
Meta-Analysis of SNPs Determining Litter Traits in Pigs
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset A
2.2. Dataset B
2.3. Gene Ontology Analysis
2.4. Gene Network Analysis
2.5. Protein–Protein Interactions
2.6. Meta-Analysis on Dataset B
2.7. Candidate Genes and Causal Variants
3. Results
3.1. SNPs and Candidate Genes from Dataset A
3.2. GO Term Analysis Results
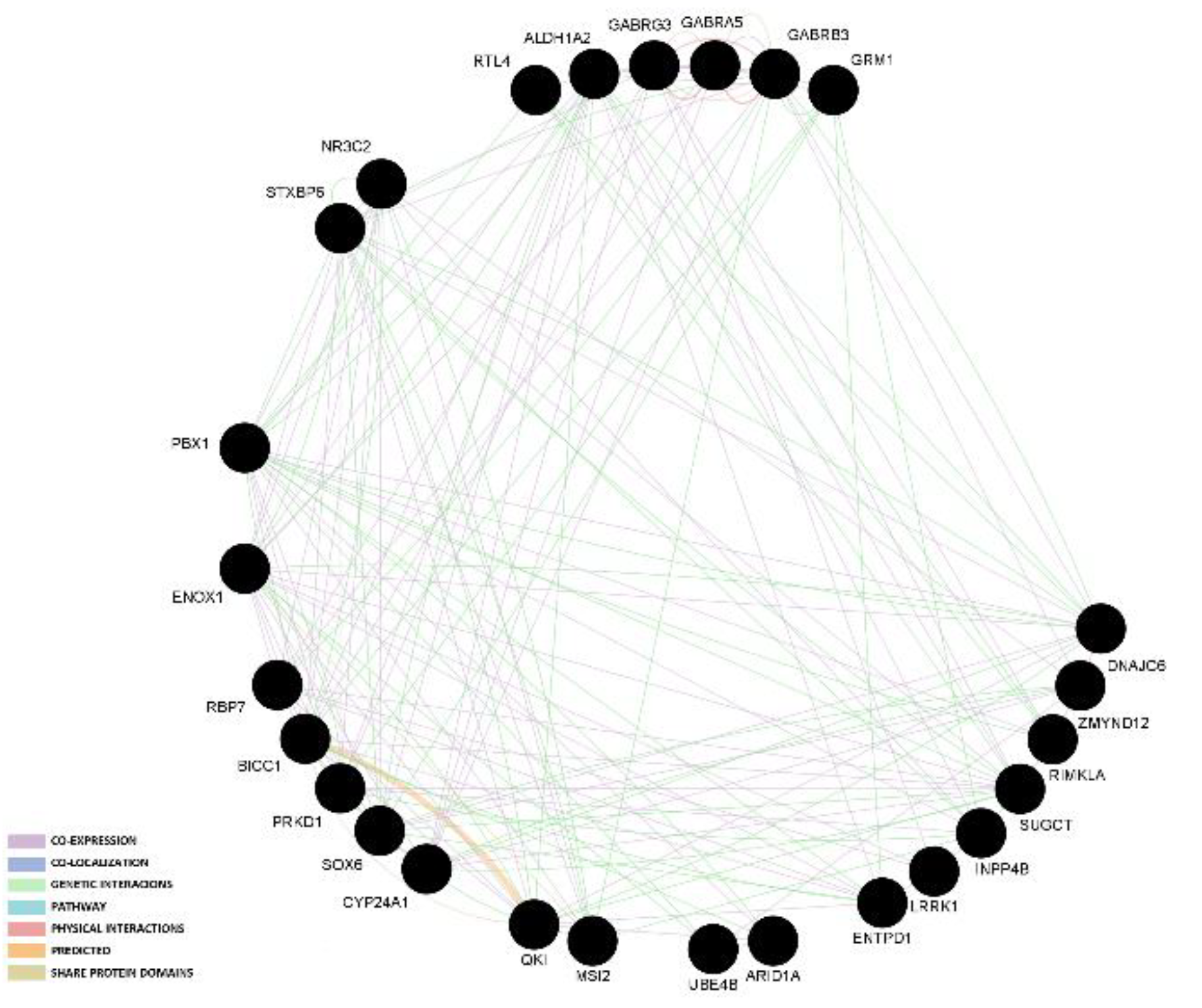
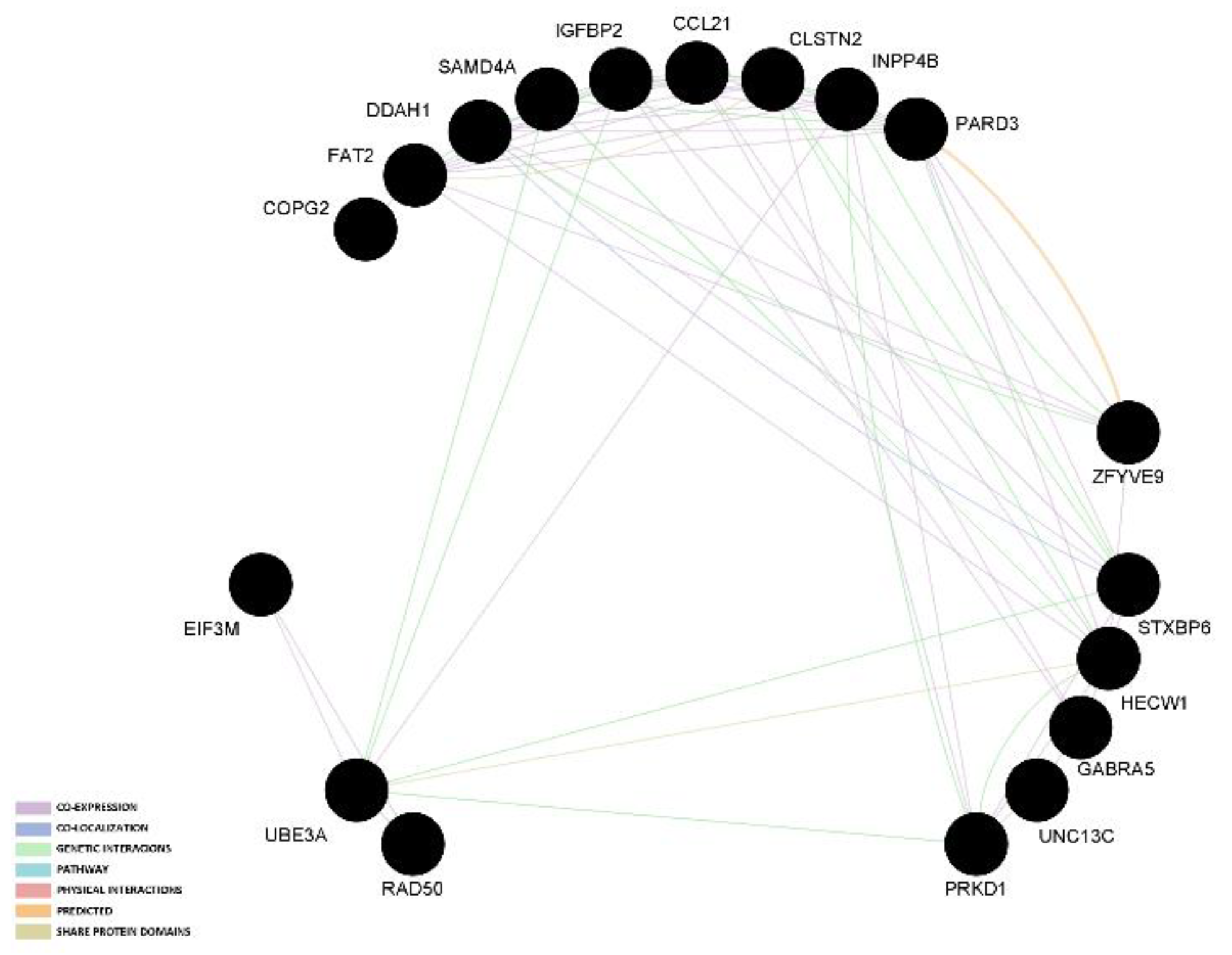
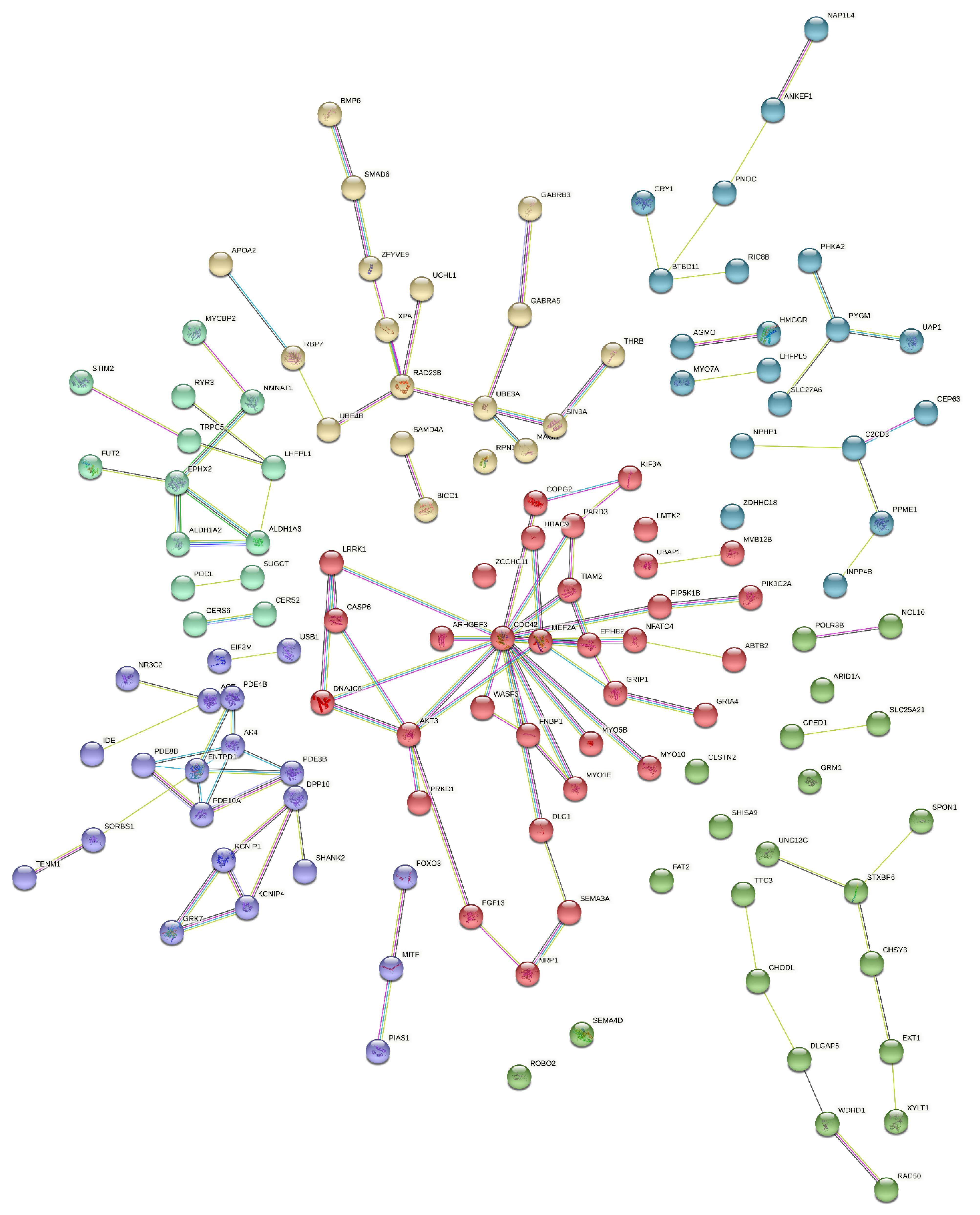
3.3. Gene Network and Protein–Protein Interactions
3.4. Meta-Analysis of Five GWA Studies
3.5. Candidate Genes and Causal Variation
4. Discussion
4.1. Genetic Relationship between Litter Traits
4.2. Connections among Candidate Genes
4.3. The New Genomic Region Associated with Litter Size
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rothschild, M.; Jacobson, C.; Vaske, D.; Tuggle, C.; Wang, L.; Short, T.; Eckardt, G.; Sasaki, S.; Vincent, A.; McLaren, D.; et al. The estrogen receptor locus is associated with a major gene influencing litter size in pigs. Proc. Natl. Acad. Sci. USA 1996, 93, 201–205. [Google Scholar] [CrossRef]
- Ernst, C.W.; Steibel, J.P. Molecular advances in QTL discovery and application in pig breeding. Trends Genet. 2013, 29, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Blaj, I.; Tetens, J.; Preuss, S.; Bennewitz, J.; Thaller, G. Genome-wide association studies and meta-analysis uncovers new candidate genes for growth and carcass traits in pigs. PLoS ONE 2018, 13, e0205576. [Google Scholar] [CrossRef]
- Rothschild, M.F.; Hu, Z.-L.; Jiang, Z. Advances in QTL Mapping in Pigs. Int. J. Biol. Sci. 2007, 3, 192–197. [Google Scholar] [CrossRef]
- Sosa-Madrid, B.S.; Santacreu, M.A.; Blasco, A.; Fontanesi, L.; Pena, R.N.; Ibáñez-Escriche, N. A genomewide association study in divergently selected lines in rabbits reveals novel genomic regions associated with litter size traits. J. Anim. Breed. Genet. 2020, 137, 123–138. [Google Scholar] [CrossRef]
- Casto-Rebollo, C.; Argente, M.J.; Garciá, M.L.; Pena, R.; Ibáñez-Escriche, N. Identification of functional mutations associated with environmental variance of litter size in rabbits. Genet. Sel. Evol. 2020, 52, 22. [Google Scholar] [CrossRef]
- Tao, L.; He, X.Y.; Jiang, Y.T.; Lan, R.; Li, M.; Li, Z.M.; Yang, W.F.; Hong, Q.H.; Chu, M.X. Combined approaches to reveal genes associated with litter size in Yunshang black goats. Anim. Genet. 2020, 51, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Liu, X.; Gebreselassie, G.; Abied, A.; Ma, Q.; Ma, Y. Genome-wide association analysis reveals the genetic locus for high reproduction trait in Chinese Arbas Cashmere goat. Genes Genom. 2020, 42, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ma, L.; Prakapenka, D.; VanRaden, P.M.; Cole, J.B.; Da, Y. A Large-Scale Genome-Wide Association Study in U.S. Holstein Cattle. Front. Genet. 2019, 10, 412. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Z.; Ye, S.; He, Y.; Huang, S.; Yuan, X.; Li, J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals 2019, 9, 732. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Ye, S.; Teng, J.; Diao, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J.; Zhang, Z. Genome-wide association studies for the number of animals born alive and dead in duroc pigs. Theriogenology 2019, 139, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Bakoev, S.; Getmantseva, L.; Bakoev, F.; Kolosova, M.; Gabova, V.; Kolosov, A.; Kostyunina, O. Survey of SNPs Associated with Total Number Born and Total Number Born Alive in Pig. Genes 2020, 11, 491. [Google Scholar] [CrossRef] [PubMed]
- Animal Genome Project. Pig QTL Data Base. Available online: http//www.animalgenome.org/QTLdb/pig.html (accessed on 21 March 2021).
- Van Son, M.; Enger, E.G.; Grove, H.; Ros-Freixedes, R.; Kent, M.P.; Lien, S.; Grindflek, E. Genome-wide association study confirm major QTL for backfat fatty acid composition on SSC14 in Duroc pigs. BMC Genom. 2017, 18, 369. [Google Scholar] [CrossRef]
- Jungerius, B.J.; Van Laere, A.S.; Te Pas, M.F.W.; Van Oost, B.A.; Andersson, L.; Groenen, M.A.M. The IGF2-intron3-G3072A substitution explains a major imprinted QTL effect on backfat thickness in a Meishan x European white pig intercross. Genet. Res. 2004, 84, 95–101. [Google Scholar] [CrossRef]
- Van Laere, A.-S.; Nguyen, M.; Braunschweig, M.; Nezer, C.; Collette, C.; Moreau, L.; Archibald, A.; Haley, C.; Buys, N.; Tally, M.; et al. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 2003, 425, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, L. Use of meta-analysis to combine candidate gene association studies: Application to study the relationship between the ESR PvuII polymorphism and sow litter size. Genet. Sel. Evol. 2005, 37, 417–435. [Google Scholar] [CrossRef]
- Bouwman, A.C.; Daetwyler, H.D.; Chamberlain, A.J.; Ponce, C.H.; Sargolzaei, M.; Schenkel, F.S.; Sahana, G.; Govignon-Gion, A.; Boitard, S.; Dolezal, M.; et al. Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 2018, 50, 362–367. [Google Scholar] [CrossRef]
- Duarte, D.A.S.; Newbold, C.J.; Detmann, E.; Silva, F.F.; Freitas, P.H.F.; Veroneze, R.; Duarte, M.S. Genome-wide association studies pathway-based meta-analysis for residual feed intake in beef cattle. Anim. Genet. 2019, 50, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Tropf, F.C.; Lee, S.H.; Verweij, R.M.; Stulp, G.; van der Most, P.J.; de Vlaming, R.; Bakshi, A.; Briley, D.A.; Rahal, C.; Hellpap, R.; et al. Hidden heritability due to heterogeneity across seven populations. Nat. Hum. Behav. 2017, 1, 757–765. [Google Scholar] [CrossRef]
- Sauvant, D.; Letourneau-Montminy, M.P.; Schmidely, P.; Boval, M.; Loncke, C.; Daniel, J.B. Review: Use and misuse of meta-analysis in Animal Science. Animal 2020, 14, s207–s222. [Google Scholar] [CrossRef]
- Uimari, P.; Sironen, A.; Sevón-Aimonen, M.-L. Whole-genome SNP association analysis of reproduction traits in the Finnish Landrace pig breed. Genet. Sel. Evol. 2011, 43, 42. [Google Scholar] [CrossRef] [PubMed]
- Sell-Kubiak, E.; Duijvesteijn, N.; Lopes, M.S.; Janss, L.L.G.; Knol, E.F.; Bijma, P.; Mulder, H.A. Genome-wide association study reveals novel loci for litter size and its variability in a Large White pig population. BMC Genom. 2015, 16, 1049. [Google Scholar] [CrossRef]
- Balogh, E.E.; Gábor, G.; Bodó, S.; Rózsa, L.; Rátky, J.; Zsolnai, A.; Anton, I. Effect of single-nucleotide polymorphisms on specific reproduction parameters in Hungarian Large White sows. Acta Vet. Hung. 2019, 67, 256–273. [Google Scholar] [CrossRef]
- Ma, X.; Li, P.H.; Zhu, M.X.; He, L.C.; Sui, S.P.; Gao, S.; Su, G.S.; Ding, N.S.; Huang, Y.; Lu, Z.Q.; et al. Genome-wide association analysis reveals genomic regions on Chromosome 13 affecting litter size and candidate genes for uterine horn length in Erhualian pigs. Animal 2018, 12, 2453–2461. [Google Scholar] [CrossRef]
- Thompson, J.R.; Attia, J.; Minelli, C. The meta-analysis of genome-wide association studies. Brief. Bioinform. 2011, 12, 259–269. [Google Scholar] [CrossRef]
- An, S.M.; Hwang, J.H.; Kwon, S.; Yu, G.E.; Park, D.H.; Kang, D.G.; Kim, T.W.; Park, H.C.; Ha, J.; Kim, C.W. Effect of Single Nucleotide Polymorphisms in IGFBP2 and IGFBP3 Genes on Litter Size Traits in Berkshire Pigs. Anim. Biotechnol. 2018, 29, 301–308. [Google Scholar] [CrossRef]
- Coster, A.; Madsen, O.; Heuven, H.C.M.; Dibbits, B.; Groenen, M.A.M.; van Arendonk, J.A.M.; Bovenhuis, H. The Imprinted Gene DIO3 Is a Candidate Gene for Litter Size in Pigs. PLoS ONE 2012, 7, e31825. [Google Scholar] [CrossRef]
- He, L.C.; Li, P.H.; Ma, X.; Sui, S.P.; Gao, S.; Kim, S.W.; Gu, Y.Q.; Huang, Y.; Ding, N.S.; Huang, R.H. Identification of new single nucleotide polymorphisms affecting total number born and candidate genes related to ovulation rate in Chinese Erhualian pigs. Anim. Genet. 2017, 48, 48–54. [Google Scholar] [CrossRef]
- Kumchoo, T.; Mekchay, S. Association of non-synonymous SNPs of OPN gene with litter size traits in pigs. Arch. Anim. Breed. 2015, 58, 317–323. [Google Scholar] [CrossRef]
- Li, X.; Ye, J.; Han, X.; Qiao, R.; Li, X.; Lv, G.; Wang, K. Whole-genome sequencing identifies potential candidate genes for reproductive traits in pigs. Genomics 2020, 112, 199–206. [Google Scholar] [CrossRef]
- Liu, C.; Ran, X.; Yu, C.; Xu, Q.; Niu, X.; Zhao, P.; Wang, J. Whole-genome analysis of structural variations between Xiang pigs with larger litter sizes and those with smaller litter sizes. Genomics 2018, 111, 310–319. [Google Scholar] [CrossRef]
- Sato, S.; Kikuchi, T.; Uemoto, Y.; Mikawa, S.; Suzuki, K. Effect of candidate gene polymorphisms on reproductive traits in a Large White pig population. Anim. Sci. J. 2016, 87, 1455–1463. [Google Scholar] [CrossRef]
- Uzzaman, M.R.; Park, J.E.; Lee, K.T.; Cho, E.S.; Choi, B.H.; Kim, T.H. A genome-wide association study of reproductive traits in a Yorkshire pig population. Livest. Sci. 2018, 209, 67–72. [Google Scholar] [CrossRef]
- Wang, H.; Wu, S.; Wu, J.; Sun, S.; Wu, S.; Bao, W. Association analysis of the SNP (rs345476947) in the FUT2 gene with the production and reproductive traits in pigs. Genes Genom. 2018, 40, 199–206. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, X.; Tan, Z.; Xing, K.; Yang, T.; Pan, Y.; Mi, S.; Sun, D.; Wang, C. Genome-wide association study for reproductive traits in a Large White pig population. Anim. Genet. 2018, 49, 127–131. [Google Scholar] [CrossRef]
- Wu, P.; Wang, K.; Yang, Q.; Zhou, J.; Chen, D.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; Jiang, A.; et al. Identifying SNPs and candidate genes for three litter traits using single-step GWAS across six parities in Landrace and Large White pigs. Physiol. Genom. 2018, 50, 1026–1035. [Google Scholar] [CrossRef]
- Wu, P.; Yang, Q.; Wang, K.; Zhou, J.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; Jiang, A.; Jiang, Y.; et al. Single step genome-wide association studies based on genotyping by sequence data reveals novel loci for the litter traits of domestic pigs. Genomics 2018, 110, 171–179. [Google Scholar] [CrossRef]
- Bergfelder-Drüing, S.; Grosse-Brinkhaus, C.; Lind, B.; Erbe, M.; Schellander, K.; Simianer, H.; Tholen, E. A Genome-Wide Association Study in Large White and Landrace Pig Populations for Number Piglets Born Alive. PLoS ONE 2015, 10, e0117468. [Google Scholar] [CrossRef]
- Suwannasing, R.; Duangjinda, M.; Boonkum, W.; Taharnklaew, R.; Tuangsithtanon, K. The identification of novel regions for reproduction trait in Landrace and Large White pigs using a single step genome-wide association study. Asian-Australas. J. Anim. Sci. 2018, 31, 1852–1862. [Google Scholar] [CrossRef]
- Onteru, S.K.; Fan, B.; Du, Z.-Q.; Garrick, D.J.; Stalder, K.J.; Rothschild, M.F. A whole-genome association study for pig reproductive traits. Anim. Genet. 2011, 43, 18–26. [Google Scholar] [CrossRef]
- Schneider, J.F.; Miles, J.R.; Brown-Brandl, T.M.; Nienaber, J.A.; Rohrer, G.A.; Vallet, J.L. Genomewide association analysis for average birth interval and stillbirth in swine. J. Anim. Sci. 2015, 93, 529–540. [Google Scholar] [CrossRef]
- Verardo, L.L.; Silva, F.F.; Lopes, M.S.; Madsen, O.; Bastiaansen, J.W.M.; Knol, E.F.; Kelly, M.; Varona, L.; Lopes, P.S.; Guimarães, S.E.F. Revealing new candidate genes for reproductive traits in pigs: Combining Bayesian GWAS and functional pathways. Genet. Sel. Evol. 2016, 48, 9. [Google Scholar] [CrossRef]
- Schneider, J.F.; Nonneman, D.J.; Wiedmann, R.T.; Vallet, J.L.; Rohrer, G.A. Genomewide association and identification of candidate genes for ovulation rate in swine. J. Anim. Sci. 2014, 92, 3792–3803. [Google Scholar] [CrossRef]
- Carbon, S.; Mungall, C. Gene Ontology Data Archive; Zenodo, Maryland, United States of America: 2021. Available online: https://doi.org/10.5281/zenodo.5228828 (accessed on 29 August 2022).
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2020, 49, D394–D403. [Google Scholar] [CrossRef]
- Keel, B.N.; Snelling, W.M.; Lindholm-Perry, A.K.; Oliver, W.T.; Kuehn, L.A.; Rohrer, G. Using SNP Weights Derived From Gene Expression Modules to Improve GWAS Power for Feed Efficiency in Pigs. Front. Genet. 2020, 10, 1339. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38 (Suppl. 2), 214–220. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks.; made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Bozhilova, L.V.; Whitmore, A.V.; Wray, J.; Reinert, G.; Deane, C.M. Measuring rank robustness in scored protein interaction networks. BMC Bioinform. 2019, 20, 446. [Google Scholar] [CrossRef]
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef] [Green Version]
- Willer, C.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef] [PubMed]
- Berg, I.V.D.; Boichard, D.; Lund, M.S. Comparing power and precision of within-breed and multibreed genome-wide association studies of production traits using whole-genome sequence data for 5 French and Danish dairy cattle breeds. J. Dairy Sci. 2016, 99, 8932–8945. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analysis. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Derks, M.F.; Gross, C.; Lopes, M.S.; Reinders, M.J.; Bosse, M.; Gjuvsland, A.B.; de Ridder, D.; Megens, H.-J.; Groenen, M.A. Accelerated discovery of functional genomic variation in pigs. Genomics 2021, 113, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Groß, C.; Derks, M.; Megens, H.-J.; Bosse, M.; Groenen, M.A.M.; Reinders, M.; De Ridder, D. pCADD: SNV prioritisation in Sus scrofa. Genet. Sel. Evol. 2020, 52, 4. [Google Scholar] [CrossRef]
- Holm, B.; Bakken, M.; Klemetsdal, G.; Vangen, O. Genetic correlations between reproduction and production traits in swine. J. Anim. Sci. 2004, 82, 3458–3464. [Google Scholar] [CrossRef]
- Sell-Kubiak, E. Selection for litter size and litter birthweight in Large White pigs: Maximum, mean and variability of reproduction traits. Animal 2021, 15, 100352. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, M.; Li, W.; He, L.; Zhou, Y.; Zhu, J.; Che, R.; Warburton, M.L.; Yang, X.; Yan, J. Genetic variants and underlying mechanisms influencing variance heterogeneity in maize. Plant J. 2020, 103, 1089–1102. [Google Scholar] [CrossRef]
- Lu, S.; Liu, S.; Wietelmann, A.; Kojonazarov, B.; Atzberger, A.; Tang, C.; Schermuly, R.; Gröne, H.-J.; Offermanns, S. Developmental vascular remodeling defects and postnatal kidney failure in mice lacking Gpr116 (Adgrf5) and Eltd1 (Adgrl4). PLoS ONE 2017, 12, e0183166. [Google Scholar] [CrossRef]
- Shaheen, R.; al Hashem, A.; Alghamdi, M.H.; Seidahmad, M.Z.; Wakil, S.M.; Dagriri, K.; Keavney, B.; Goodship, J.; Alyousif, S.; Al-Habshan, F.M.; et al. Positional mapping of PRKD1.; NRP1 and PRDM1 as novel candidate disease genes in truncus arteriosus. J. Med. Genet. 2015, 52, 322–329. [Google Scholar] [CrossRef]
- Massadeh, S.; Albeladi, M.; Albesher, N.; Alhabshan, F.; Kampe, K.; Chaikhouni, F.; Kabbani, M.; Beetz, C.; Alaamery, M. Novel Autosomal Recessive Splice-Altering Variant in PRKD1 Is Associated with Congenital Heart Disease. Genes 2021, 12, 612. [Google Scholar] [CrossRef]
- Nonneman, D.; Lents, C.; Rohrer, G.; Rempel, L.; Vallet, J. Genome-wide association with delayed puberty in swine. Anim. Genet. 2013, 45, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Watschinger, K.; Werner, E.R. Alkylglycerol monooxygenase. IUBMB Life 2013, 65, 366–372. [Google Scholar] [CrossRef]
- Sailer, S.; Keller, M.A.; Werner, E.R.; Watschinger, K. The Emerging Physiological Role of AGMO 10 Years after Its Gene Identification. Life 2021, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Okur, V.; Watschinger, K.; Niyazov, D.; McCarrier, J.; Basel, D.; Hermann, M.; Werner, E.R.; Chung, W.K. Biallelic variants in AGMO with diminished enzyme activity are associated with a neurodevelopmental disorder. Hum. Genet. 2019, 138, 1259–1266. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong Association of De Novo Copy Number Mutations with Autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef]
- Fontaine-Bisson, B.; The MAGIC investigators; Renström, F.; Rolandsson, O.; Payne, F.; Hallmans, G.; Barroso, I.; Franks, P.W. Evaluating the discriminative power of multi-trait genetic risk scores for type 2 diabetes in a northern Swedish population. Diabetologia 2010, 53, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Boesgaard, T.W.; Grarup, N.; Jørgensen, T.; Borch-Johnsen, K.; Hansen, T.; Pedersen, O. Variants at DGKB/TMEM195.; ADRA2A.; GLIS3 and C2CD4B loci are associated with reduced glucose-stimulated beta cell function in middle-aged Danish people. Diabetologia 2010, 53, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. Erratum: New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 464. [Google Scholar] [CrossRef]
- Pradhan, B.; Bhattacharya, I.; Sarkar, R.; Majumdar, S.S. Downregulation of Sostdc1 in Testicular Sertoli Cells is Prerequisite for Onset of Robust Spermatogenesis at Puberty. Sci. Rep. 2019, 9, 11458. [Google Scholar] [CrossRef] [Green Version]
- Myromslien, F.D.; Tremoen, N.H.; Andersen-Ranberg, I.; Fransplass, R.; Stenseth, E.-B.; Zeremichael, T.T.; Van Son, M.; Grindflek, E.; Gaustad, A.H. Sperm DNA integrity in Landrace and Duroc boar semen and its relationship to litter size. Reprod. Domest. Anim. 2018, 54, 160–166. [Google Scholar] [CrossRef]
- Tremoen, N.H.; Van Son, M.; Andersen-Ranberg, I.; Grindflek, E.; Myromslien, F.D.; Gaustad, A.H.; Våge, D.I. Association between single-nucleotide polymorphisms within candidate genes and fertility in Landrace and Duroc pigs. Acta Vet. Scand. 2019, 61, 58. [Google Scholar] [CrossRef] [PubMed]
- Burdelski, C.; Borcherding, L.; Kluth, M.; Hube-Magg, C.; Melling, N.; Simon, R.; Möller-Koop, C.; Weigand, P.; Minner, S.; Haese, A.; et al. Family with sequence similarity 13C (FAM13C) overexpression is an independent prognostic marker in prostate cancer. Oncotarget 2017, 8, 31494–31508. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Cao, J.; Guo, G.; Li, X.; Wang, Y.; Yu, Y.; Zhang, S.; Zhang, Q.; Zhang, Y. Genome-wide association study identifies QTLs for displacement of abomasum in Chinese Holstein cattle1. J. Anim. Sci. 2019, 97, 1133–1142. [Google Scholar] [CrossRef]
- Georges, H.; Bishop, J.; Van Campen, H.; Barfield, J.; Hansen, T. 102 A delay in maternal zygotic transition may lead to early embryonic loss in poor-quality bovine blastocysts. Reprod. Fertil. Dev. 2020, 32, 177. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Source of Data | Uimari et al. [22] | Sell-Kubiak et al. [23] | Ma et al. [25] | Zhang et al. [10] | Balogh et al. [24] |
|---|---|---|---|---|---|
| Population | Finnish Landrace | Large White | Erhualian | Duroc | Hungarian Large White |
| Phenotype | deEBV 1 | deEBV 1 | EBV 2 | deEBV 1 | TNB |
| BeadChip | Porcine SNP60 Bead Chip | ||||
| Individuals | 328 | 2351 | 48 | 1067 | 290 |
| Available SNPs | 57,868 | 40,969 | 28,020 | 32,147 | 56,592 |
| Method | single-SNP mixed model with pedigree relationship matrix | single-SNP mixed model with genomic relationship matrix | single-SNP mixed model with pedigree relationship matrix | single-SNP mixed model with genomic relationship matrix | multi-SNP mixed model with genomic relationship matrix |
| Detected SNPs | 5 | 0 | 5 | 7 | 3 |
| Threshold | −log10(p-value) ≤ 5.7 3 | −log10(p-value) ≥ 5 | −log10(p-value) ≤ 5.75 3 | −log10(p-value) ≥ 4 | −log10(p-value) ≥ 5 |
| Trait | Number of SNPs | Number of Genes | Publication | |
|---|---|---|---|---|
| TNB | 1 | 1 | An et al. | [27] |
| 13 | 7 | Coster et al. | [28] | |
| 145 | 83 | He et al. | [29] | |
| 3 | 0 | Kumchoo and Mekchay | [30] | |
| 1 | 1 | Li et al. | [31] | |
| 1 | 1 | Liu et al. | [32] | |
| 7 | 5 | Ma et al. | [25] | |
| 4 | 4 | Sato et al. | [33] | |
| 10 | 7 | Sell-Kubiak et al. | [23] | |
| 5 | 2 | Uimari et al. | [22] | |
| 2 | 2 | Uzzaman et al. | [34] | |
| 10 | 5 | H. Wang et al. | [35] | |
| 1 | 1 | Y. Wang et al. | [36] | |
| 40 | 6 | Wu et al. | [37] | |
| 5 | 3 | Wu et al. | [38] | |
| 7 | 4 | Zhang et al. | [10] | |
| NBA | 1 | 1 | An et al. | [27] |
| 17 | 8 | Bergfelder-Drüing et al. | [39] | |
| 11 | 6 | Chen et al. | [11] | |
| 3 | 3 | Coster et al. | [28] | |
| 3 | 3 | Kumchoo and Mekchay | [30] | |
| 1 | 1 | Li et al. | [31] | |
| 9 | 7 | Ma et al. | [25] | |
| 2 | 2 | Sato et al. | [33] | |
| 27 | 11 | Suwannasing et al. | [40] | |
| 1 | 0 | Uzzaman et al. | [34] | |
| 5 | 3 | Y. Wang et al. | [36] | |
| 101 | 19 | Wu et al. | [37] | |
| 15 | 6 | Wu et al. | [38] | |
| SB | 46 | 22 | Chen et al. | [11] |
| 6 | 3 | Onteru et al. | [41] | |
| 13 | 11 | Schneider et al. | [42] | |
| 2 | 1 | Uimari et al. | [22] | |
| 22 | 15 | Verardo et al. | [43] | |
| LWT | 1 | 1 | Coster et al. | [28] |
| 1 | 1 | Liu et al. | [32] | |
| 10 | 2 | Zhang et al. | [10] | |
| CLN | 24 | 12 | Schneider et al. | [44] |
| Trait | Gene | Number of SNPs | Publication | |
|---|---|---|---|---|
| TNB | GABRG3 | 5 | Coster et al. | [28] |
| MSI2 | 3 | He et al. | [29] | |
| BICC1 | 2 | Zhang et al. | [26] | |
| ENTPD1 | 2 | He et al. | [29] | |
| ENOX1 | 2 | Sell-Kubiak et al. | [23] | |
| SUGCT | 2 | Sell-Kubiak et al. | [23] | |
| NR3C2 | 2 | Wu et al. | [38] | |
| GABRB3 | 2 | Coster et al. | [28] | |
| NBA | RBP7 | 5 | Suwannasing et al. | [40] |
| ARID1A | 3 | Chen et al. | [11] | |
| LRRK1 | 2 | Suwannasing et al. | [40] | |
| ZMYND12 | 2 | Suwannasing et al. | [40] | |
| RIMKLA | 2 | Suwannasing et al. | [40] | |
| RTL4 | 2 | Suwannasing et al. | [40] | |
| UBE4B | 3 | Suwannasing et al. | [40] | |
| ALDH1A2 | 2 | Wu et al. | [37] | |
| GABRA5 | 2 | Wu et al. | [37] | |
| INPP4B | 2 | Wu et al. | [37] | |
| DNAJC6 | 2 | Wu et al. | [37] | |
| QKI | 2 | Wu et al. | [37] | |
| SOX6 | 2 | Wu et al. | [37] | |
| SB | PRKD1 | 5 | Chen et al. | [11] |
| STXBP6 | 4 | Chen et al. | [11] | |
| PBX1 | 2 | Chen et al. | [11] | |
| GRM1 | 2 | Chen et al. | [11] | |
| CYP24A1 | 2 | Verardo et al. | [43] | |
| Candidate Gene | Publication and Population | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| An et al. [27] | Chen et al. [11] | Coster et al. [28] | He et al. [29] | Li et al. [31] | Ma et al. [25] | Sato et al. [33] | Schneider et al. [44] | Schneider et al. [42] | Verardo et al. [43] | Wu et al. [37] | Y. Wang et al. [36] | |
| Berkshire | Duroc | Large White | Erhualian | Yorkshire | Erhualian | Large White | Landrace x Duroc | Duroc, Yorkshire, Landrace | Large White | Yorkshire | Large White | |
| ASIC2 | TNB | TNB | ||||||||||
| CCL21 | TNB, NBA | |||||||||||
| CLSTN2 | TNB, NBA | |||||||||||
| COPG2 | TNB, LBW | |||||||||||
| DDAH1 | TNB | TNB, NBA | ||||||||||
| EIF3M | TNB, NBA | |||||||||||
| FAT2 | TNB, NBA | |||||||||||
| GABRA5 | TNB | TNB | ||||||||||
| HECW1 | TNB, NBA | |||||||||||
| IGFBP2 | TNB | |||||||||||
| INPP4B | TNB, NBA | |||||||||||
| MAPK1IP1L | SB | SB | ||||||||||
| NEK10 | TNB | TNB | ||||||||||
| PARD3 | TNB, NBA | |||||||||||
| PRKD1 | SB | NBA | ||||||||||
| RAD50 | TNB, NBA | |||||||||||
| SAMD4A | CLN | SB | ||||||||||
| STXBP6 | SB | TNB | ||||||||||
| UBE3A | TNB, NBA | |||||||||||
| UNC13C | TNB, NBA | |||||||||||
| ZFYVE9 | TNB, NBA | CLN | ||||||||||
| GO Term | Biological Processes (with Hierarchy When Applicable) | Observed Genes | Expected Genes | FDR | ||||
|---|---|---|---|---|---|---|---|---|
| 9987 | cellular process | 177 | 148.91 | 0.019 | ||||
| 7399 |  | nervous system development | 32 | 15.2 | 0.027 | |||
| 21785 |  | branchiomotor neuron axon guidance | 3 | 0.06 | 0.02 | |||
| 48518 | positive regulation of the biological process | 77 | 49.5 | 0.012 | ||||
| 31344 |  | regulation of cell projection organization | 17 | 4.36 | 0.003 | |||
| 120035 |  | regulation of plasma membrane-bounded cell projection organization | 17 | 4.29 | 0.003 | |||
| 51489 |  | regulation of filopodium assembly | 5 | 0.32 | 0.012 | |||
| 51491 |  | positive regulation of filopodium assembly | 4 | 0.17 | 0.016 | |||
| 51239 | regulation of multicellular organismal process | 41 | 20.21 | 0.009 | ||||
| 43536 |  | positive regulation of blood vessel endothelial cell migration | 5 | 0.45 | 0.04 | |||
| 90050 |  | positive regulation of cell migration involved in sprouting angiogenesis | 4 | 0.2 | 0.026 | |||
| 8299 | isoprenoid biosynthetic process | 4 | 0.24 | 0.043 | ||||
| 48813 | dendrite morphogenesis | 5 | 0.46 | 0.042 | ||||
| 22603 | regulation of anatomical structure morphogenesis | 19 | 6.88 | 0.033 | ||||
| 10975 |  | regulation of neuron projection development | 14 | 2.84 | 0.002 | |||
| 50770 |  | regulation of axonogenesis | 8 | 1.13 | 0.013 | |||
| 50771 |  | negative regulation of axonogenesis | 5 | 0.48 | 0.048 | |||
| 34765 | regulation of ion transmembrane transport | 12 | 3.25 | 0.044 | ||||
| 1904062 |  | regulation of cation transmembrane transport | 12 | 2.25 | 0.004 | |||
| 48583 | regulation of response to stimulus | 54 | 30.14 | 0.009 | ||||
| 50920 |  | regulation of chemotaxis | 9 | 1.78 | 0.037 | |||
| 32409 | regulation of transporter activity | 9 | 1.8 | 0.039 | ||||
| 1655 | urogenital system development | 12 | 2.55 | 0.01 | ||||
| 72001 |  | renal system development | 10 | 2.29 | 0.044 | |||
| 44057 | regulation of system process | 14 | 3.57 | 0.013 | ||||
| 65008 | regulation of biological quality | 56 | 30.03 | 0.003 | ||||
| 42592 |  | homeostatic process | 29 | 12.43 | 0.013 | |||
| 48878 |  | chemical homeostasis | 23 | 8.89 | 0.019 | |||
| 51240 | positive regulation of multicellular organismal process | 25 | 10.62 | 0.033 | ||||
| 10646 | regulation of cell communication | 45 | 25.74 | 0.048 | ||||
| 9966 |  | regulation of signal transduction | 42 | 22.78 | 0.033 | |||
| 51179 | localization | 74 | 44.32 | 0.003 | ||||
| 51234 |  | establishment of localization | 60 | 33.77 | 0.004 | |||
| 6810 |  | transport | 57 | 32.4 | 0.009 | |||
| Line | Genotype | N | TNB | NBA | SB | Mummies |
|---|---|---|---|---|---|---|
| Synthetic boar line ♀ | 0/0 | 17,527 | 10.13 (3.10) | 9.28 (3.10) | 0.85 | 0.30 |
| 0/1 | 3247 | 10.07 (3.05) | 9.29 (3.02) | 0.78 | 0.32 | |
| 1/1 | 115 | 10.03 (2.95) | 9.46 (2.85) | 0.57 | 0.39 | |
| Synthetic boar line ♂ | 0/0 | 25,500 | 10.08 (3.10) | 9.23 (3.10) | 0.85 | 0.33 |
| 0/1 | 4333 | 9.95 (3.18) | 9.12 (3.17) | 0.83 | 0.37 | |
| 1/1 | 231 | 9.64 (3.01) | 8.86 (2.94) | 0.78 | 0.34 | |
| Crossbreed 1 ♀ | 0/0 | 32,015 | 16.56 (3.89) | 15.39 (3.74) | 1.17 | 0.39 |
| 0/1 | 5290 | 16.52 (3.86) | 15.30 (3.75) | 1.21 | 0.35 | |
| 1/1 | 206 | 16.41 (4.08) | 15.37 (3.84) | 1.04 | 0.43 | |
| Crossbreed 2 ♂ | 0/0 | 54,824 | 16.00 (3.93) | 15.06 (3.85) | 0.94 | 0.36 |
| 0/1 | 9714 | 16.02 (3.80) | 15.05 (3.78) | 0.97 | 0.36 | |
| 1/1 | 172 | 15.83 (4.08) | 15.02 (3.92) | 0.80 | 0.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sell-Kubiak, E.; Dobrzanski, J.; Derks, M.F.L.; Lopes, M.S.; Szwaczkowski, T. Meta-Analysis of SNPs Determining Litter Traits in Pigs. Genes 2022, 13, 1730. https://doi.org/10.3390/genes13101730
Sell-Kubiak E, Dobrzanski J, Derks MFL, Lopes MS, Szwaczkowski T. Meta-Analysis of SNPs Determining Litter Traits in Pigs. Genes. 2022; 13(10):1730. https://doi.org/10.3390/genes13101730
Chicago/Turabian StyleSell-Kubiak, Ewa, Jan Dobrzanski, Martijn F. L. Derks, Marcos S. Lopes, and Tomasz Szwaczkowski. 2022. "Meta-Analysis of SNPs Determining Litter Traits in Pigs" Genes 13, no. 10: 1730. https://doi.org/10.3390/genes13101730