
Construction of a ceRNA Network and Comprehensive Analysis of lncRNA in Hepatocellular Carcinoma



Abstract
:1. Introduction
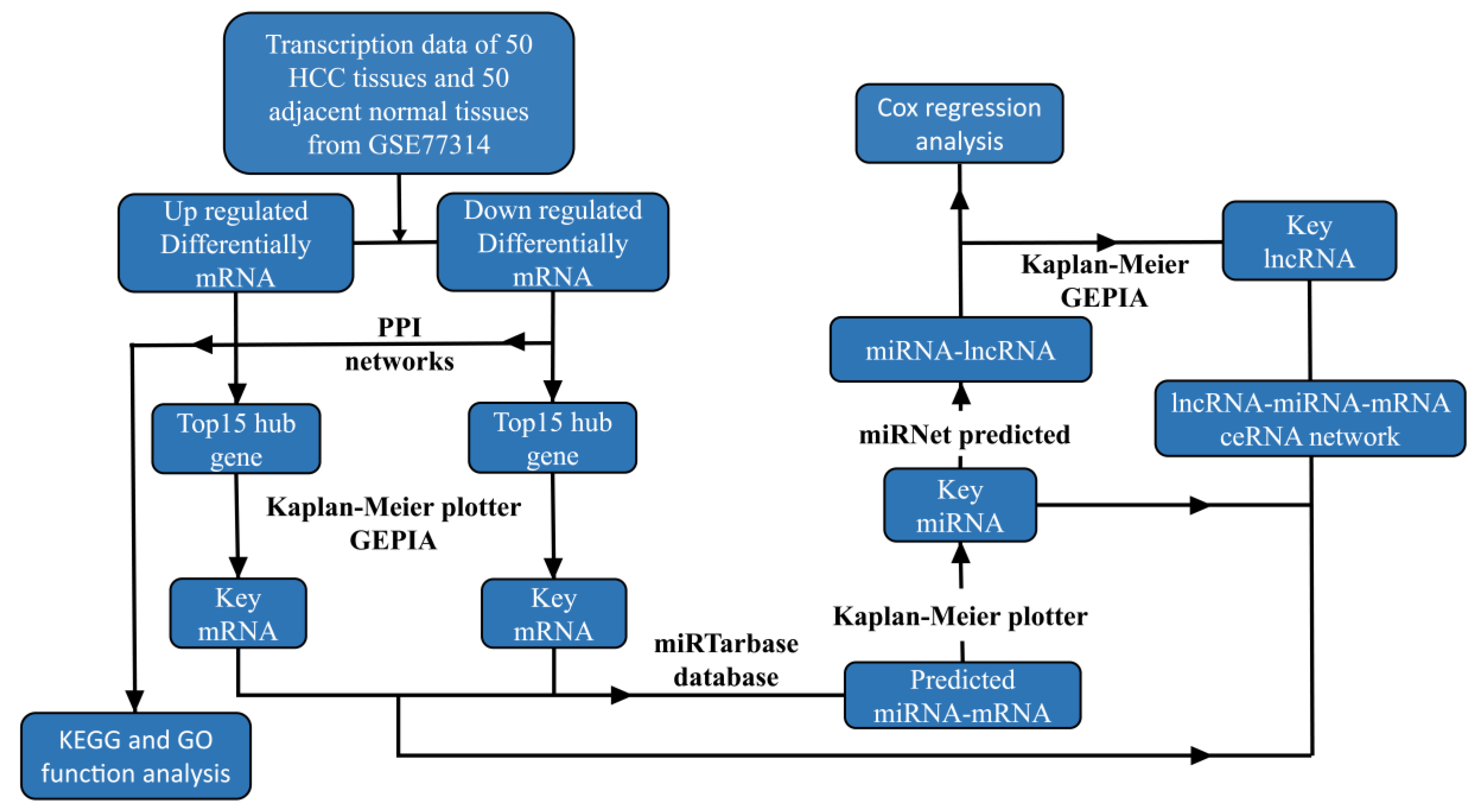
2. Materials and Methods
2.1. Screening of the Original Data
2.2. Screening of Differential Genes
2.3. Functional Enrichment Analysis of DE mRNA
2.4. Construction of PPI Network and Screening of Key mRNAs
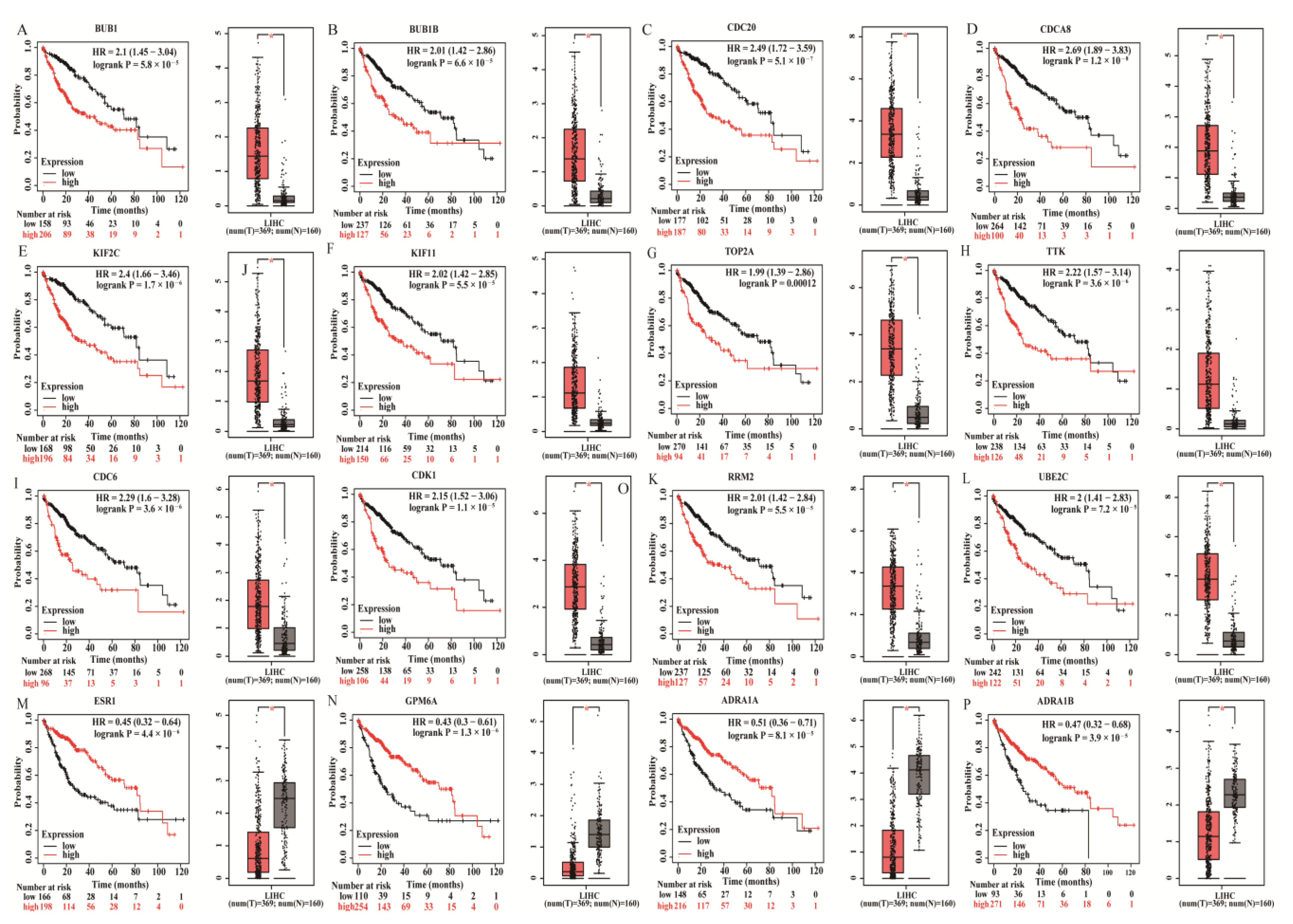
2.5. Survival Analysis
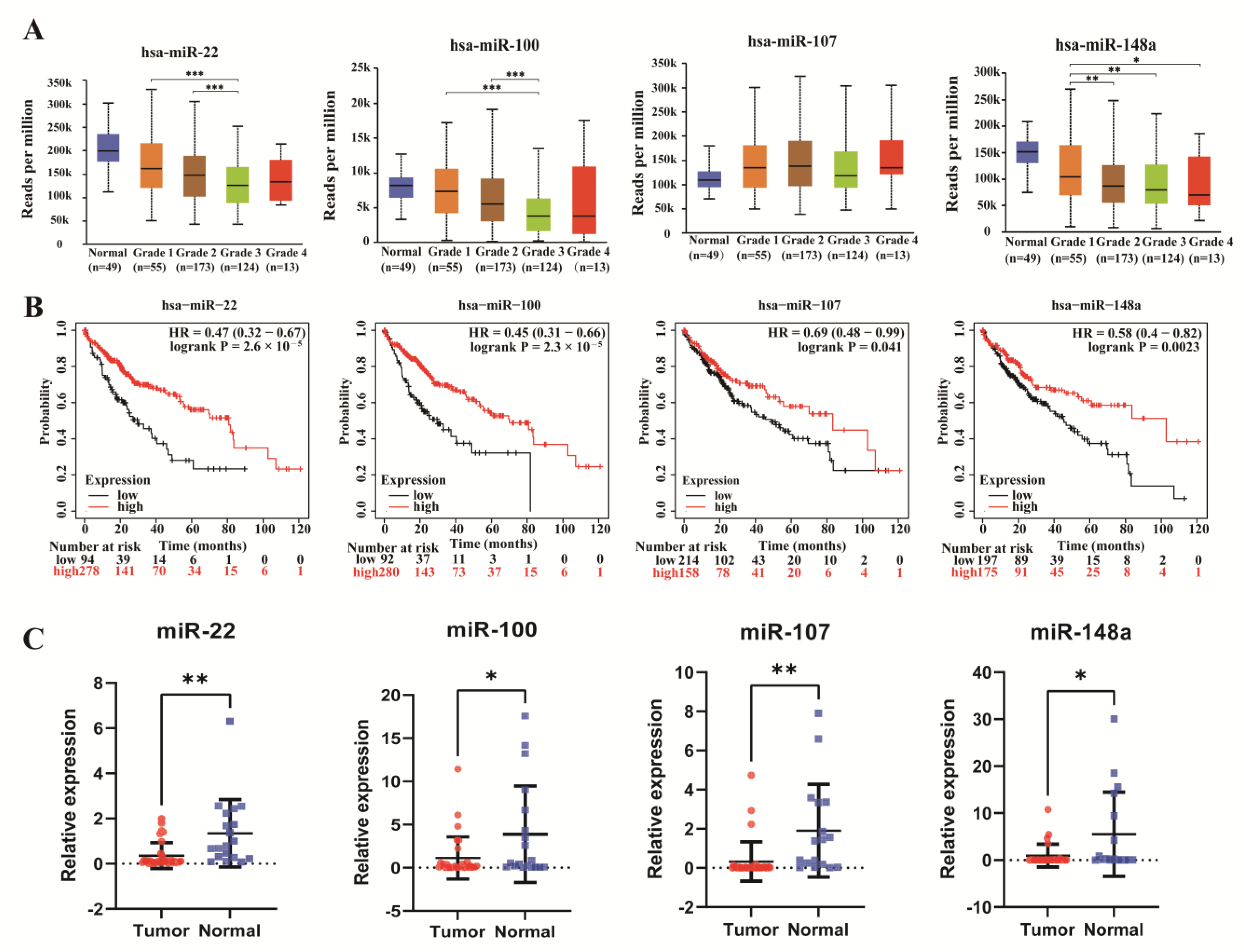
2.6. Gene Expression Analysis
2.7. Construction of ceRNA Network
2.8. Tissue Samples and Serum Collection
2.9. RNA Expression Analyses
2.10. Statistical Analysis
3. Results
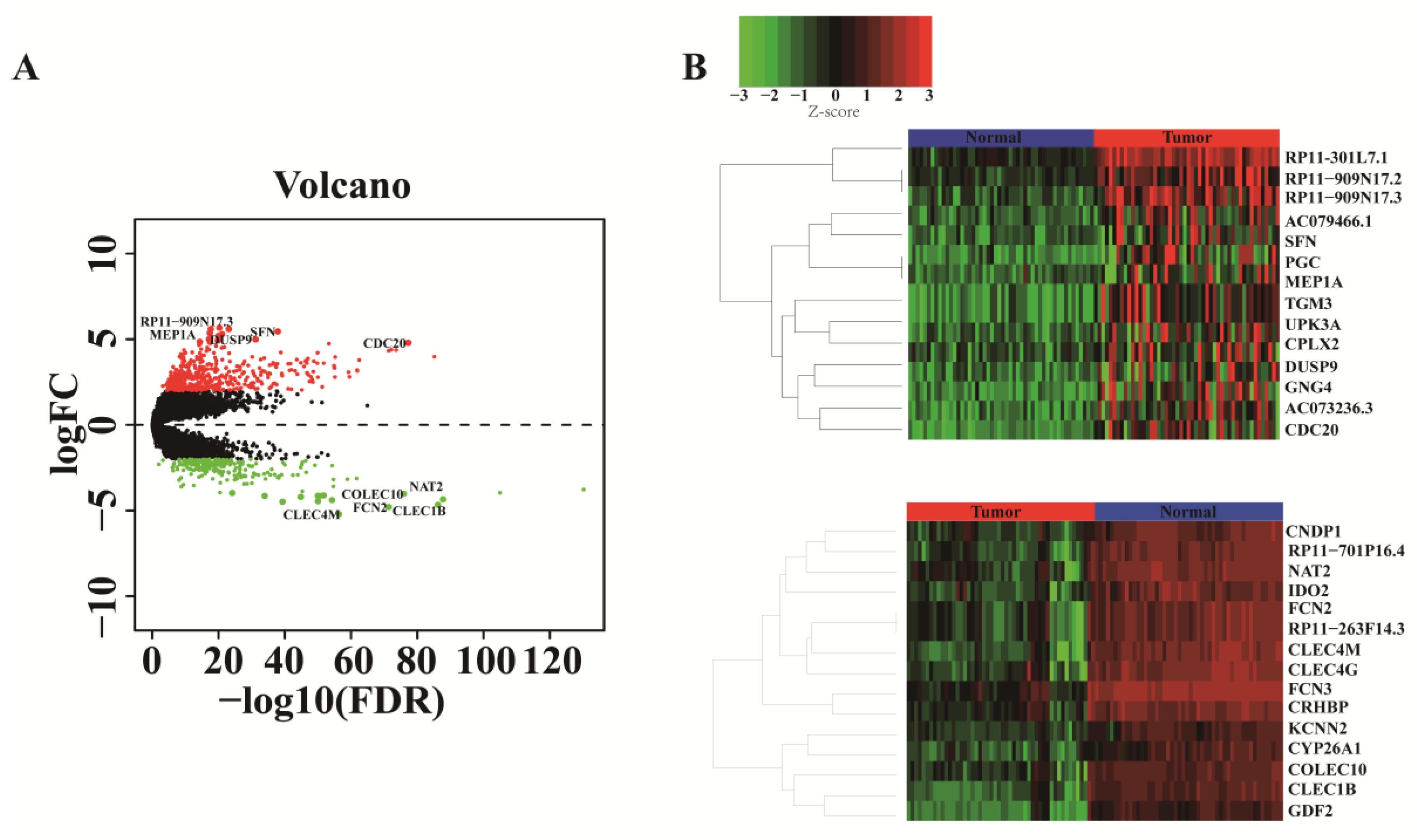
3.1. Screening of Differential Genes for Liver Cancer
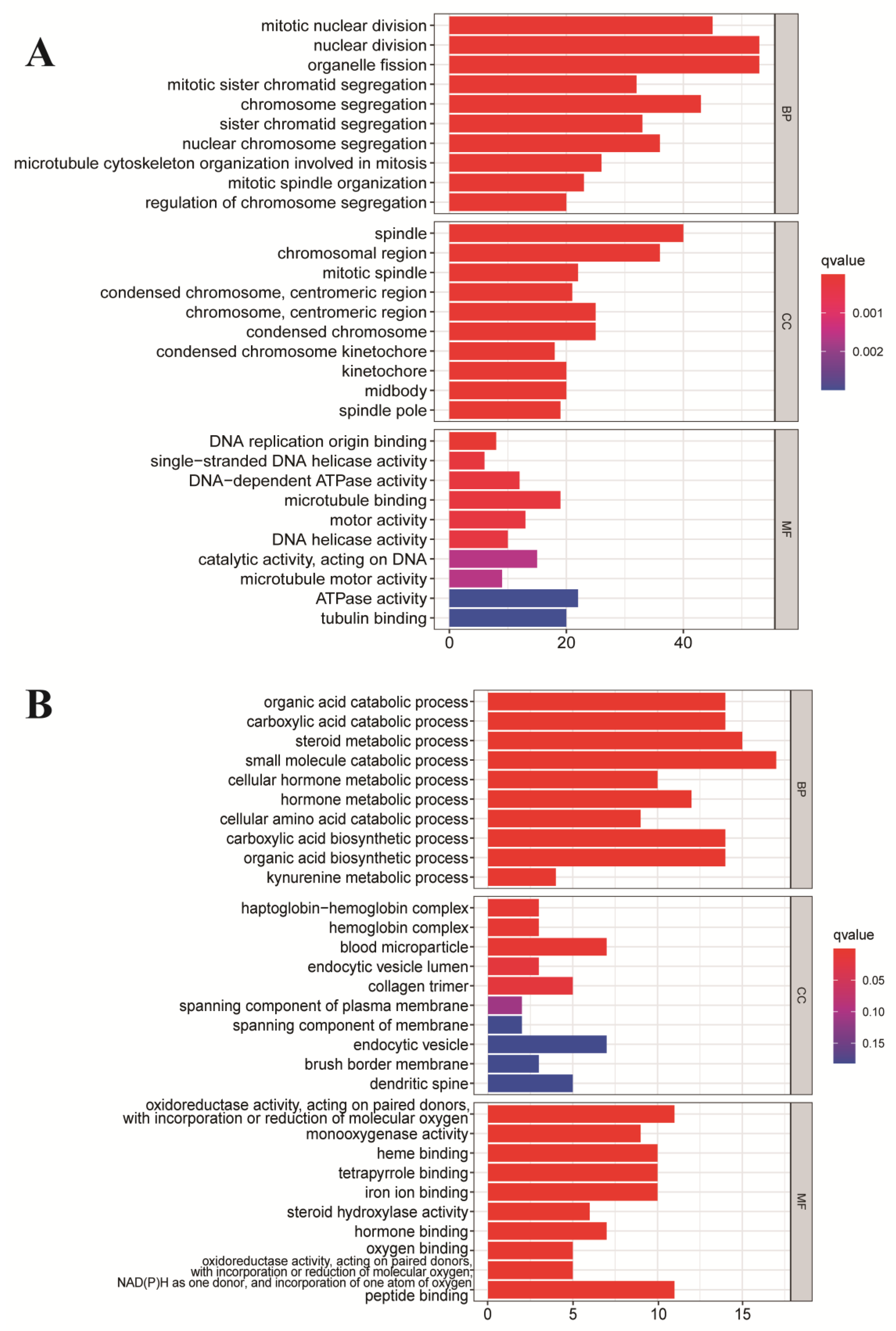
3.2. Functional Enrichment Analysis of DEmRNAs
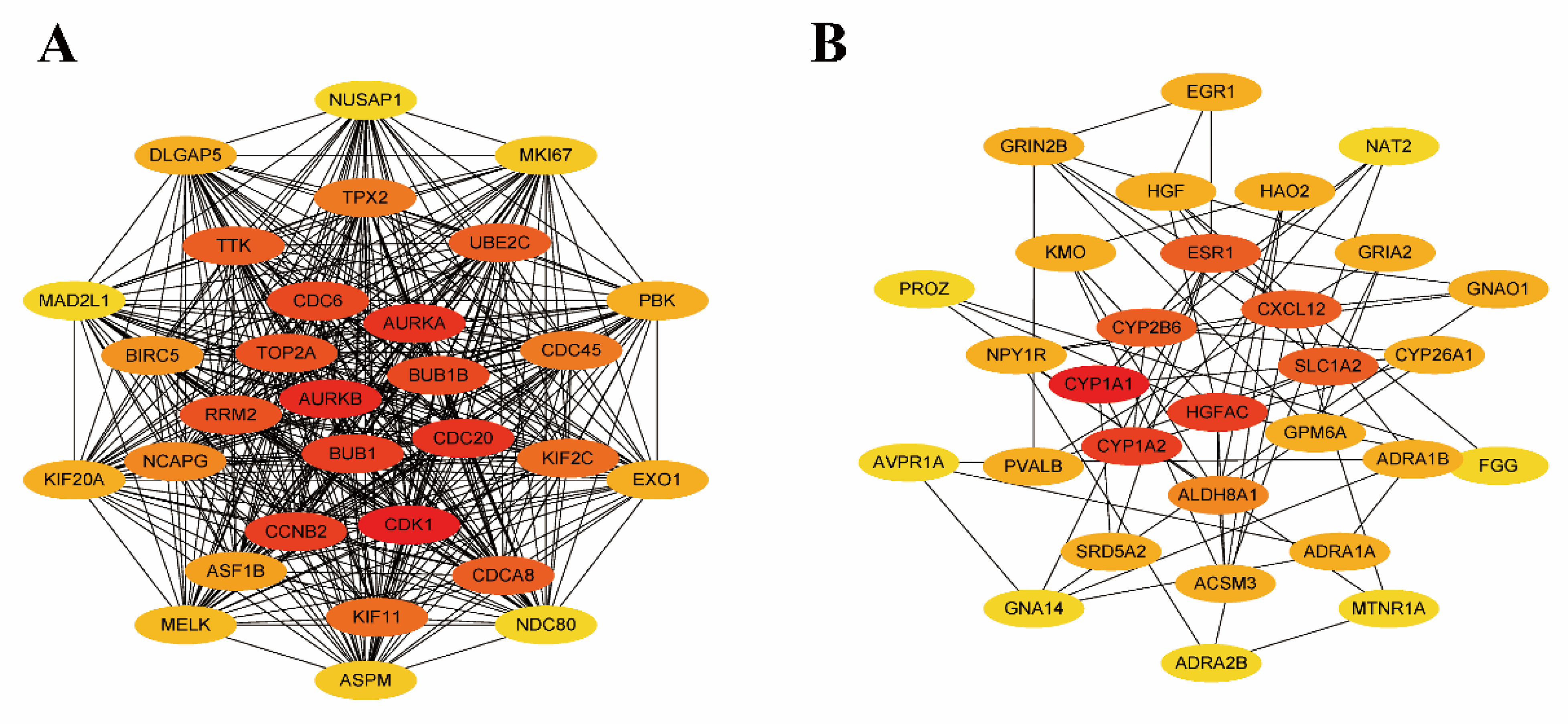
3.3. Establishing the PPI Network and Screening of Hub Genes
3.4. Integrated Analysis of miRNAs in the ceRNA Network
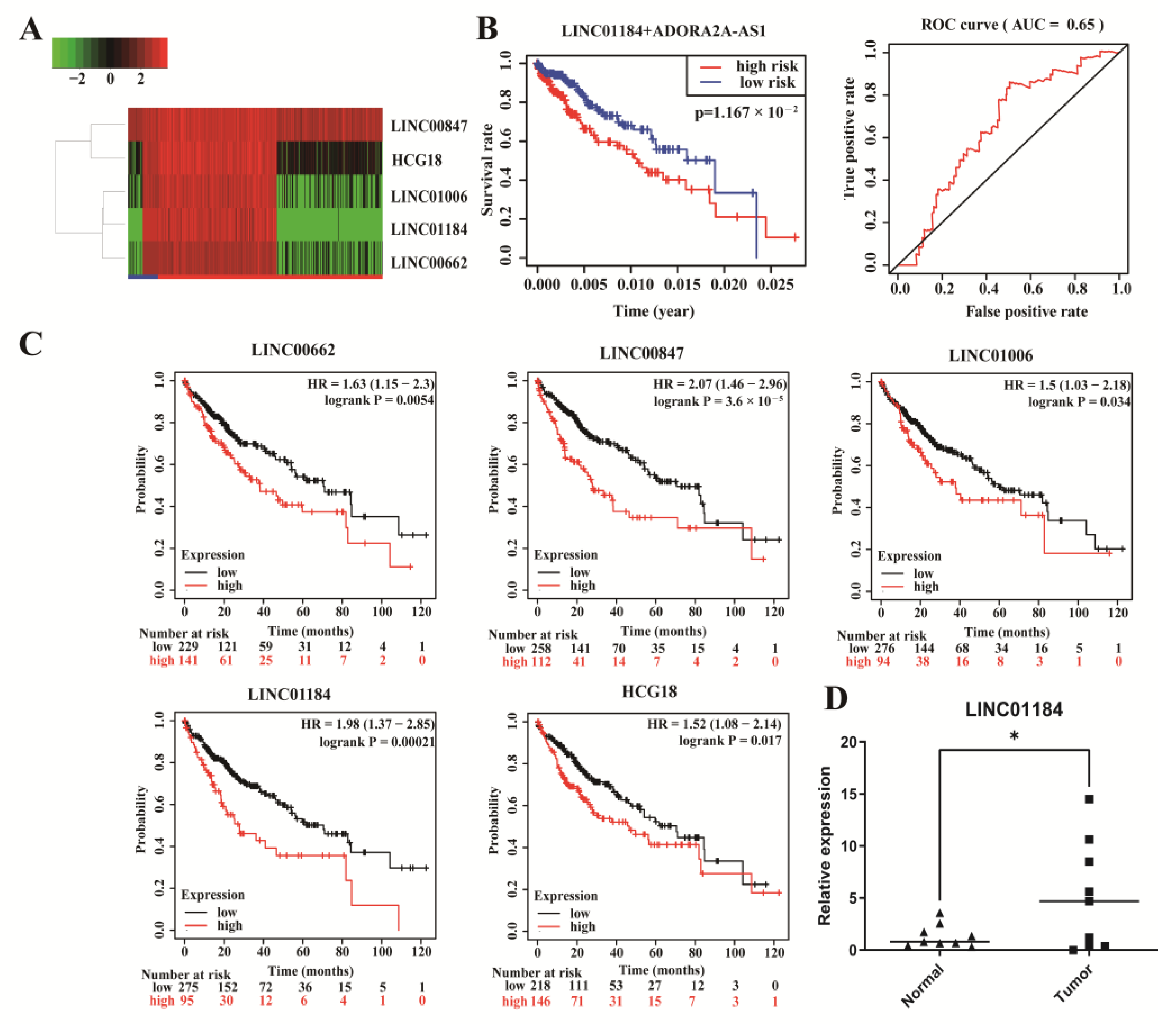
3.5. Integrated Analysis of lncRNAs in the ceRNA Network
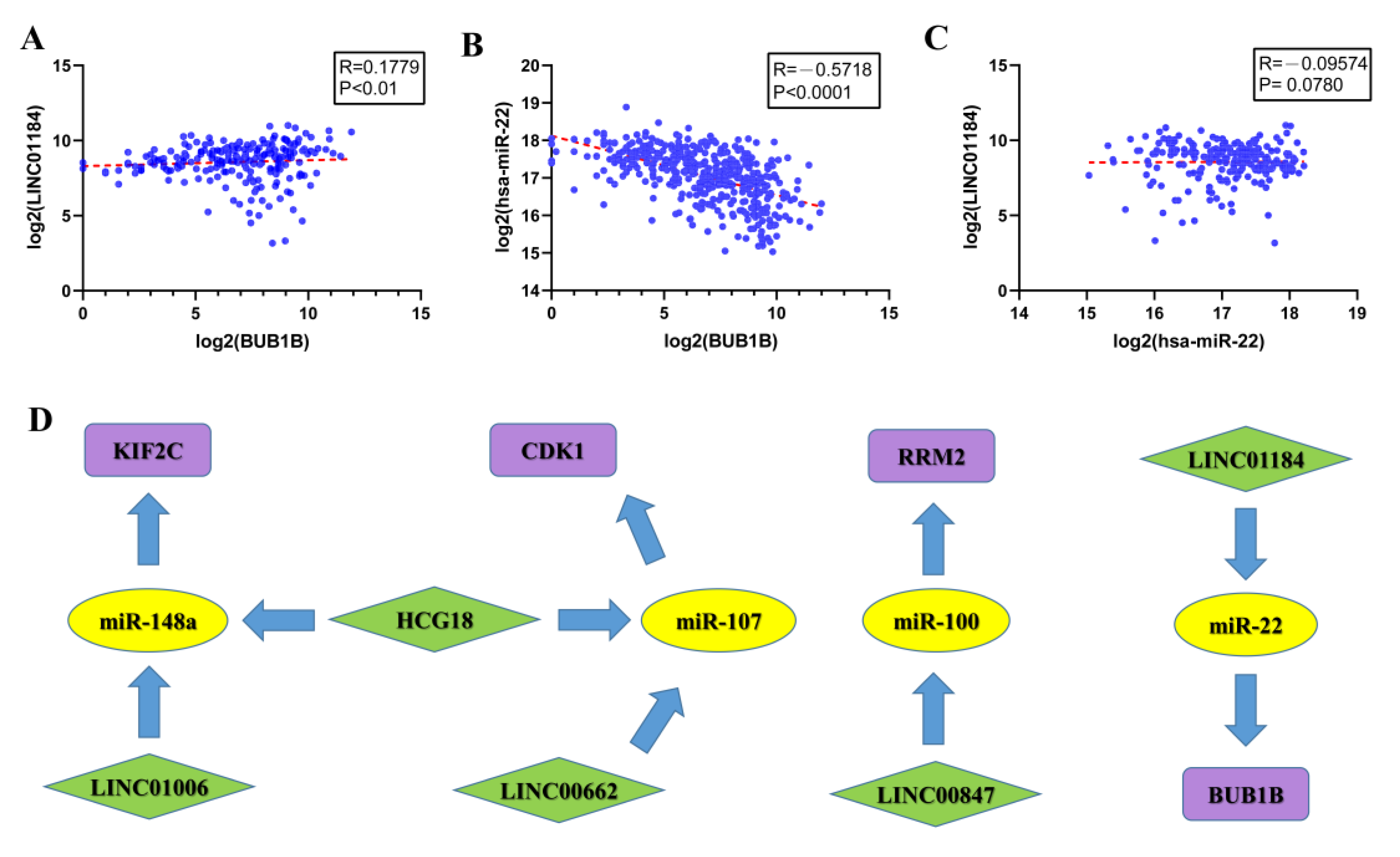
3.6. Construction of ceRNA Sub-Network
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Agarwal, P.; Villanueva, A.; Rao, S.; Dawson, L.A.; Karasic, T.; Llovet, J.M.; Finn, R.S.; Groopman, J.D.; El-Serag, H.B.; et al. Recent developments and therapeutic strategies against hepatocellular carcinoma. Cancer Res. 2019, 79, 4326–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tampaki, M.; Papatheodoridis, G.V.; Cholongitas, E. Intrahepatic recurrence of hepatocellular carcinoma after resection: An update. Clin. J. Gastroenterol. 2021, 14, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Huang, J.; Wang, G.; Huang, J.; Wu, X.; Li, J.; Yi, W.; Qin, F.; Huang, D. Integrated analysis of clinical significance and functional involvement of microRNAs in hepatocellular carcinoma. J. Cell. Physiol. 2019, 234, 23581–23595. [Google Scholar] [CrossRef]
- Chan, J.J.; Kwok, Z.H.; Chew, X.H.; Zhang, B.; Liu, C.; Soong, T.W.; Yang, H.; Tay, Y. A FTH1 gene: Pseudogene: MicroRNA network regulates tumorigenesis in prostate cancer. Nucleic Acids Res. 2018, 46, 1998–2011. [Google Scholar] [CrossRef]
- Luo, Z.; Rong, Z.; Zhang, J.; Zhu, Z.; Yu, Z.; Li, T.; Fu, Z.; Qiu, Z.; Huang, C. Circular RNA circCCDC9 acts as a miR-6792-3p sponge to suppress the progression of gastric cancer through regulating CAV1 expression. Mol. Cancer 2020, 19, 1–21. [Google Scholar] [CrossRef]
- Karreth, F.A.; Pandolfi, P.P. ceRNA cross-talk in cancer: When ce-bling rivalries go awry. Cancer Discov. 2013, 3, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhu, J.; Ma, X.; Han, S.; Xiao, D.; Jia, Y.; Wang, Y. ceRNA network construction and comparison of gastric cancer with or without Helicobacter pylori infection. J. Cell. Physiol. 2019, 234, 7128–7140. [Google Scholar] [CrossRef]
- Lin, A.; Ji, P.; Niu, X.; Zhao, X.; Chen, Y.; Liu, W.; Liu, Y.; Fan, W.; Sun, Y.; Miao, C. CstF64-induced shortening of the BID 3′UTR promotes esophageal squamous cell carcinoma progression by disrupting ceRNA crosstalk with ZFP36L2. Cancer Res. 2021, 81, 5638–5651. [Google Scholar] [CrossRef]
- Hu, G.; Ma, J.; Zhang, J.; Chen, Y.; Liu, H.; Huang, Y.; Zheng, J.; Xu, Y.; Xue, W.; Zhai, W. Hypoxia-induced lncHILAR promotes renal cancer metastasis via ceRNA for the miR-613/206/1-1-3p/Jagged-1/Notch/CXCR4 signaling pathway. Mol. Ther. 2021, 29, 2979–2994. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhuang, S.; Chen, X.; Du, J.; Zhong, L.; Ding, J.; Wang, L.; Yi, J.; Hu, G.; Tang, G. LncRNA ITGB8-AS1 functions as a ceRNA to promote colorectal cancer growth and migration through integrin-mediated focal adhesion signaling. Mol. Ther. 2022, 30, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mao, X.; Ma, Z.; Chen, W.; Guo, X.; Yu, L.; Deng, X.; Jiang, F.; Li, T.; Lin, N. Aberrant regulation of LncRNA TUG1-microRNA-328-3p-SRSF9 mRNA Axis in hepatocellular carcinoma: A promising target for prognosis and therapy. Mol. Cancer 2022, 21, 36. [Google Scholar] [CrossRef]
- Zuo, Z.; Chen, T.; Zhang, Y.; Han, L.; Liu, B.; Yang, B.; Han, T.; Zheng, Z. Construction of a ceRNA network in hepatocellular carcinoma and comprehensive analysis of immune infiltration patterns. Am. J. Transl. Res. 2021, 13, 13356. [Google Scholar] [PubMed]
- Luo, Y.; Li, H.; Huang, H.; Xue, L.; Li, H.; Liu, L.; Fu, H. Integrated analysis of ceRNA network in hepatocellular carcinoma using bioinformatics analysis. Medicine 2021, 100, e26194. [Google Scholar] [CrossRef]
- Long, J.; Bai, Y.; Yang, X.; Lin, J.; Yang, X.; Wang, D.; He, L.; Zheng, Y.; Zhao, H. Construction and comprehensive analysis of a ceRNA network to reveal potential prognostic biomarkers for hepatocellular carcinoma. Cancer Cell Int. 2019, 19, 90. [Google Scholar] [CrossRef]
- Tao, H.; Li, J.; Liu, J.; Yuan, T.; Zhang, E.; Liang, H.; Huang, Z. Construction of a ceRNA network and a prognostic lncRNA signature associated with vascular invasion in hepatocellular carcinoma based on weighted gene co-expression network analysis. J. Cancer 2021, 12, 3754. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-based survival analysis tool tailored for medical research (KMplot): Development and implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z.J.N. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.P.; Shen, S.L.; Yu, Y.; Yue, X.; Hu, W.J.; Li, S.Q. Kinesin family member 2C aggravates the progression of hepatocellular carcinoma and interacts with competing endogenous RNA. J. Cell. Biochem. 2020, 121, 4419–4430. [Google Scholar] [CrossRef]
- Chan, T.H.M.; Chen, L.; Liu, M.; Hu, L.; Zheng, B.-J.; Poon, V.K.-M.; Huang, P.; Yuan, Y.-F.; Huang, J.-D.; Yang, J.; et al. Translationally controlled tumor protein induces mitotic defects and chromosome missegregation in hepatocellular carcinoma development. Hepatology 2012, 55, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.X.; Wang, X.Q.; Chok, S.H.; Man, K.; Tsang, S.H.Y.; Chan, A.C.Y.; Ma, K.W.; Xia, W.; Cheung, T.T. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics 2018, 8, 3737–3750. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.-Y.; Li, Y.-S.; Hsu, H.-W.; Lin, H.; Wang, H.-Y.; Wo, R.R.; Cheng, A.-L.; Hsu, C.-H. Potent Activity of Composite Cyclin Dependent Kinase Inhibition against Hepatocellular Carcinoma. Cancers 2019, 11, 1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satow, R.; Shitashige, M.; Kanai, Y.; Takeshita, F.; Ojima, H.; Jigami, T.; Honda, K.; Kosuge, T.; Ochiya, T.; Hirohashi, S.; et al. Combined Functional Genome Survey of Therapeutic Targets for Hepatocellular Carcinoma. Clin. Cancer Res. 2010, 16, 2518–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Zhang, S.; Wang, P.; Wang, H.; Sha, B.; Peng, H.; Ju, Z.; Rao, J.; Lu, L. BUB1B promotes hepatocellular carcinoma progression via activation of the mTORC1 signaling pathway. Cancer Med. 2020, 9, 8159–8172. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, J.; Qian, Y.; Liu, W.; Liang, J.; Huang, Z.; Wang, S.; Zhao, C. Identification of plasma miR-148a as a noninvasive biomarker for hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, F.; Zhao, M.; Yang, Z.; Zhang, S.; Ye, L.; Gao, H.; Zhang, X. MiR-107 suppresses proliferation of hepatoma cells through targeting HMGA2 mRNA 3′UTR. Biochem. Biophys. Res. Commun. 2016, 480, 455–460. [Google Scholar] [CrossRef]
- Petrelli, A.; Perra, A.; Schernhuber, K.; Cargnelutti, M.; Salvi, A.; Migliore, C.; Ghiso, E.; Benetti, A.; Barlati, S.; Ledda-Columbano, G. Sequential analysis of multistage hepatocarcinogenesis reveals that miR-100 and PLK1 dysregulation is an early event maintained along tumor progression. Oncogene 2012, 31, 4517–4526. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Tang, S.; Yang, Y.; Yang, F.; Jiang, W.; Liu, Y.; Zhang, F.; Fang, H.; Wang, S.; Zhang, Y.J.F. Generation and Validation of miR-100 Hepatocyte-Specific Knock-Out Mice. Front. Oncol. 2019, 9, 535. [Google Scholar] [CrossRef]
- Chen, S.; Pu, J.; Bai, J.; Yin, Y.; Wu, K.; Wang, J.; Shuai, X.; Gao, J.; Tao, K.; Wang, G.; et al. EZH2 promotes hepatocellular carcinoma progression through modulating miR-22/galectin-9 axis. J. Exp. Clin. Cancer Res. 2018, 37, 3. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Gong, C.; Wu, P.; Battaglia-Hsu, S.-F.; Feng, J.; Liu, P.; Wang, H.; Guo, D.; Yao, Y.; Chen, B. LINC00662 promotes hepatocellular carcinoma progression via altering genomic methylation profiles. Cell Death Differ. 2020, 27, 2191–2205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.; Li, M.; Sun, P.; Bai, T.; Wang, W.; Bai, H.; Gou, J.; Wang, Z. HCG18 Participates in Vascular Invasion of Hepatocellular Carcinoma by Regulating Macrophages and Tumor Stem Cells. Front. Cell Dev. Biol. 2021, 9, 2335. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, S.; Cheng, X. LINC01006 regulates the proliferation, migration and invasion of hepatocellular carcinoma cells through regulating miR-433-3p/CBX3 axis. Ann. Hepatol. 2021, 25, 100343. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Lv, M.; Li, L.; Sun, Y.; Liu, S.; Shen, Y.; Liu, Z.; Luo, S. Long intergenic noncoding RNA 00641 inhibits breast cancer cell proliferation, migration, and invasion by sponging miR-194-5p. J. Cell. Physiol. 2020, 235, 2668–2675. [Google Scholar] [CrossRef]
- Li, H.; Chen, Y.-k.; Wan, Q.; Shi, A.-q.; Wang, M.; He, P.; Tang, L.-x. Long Non-coding RNA LINC00847 Induced by E2F1 Accelerates Non-small Cell Lung Cancer Progression Through Targeting miR-147a/IFITM1 Axis. Front. Med. 2021, 8, 528. [Google Scholar] [CrossRef]
- Sui, Y.-X.; Zhao, D.-L.; Yu, Y.; Wang, L.-C. The Role, Function, and Mechanism of Long Intergenic Noncoding RNA1184 (linc01184) in Colorectal Cancer. Dis. Markers 2021, 2021, 8897906. [Google Scholar] [CrossRef]
- Tian, X.; Wu, Y.; Yang, Y.; Wang, J.; Niu, M.; Gao, S.; Qin, T.; Bao, D. Long noncoding RNA LINC00662 promotes M2 macrophage polarization and hepatocellular carcinoma progression via activating Wnt/β-catenin signaling. Mol. Oncol. 2020, 14, 462–483. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Hou, G.; Li, L.; Li, Y.; Zhou, W.; Liu, L. Potential diagnostic and prognostic marker dimethylglycine dehydrogenase (DMGDH) suppresses hepatocellular carcinoma metastasis in vitro and in vivo. Oncotarget 2016, 7, 32607. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Variable | Numbers (n = 40) | Percentage |
|---|---|---|
| Age, n | ||
| <60 | 21 | 52.5% |
| >60 | 19 | 47.5% |
| Gender, n | ||
| Female | 16 | 40% |
| Male | 24 | 60% |
| ALT(IU/L), n | ||
| Low (<50) | 18 | 45% |
| High (>50) | 22 | 55% |
| AFP(ng/mL), n | ||
| Low (<7) | 13 | 32.5% |
| High (>7) | 27 | 67.5% |
| Tumor size, n | ||
| Small (<3 cm) | 13 | 32.5% |
| Large (>3 cm) | 24 | 60% |
| NA | 3 | 7.5% |
| Clinical Variable | Numbers (n = 9) | Percentage |
|---|---|---|
| Age, n | ||
| <60 | 7 | 77.8% |
| >60 | 2 | 22.2% |
| Gender, n | ||
| Female | 1 | 11.1% |
| Male | 8 | 88.9% |
| Grade, n | ||
| Grade 1 | 1 | 11.1% |
| Grade 2 | 3 | 33.3% |
| Grade 3 | 5 | 55.6% |
| Grade 4 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhao, J.; Zhu, C.; Yang, K.; Zhu, L.; Liu, Y. Construction of a ceRNA Network and Comprehensive Analysis of lncRNA in Hepatocellular Carcinoma. Genes 2022, 13, 785. https://doi.org/10.3390/genes13050785
Wang L, Zhao J, Zhu C, Yang K, Zhu L, Liu Y. Construction of a ceRNA Network and Comprehensive Analysis of lncRNA in Hepatocellular Carcinoma. Genes. 2022; 13(5):785. https://doi.org/10.3390/genes13050785
Chicago/Turabian StyleWang, Lin, Jun Zhao, Cancan Zhu, Ke Yang, Ling Zhu, and Yong Liu. 2022. "Construction of a ceRNA Network and Comprehensive Analysis of lncRNA in Hepatocellular Carcinoma" Genes 13, no. 5: 785. https://doi.org/10.3390/genes13050785