestone, J.A.; Miller, S.D. CTLA-4 downregulates epitope spreading and mediates remission in relapsing experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2000, 109, 173–180. [Google Scholar] [CrossRef]
- Verhagen, J.; Gabryšová, L.; Minaee, S.; Sabatos, C.A.; Anderson, G.; Sharpe, A.H.; Wraith, D.C. Enhanced selection of FoxP3+ T-regulatory cells protects CTLA-4-deficient mice from CNS autoimmune disease. Proc. Natl. Acad. Sci. 2009, 106, 3306–3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, A.M.; Lovitch, S.B.; Sage, P.T.; Juneja, V.R.; Lee, Y.; Trombley, J.D.; Arancibia-Cárcamo, C.V.; Sobel, R.A.; Rudensky, A.Y.; Kuchroo, V.K.; et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J. Exp. Med. 2015, 212, 1603–1621. [Google Scholar] [CrossRef]
- Klocke, K.; Sakaguchi, S.; Holmdahl, R.; Wing, K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc. Natl. Acad. Sci. USA 2016, 113, E2383–E2392. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Kim, W.J.; Kim, Y.H.; Lee, S.; Koo, J.H.; Lee, J.A.; Yoon, H.; Kim, D.H.; Park, H.J.; Kim, H.M.; et al. dNP2 is a blood–brain barrier-permeable peptide enabling ctCTLA-4 protein delivery to ameliorate experimental autoimmune encephalomyelitis. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.R.; Kim, W.J.; Lim, S.; Lee, H.G.; Koo, J.H.; Nam, K.H.; Kim, S.M.; Park, S.D.; Choi, J.M. In Vivo Induction of Regulatory T Cells Via CTLA-4 Signaling Peptide to Control Autoimmune Encephalomyelitis and Prevent Disease Relapse. Adv. Sci. 2021, 8, 2004973. [Google Scholar] [CrossRef]
- Spanier, J.A.; Nashold, F.E.; Nelson, C.D.; Praska, C.E.; Hayes, C.E. Vitamin D 3-mediated resistance to a multiple sclerosis model disease depends on myeloid cell 1,25-dihydroxyvitamin D 3 synthesis and correlates with increased CD4 + T cell CTLA-4 expression. J. Neuroimmunol. 2020, 338, 577105. [Google Scholar] [CrossRef]
- Kim, W.J.; Kim, G.R.; Cho, H.J.; Choi, J.M. The Cysteine-Containing Cell-Penetrating Peptide AP Enables Efficient Macromolecule Delivery to T Cells and Controls Autoimmune Encephalomyelitis. Pharmaceutics 2021, 13, 1134. [Google Scholar] [CrossRef]
- Van Der Star, B.J.; Vogel, D.Y.; Kipp, M.; Puentes, F.; Baker, D.; Amor, S. In Vitro and In Vivo Models of Multiple Sclerosis. CNS Neurol. Disord.—Drug Targets 2012, 11, 570–588. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.; Babron, M.C.; Birebent, B.; Matsuda, F.; Quelvennec, E.; Liblau, R.; Cournu-Rebeix, I.; Momigliano-Richiardi, P.; Sequeiros, J.; Yaouanq, J.; et al. Genetic interaction of CTLA-4 with HLA-DR15 in multiple sclerosis patients. Ann. Neurol. 2003, 54, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Pham, K.; James, I.; Nolan, D.; Castley, A.; Christiansen, F.T.; Czarniak, P.; Luo, Y.; Wu, J.; Garlepp, M.; et al. The influence of non-HLA gene polymorphisms and interactions on disease risk in a Western Australian multiple sclerosis cohort. J. Neuroimmunol. 2013, 261, 92–97. [Google Scholar] [CrossRef]
- Wagner, M.; Sobczyński, M.; Karabon, L.; Bilińska, M.; Pokryszko-Dragan, A.; Pawlak-Adamska, E.; Cyrul, M.; Kuśnierczyk, P.; Jasek, M. Polymorphisms in CD28, CTLA-4, CD80 and CD86 genes may influence the risk of multiple sclerosis and its age of onset. J. Neuroimmunol. 2015, 288, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Harbo, H.F.; Celius, E.G.; Vartdal, F.; Spurkland, A. CTLA4 promoter and exon 1 dimorphisms in multiple sclerosis. Tissue Antigens 1999, 53, 106–110. [Google Scholar] [CrossRef]
- Kantarci, O.H.; Hebrink, D.D.; Achenbach, S.J.; Atkinson, E.J.; Waliszewska, A.; Buckle, G.; McMurray, C.T.; De Andrade, M.; Hafler, D.A.; Weinshenker, B.G. CTLA4 is associated with susceptibility to multiple sclerosis. J. Neuroimmunol. 2003, 134, 133–141. [Google Scholar] [CrossRef]
- Ligers, A.; Xu, C.; Saarinen, S.; Hillert, J. Olle Olerup The CTLA-4 gene is associated with multiple sclerosis. J. Neuroimmunol. 1999, 97, 182–190. [Google Scholar] [CrossRef]
- Ligers, A.; Teleshova, N.; Masterman, T.; Huang, W.X.; Hillert, J. CTLA-4 gene expression is influenced by promoter and exon 1 polymorphisms. Genes Immun. 2001, 2, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Mkhikian, H.; Grigorian, A.; Li, C.F.; Chen, H.L.; Newton, B.; Zhou, R.W.; Beeton, C.; Torossian, S.; Tatarian, G.G.; Lee, S.U.; et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat. Commun. 2011, 2, 334. [Google Scholar] [CrossRef] [Green Version]
- Barcellos, L.F.; Kamdar, B.B.; Ramsay, P.P.; DeLoa, C.; Lincoln, R.R.; Caillier, S.; Schmidt, S.; Haines, J.L.; Pericak-Vance, M.A.; Oksenberg, J.R.; et al. Clustering of autoimmune diseases in families with a high-risk for multiple sclerosis: A descriptive study. Lancet. Neurol. 2006, 5, 924–931. [Google Scholar] [CrossRef]
- Bilińska, M.; Frydecka, I.; Noga, L.; Dobosz, T.; Żołȩdziewska, M.; Suwalska, K.; Tutak, A.; Pokryszko-Dragan, A. Progression of multiple sclerosis is associated with exon 1 CTLA-4 gene polymorphism. Acta Neurol. Scand. 2004, 110, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, T.; Kikuchi, S.; Niino, M.; Yabe, I.; Miyagishi, R.; Fukaura, H.; Hamada, T.; Tashiro, K.; Sasaki, H. Attack-related severity: A key factor in understanding the spectrum of idiopathic inflammatory demyelinating disorders. J. Neurol. Sci. 2004, 225, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Dinčić, E.; Živković, M.; Stanković, A.; Obradović, D.; Alavantić, D.; Kostić, V.; Raičević, R. Association of polymorphisms in CTLA-4, IL-1ra and IL-1beta genes with multiple sclerosis in Serbian population. J. Neuroimmunol. 2006, 177, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, V.; Alloza, I.; Heggarty, S.; Goris, A.; Dubois, B.; Carton, H.; Vandenbroeck, K. The CTLA4 +49 A/G*G-CT60*G haplotype is associated with susceptibility to multiple sclerosis in Flanders. J. Neuroimmunol. 2005, 164, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Malferrari, G.; Stella, A.; Monferini, E.; Saltini, G.; Proverbio, M.C.; Grimaldi, L.M.; Rossi-Bernardi, L.; Biunno, I. Ctla4 and multiple sclerosis in the Italian population. Exp. Mol. Pathol. 2005, 78, 55–57. [Google Scholar] [CrossRef]
- Yousefipour, G.; Erfani, N.; Momtahan, M.; Moghaddasi, H.; Ghaderi, A. CTLA4 exon 1 and promoter polymorphisms in patients with multiple sclerosis. Acta Neurol. Scand. 2009, 120, 424–429. [Google Scholar] [CrossRef]
- Heggarty, S.; Suppiah, V.; Silversides, J.; O’Doherty, C.; Droogan, A.; McDonnell, G.; Hawkins, S.; Graham, C.; Vandenbroeck, K. CTLA4 gene polymorphisms and multiple sclerosis in Northern Ireland. J. Neuroimmunol. 2007, 187, 187–191. [Google Scholar] [CrossRef]
- Masterman, T.; Ligers, A.; Zhang, Z.; Hellgren, D.; Salter, H.; Anvret, M.; Hillert, J. CTLA4 dimorphisms and the multiple sclerosis phenotype. J. Neuroimmunol. 2002, 131, 208–212. [Google Scholar] [CrossRef]
- Mäurer, M.; Ponath, A.; Kruse, N.; Rieckmann, P. CTLA4 exon 1 dimorphism is associated with primary progressive multiple sclerosis. J. Neuroimmunol. 2002, 131, 213–215. [Google Scholar] [CrossRef]
- Fransen, N.L.; Crusius, J.B.A.; Smolders, J.; Mizee, M.R.; van Eden, C.G.; Luchetti, S.; Remmerswaal, E.B.M.; Hamann, J.; Mason, M.R.J.; Huitinga, I. Post-mortem multiple sclerosis lesion pathology is influenced by single nucleotide polymorphisms. Brain Pathol. 2020, 30, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Karabon, L.; Kosmaczewska, A.; Bilinska, M.; Pawlak, E.; Ciszak, L.; Jedynak, A.; Jonkisz, A.; Noga, L.; Pokryszko-Dragan, A.; Koszewicz, M.; et al. The CTLA-4 gene polymorphisms are associated with CTLA-4 protein expression levels in multiple sclerosis patients and with susceptibility to disease. Immunology 2009, 128, e787–e796. [Google Scholar] [CrossRef] [PubMed]
- Čizmarević, N.S.; Gašparović, I.; Peterlin, B.; Sepčić, J.; Rudolf, G.; Kapović, M.; Lavtar, P.; Ristić, S. CTLA-4 +49 A/G gene polymorphism in Croatian and Slovenian multiple sclerosis patients. Int. J. Immunogenet. 2011, 38, 419–426. [Google Scholar] [CrossRef]
- Dyment, D.A.; Steckley, J.L.; Willer, C.J.; Armstrong, H.; Sadovnick, A.D.; Risch, N.; Ebers, G.C. No evidence to support CTLA-4 as a susceptibility gene in MS families: The Canadian Collaborative Study. J. Neuroimmunol. 2002, 123, 193–198. [Google Scholar] [CrossRef]
- Roxburgh, R.H.; Sawcer, S.; Maranian, M.; Seaman, S.; Hensiek, A.; Yeo, T.; Deans, J.; Compston, A. No evidence of a significant role for CTLA-4 in multiple sclerosis. J. Neuroimmunol. 2006, 171, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Greve, B.; Simonenko, R.; Illes, Z.; Peterfalvi, A.; Hamdi, N.; Mycko, M.P.; Selmaj, K.W.; Rozsa, C.; Rajczy, K.; Bauer, P.; et al. Multiple sclerosis and the CTLA4 autoimmunity polymorphism CT60: No association in patients from Germany, Hungary and Poland. Mult. Scler. 2008, 14, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, T.; Yanagawa, T.; Kikuchi, S.; Yabe, I.; Sasaki, H.; Hamada, T.; Miyasaka, K.; Gomi, K.; Tashiro, K. CTLA-4 gene polymorphism may modulate disease in Japanese multiple sclerosis patients. J. Neurol. Sci. 1999, 171, 49–55. [Google Scholar] [CrossRef]
- Fukazawa, T.; Kikuchi, S.; Miyagishi, R.; Niino, M.; Yabe, I.; Hamada, T.; Sasaki, H. CTLA-4 gene polymorphism is not associated with conventional multiple sclerosis in Japanese. J. Neuroimmunol. 2005, 159, 225–229. [Google Scholar] [CrossRef]
- Haghighi, A.B.; Ghahramani, S.; Azarpira, N.; Pourjafar, M.; Nikseresht, A.R. Cytotoxic T lymphocyte associated antigen-4 exon 1 A/G polymorphism in Iranian patients with multiple sclerosis. Eur. J. Neurol. 2008, 15, 862–864. [Google Scholar] [CrossRef]
- Heidari, A.; Keramatipour, M.; Amirzargar, A.A.; Rashidi-Nezhad, A.; Sahmani, A.A.; Mozhdehipanah, H.; Daloii, M.R.N. CTLA-4 gene polymorphisms (-318C/T, +49A/G, +6230A/G) in Iranian patients with multiple sclerosis. Iran. J. Allergy Asthma Immunol. 2010, 9, 219–223. [Google Scholar]
- Luomala, M.; Lehtimäki, T.; Huhtala, H.; Ukkonen, M.; Koivula, T.; Hurme, M.; Elovaara, I. Promoter polymorphism of IL-10 and severity of multiple sclerosis. Acta Neurol. Scand. 2003, 108, 396–400. [Google Scholar] [CrossRef]
- Van Veen, T.; Crusius, J.B.A.; Van Winsen, L.; Xia, B.; Barkhof, F.; Peña, A.S.; Polman, C.H.; Uitdehaag, B.M.J. CTLA-4 and CD28 gene polymorphisms in susceptibility, clinical course and progression of multiple sclerosis. J. Neuroimmunol. 2003, 140, 188–193. [Google Scholar] [CrossRef]
- Wray, B.N.; Stankovich, J.; Whittock, L.; Dwyer, T.; Ponsonby, A.L.; van der Mei, I.A.F.; Taylor, B.; Dickinson, J.; Foote, S.; McMorran, B.J. CTLA-4 and multiple sclerosis: The A49G single nucleotide polymorphism shows no association with multiple sclerosis in a Southern Australian population. J. Neuroimmunol. 2008, 196, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Boćko, D.; Bilinska, M.; Dobosz, T.; Zołedziewska, M.; Suwalska, K.; Tutak, A.; Gruszka, E.; Frydecka, I. Lack of association between an exon 1 CTLA-4 gene polymorphism A(49)G and multiple sclerosis in a Polish population of the Lower Silesia region. Arch. Immunol. Ther. Exp. 2003, 51, 201–205. [Google Scholar]
- Rasmussen, H.B.; Kelly, M.A.; Francis, D.A.; Clausen, J. CTLA4 in multiple sclerosis. Lack of genetic association in a European Caucasian population but evidence of interaction with HLA-DR2 among Shanghai Chinese. J. Neurol. Sci. 2001, 184, 143–147. [Google Scholar] [CrossRef]
- Lorentzen, Å.R.; Celius, E.G.; Ekstrøm, P.O.; Wiencke, K.; Lie, B.A.; Myhr, K.M.; Ling, V.; Thorsby, E.; Vartdal, F.; Spurkland, A.; et al. Lack of association with the CD28/CTLA4/ICOS gene region among Norwegian multiple sclerosis patients. J. Neuroimmunol. 2005, 166, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Teutsch, S.M.; Booth, D.R.; Bennetts, B.H.; Heard, R.N.S.; Stewart, G.J. Association of common T cell activation gene polymorphisms with multiple sclerosis in Australian patients. J. Neuroimmunol. 2004, 148, 218–230. [Google Scholar] [CrossRef]
- Bagos, P.G.; Karnaouri, A.C.; Nikolopoulos, G.K.; Hamodrakas, S.J. No evidence for association of CTLA-4 gene polymorphisms with the risk of developing multiple sclerosis: A meta-analysis. Mult. Scler. 2007, 13, 156–168. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, H.X. CTLA-4 gene and the susceptibility of multiple sclerosis: An updated meta-analysis study including 12,916 cases and 15,455 controls. J. Neurogenet. 2014, 28, 153–163. [Google Scholar] [CrossRef]
- Haibing, X.; Xu, C.; Jifu, C.; Wenshuang, Z.; Ling, L.; Yuzhen, C.; Yanjun, H. Correlation between CTLA-4 gene rs221775A>G single nucleotide polymorphism and multiple sclerosis susceptibility. A meta-analysis. Open Med. 2016, 11, 264–269. [Google Scholar] [CrossRef]
- Song, G.G.; Lee, Y.H. CTLA-4 +49 A/G and -318 C/T polymorphisms and susceptibility to multiple sclerosis: A meta-analysis. Immunol. Invest. 2013, 42, 409–422. [Google Scholar] [CrossRef]
- Palacios, R.; Comas, D.; Elorza, J.; Villoslada, P. Genomic regulation of CTLA4 and multiple sclerosis. J. Neuroimmunol. 2008, 203, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Espejo, C.; Brieva, L.; Ruggiero, G.; Río, J.; Montalban, X.; Martínez-Cáceres, E.M. IFN-beta treatment modulates the CD28/CTLA-4-mediated pathway for IL-2 production in patients with relapsing-remitting multiple sclerosis. Mult. Scler. 2004, 10, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.W.; Hu, Y.C.; Yang, Y.H.; Chien, Y.H.; Lee, N.C.; Yu, H.H.; Chiang, B.L.; Wang, L.C. CTLA-4 gene mutation and multiple sclerosis: A case report and literature review. J. Microbiol. Immunol. Infect. 2021, 55, 545–548. [Google Scholar] [CrossRef]
- Kaninia, S.; Grammatikos, A.; Urankar, K.; Renowden, S.A.; Patel, N.K.; Gompels, M.M.; Rice, C.M. CNS demyelination associated with immune dysregulation and a novel CTLA-4 variant. Mult. Scler. 2021, 27, 1464. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| In Silico, in Vitro, and Ex Vivo Studies | |
|---|---|
| Evidence | References |
| [80] |
| [81] |
| [76] |
| [82] |
| [83] |
| [84] |
| [85] |
| [86] |
| [87] |
| [88] |
| [89] |
| [90] |
| In Vivo Studies | |
|---|---|
| Evidence | References |
| [91] |
| [92] |
| [93] |
| [94] |
| [95] |
| [96] |
| [97] |
| [98] |
| [99] |
| [100,101,102] |
| [103] |
| [104,105,106] |
| [107] |
| [108] |
| [109] |
| [110] |
| Genetic Studies | |
|---|---|
| Evidence | References |
| [113] |
| [112] |
| [114] |
| [115] |
| [116] |
| [117] |
| [118] |
| [119] |
| [120] |
| [121] |
| [122] |
| [123] |
| [124] |
| [125] |
| [126] |
| [127] |
| [128] |
| [129] |
| [130] |
| [131] |
| [132] |
| [133] |
| [134] |
| [135] |
| [136] |
| [137] |
| [138] |
| [139] |
| [140] |
| [141] |
| [142] |
| [143] |
| [144] |
| [145] |
| [146] |
| [147] |
| [148] |
| [149] |
| [150] |
| [151] |
| Clinical Studies Investigating the Effects of Common MS Therapies on CTLA-4 | |
|---|---|
| Evidence | References |
| [152] |
| Case reports | |
| Evidence | References |
| [153] |
| [154] |
| ClinicalTrials.gov Identifier | Official Title | Recruitment Status | Intervention/Treatment | Phase |
| NCT00076934 | A Phase I Study: Safety of RG2077 (CTLA4-IgG4m) in Patients with Relapsing-Remitting Multiple Sclerosis | Completed | Drug: RG2077 (CTLA4-IgG4m) | Phase 1 |
| NCT00035529 | A Phase II, Randomized, Double-Blind, Placebo Controlled Study to Evaluate the Preliminary Efficacy, Pharmacokinetics and Immunogenicity of BMS-188667 Administered to Subjects with Relapsing-Remitting Multiple Sclerosis | Terminated | Drug: Placebo Drug: BMS 188667 (Abatacept) | Phase 2 |
| NCT01116427 | A Phase II, Randomized, Double-blind, Parallel-group, Placebo-controlled, Multicenter Study to Evaluate the Safety and Efficacy of Abatacept in Adults with Relapsing-remitting Multiple Sclerosis | Completed | Biological: abatacept Drug: Placebo | Phase 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basile, M.S.; Bramanti, P.; Mazzon, E. The Role of Cytotoxic T-Lymphocyte Antigen 4 in the Pathogenesis of Multiple Sclerosis. Genes 2022, 13, 1319. https://doi.org/10.3390/genes13081319
Basile MS, Bramanti P, Mazzon E. The Role of Cytotoxic T-Lymphocyte Antigen 4 in the Pathogenesis of Multiple Sclerosis. Genes. 2022; 13(8):1319. https://doi.org/10.3390/genes13081319
Chicago/Turabian StyleBasile, Maria Sofia, Placido Bramanti, and Emanuela Mazzon. 2022. "The Role of Cytotoxic T-Lymphocyte Antigen 4 in the Pathogenesis of Multiple Sclerosis" Genes 13, no. 8: 1319. https://doi.org/10.3390/genes13081319