
Genotype-Phenotype Comparison in POGZ-Related Neurodevelopmental Disorders by Using Clinical Scoring

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Subjects and Methods
2.1. Clinical Evaluation and Severity Scoring
2.2. Variant Evaluation
2.3. Facial Gestalt Analysis
2.4. Statistical Analysis
3. Results
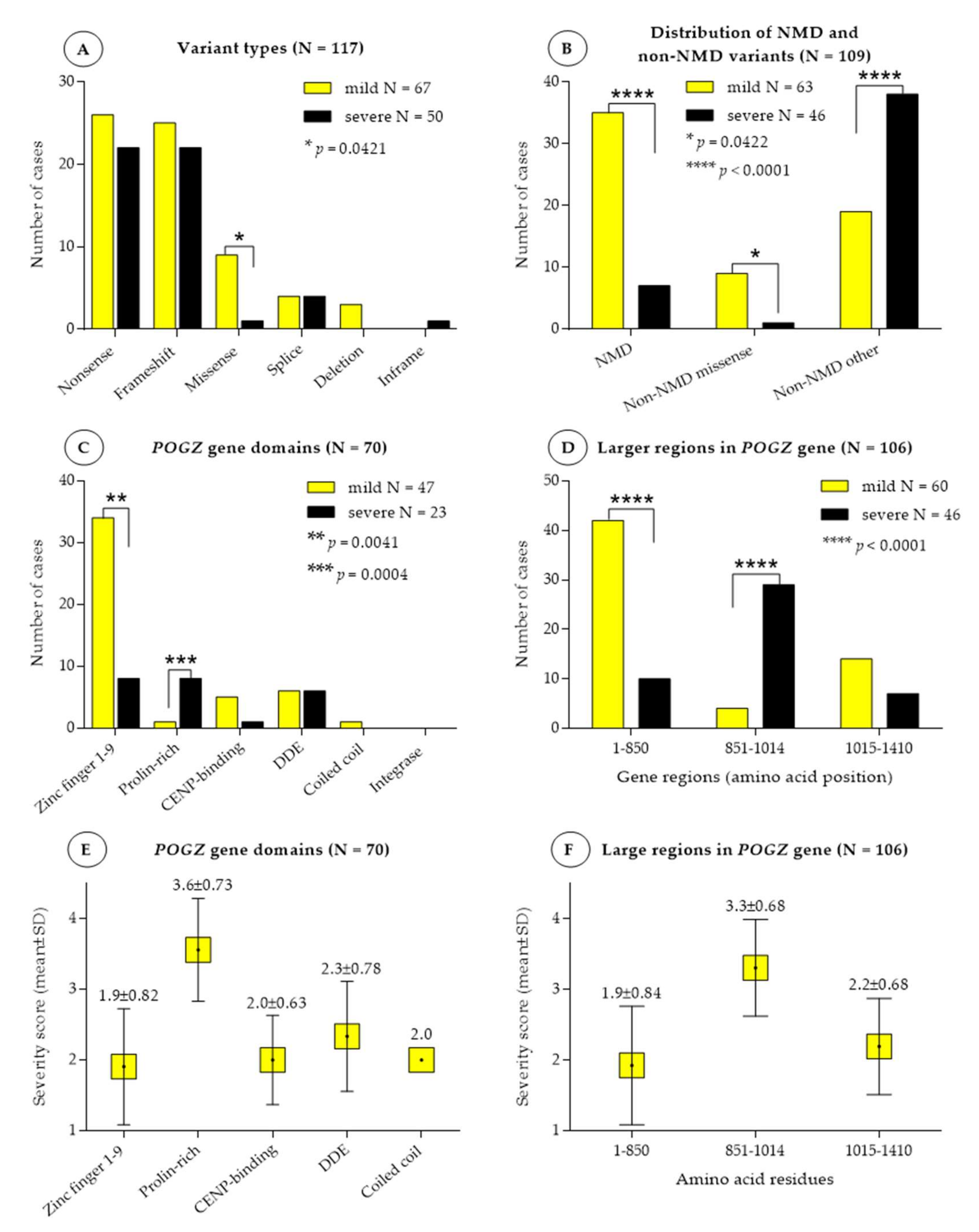
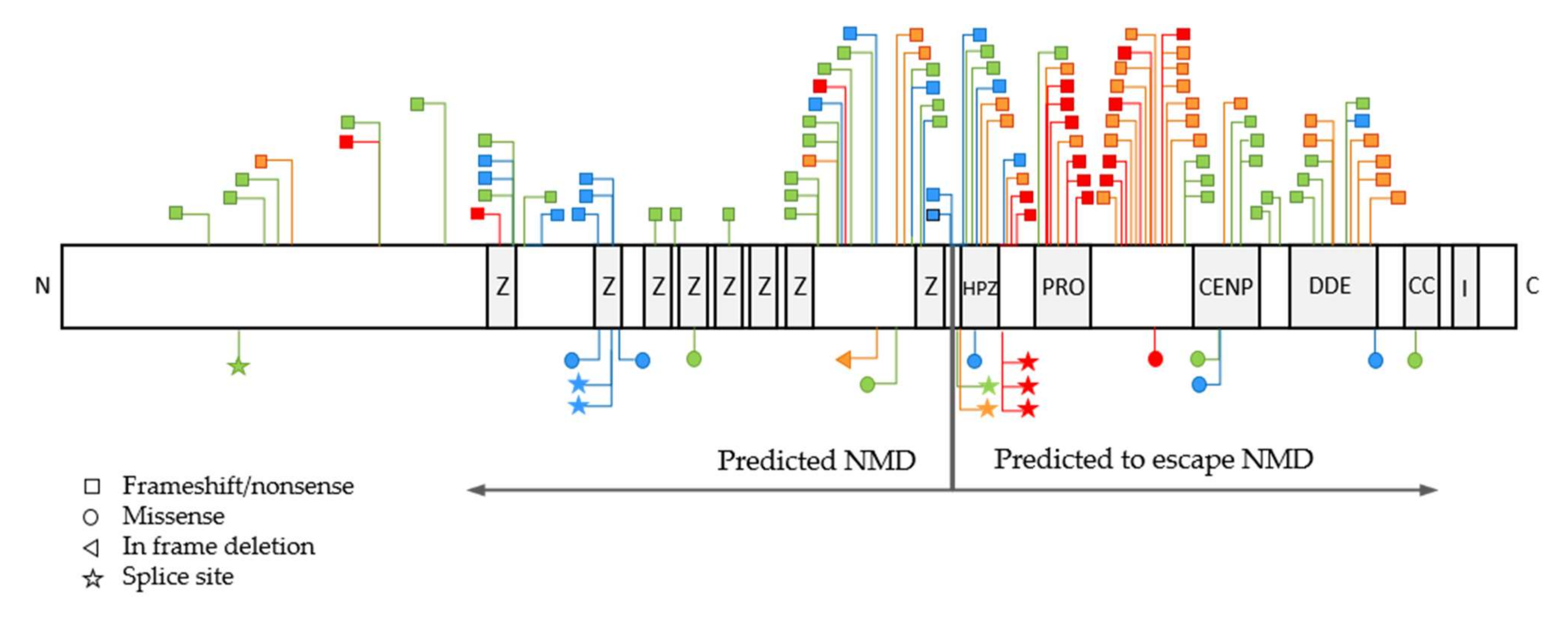
3.1. Variant Types and Their Distribution
3.2. Association between Disease Severity, Variant Types and Nonsense-Mediated RNA Decay
3.3. Association between Disease Severity and POGZ Domains and Larger Gene Regions
3.4. Frequency of POGZ-Related Symptoms
3.5. Facial Gestalt Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nozawa, R.-S.; Nagao, K.; Masuda, H.-T.; Iwasaki, O.; Hirota, T.; Nozaki, N.; Kimura, H.; Obuse, C. Human POGZ modulates dissociation of HP1α from mitotic chromosome arms through Aurora B activation. Nat. Cell Biol. 2010, 12, 719–727. [Google Scholar] [CrossRef]
- Matsumura, K.; Seiriki, K.; Okada, S.; Nagase, M.; Ayabe, S.; Yamada, I.; Furuse, T.; Shibuya, H.; Yasuda, Y.; Yamamori, H.; et al. Pathogenic POGZ mutation causes impaired cortical development and reversible autism-like phenotypes. Nat. Commun. 2020, 11, 859. [Google Scholar] [CrossRef] [Green Version]
- Markenscoff-Papadimitriou, E.; Binyameen, F.; Whalen, S.; Price, J.; Lim, K.; Ypsilanti, A.R.; Catta-Preta, R.; Pai, E.L.-L.; Mu, X.; Xu, D.; et al. Autism risk gene POGZ promotes chromatin accessibility and expression of clustered synaptic genes. Cell Rep. 2021, 37, 110089. [Google Scholar] [CrossRef]
- White, J.; Beck, C.R.; Harel, T.; Posey, J.E.; Jhangiani, S.N.; Tang, S.; Farwell, K.D.; Powis, Z.; Mendelsohn, N.J.; Baker, J.A.; et al. POGZ truncating alleles cause syndromic intellectual disability. Genome Med. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batzir, N.A.; Posey, J.E.; Song, X.; Akdemir, Z.C.; Rosenfeld, J.A.; Brown, C.W.; Chen, E.; Holtrop, S.G.; Mizerik, E.; Moreno, M.N.; et al. Phenotypic expansion of POGZ—Related intellectual disability syndrome (White-Sutton syndrome). Am. J. Med. Genet. Part A 2020, 182, 38–52. [Google Scholar] [CrossRef]
- Stessman, H.A.; Willemsen, M.H.; Fenckova, M.; Penn, O.; Hoischen, A.; Xiong, B.; Wang, T.; Hoekzema, K.; Vives, L.; Vogel, I.; et al. Disruption of POGZ Is Associated with Intellectual Disability and Autism Spectrum Disorders. Am. J. Hum. Genet. 2016, 98, 541–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokkema, I.; Taschner, P.E.M.; Schaafsma, G.; Celli, J.; Laros, J.F.; Dunnen, J.T.D. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443, Corrigendum in Nature 2021, 590, E53. [Google Scholar] [CrossRef] [PubMed]
- Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; Punetha, J.; et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Fukai, R.; Hiraki, Y.; Yofune, H.; Tsurusaki, Y.; Nakashima, M.; Saitsu, H.; Tanaka, F.; Miyake, N.; Matsumoto, N. A case of autism spectrum disorder arising from a de novo missense mutation in POGZ. J. Hum. Genet. 2015, 60, 277–279. [Google Scholar] [CrossRef]
- Tan, B.; Zou, Y.; Zhang, Y.; Zhang, R.; Ou, J.; Shen, Y.; Zhao, J.; Luo, X.; Guo, J.; Zeng, L.; et al. A novel de novo POGZ mutation in a patient with intellectual disability. J. Hum. Genet. 2016, 61, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Dentici, M.L.; Niceta, M.; Pantaleoni, F.; Barresi, S.; Bencivenga, P.; Dallapiccola, B.; Digilio, M.C.; Tartaglia, M. Expanding the phenotypic spectrum of truncating POGZ mutations: Association with CNS malformations, skeletal abnormalities, and distinctive facial dysmorphism. Am. J. Med. Genet. Part A 2017, 173, 1965–1969. [Google Scholar] [CrossRef]
- Zhao, W.; Quan, Y.; Wu, H.; Han, L.; Bai, T.; Ma, L.; Li, B.; Xun, G.; Ou, J.; Zhao, J.; et al. POGZ de novo missense variants in neuropsychiatric disorders. Mol. Genet. Genom. Med. 2019, 7, e900. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Ramakrishnaiah, R.; Schaefer, B. The neurological aspects related to POGZ mutation: Case report and review of CNS malformations and epilepsy. Acta Neurol. Belg. 2019, 120, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, A.; Barresi, S.; Trivisano, M.; Ciolfi, A.; Dentici, M.L.; Radio, F.C.; Vigevano, F.; Tartaglia, M.; Specchio, N. POGZ-related epilepsy: Case report and review of the literature. Am. J. Med. Genet. Part A 2019, 179, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Pascolini, G.; Agolini, E.; Fleischer, N.; Gulotta, E.; Cesario, C.; D’Elia, G.; Novelli, A.; Majore, S.; Grammatico, P. A novel patient with White–Sutton syndrome refines the mutational and clinical repertoire of the POGZ—Related phenotype and suggests further observations. Am. J. Med. Genet. Part A 2020, 182, 1791–1795. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.S.; Jackson, V.E.; Scerri, T.S.; van Reyk, O.; Coleman, M.; Braden, R.O.; Turner, S.; Rigbye, K.A.; Boys, A.; Barton, S.; et al. Severe childhood speech disorder: Gene discovery highlights transcriptional dysregulation. Neurology 2020, 94, e2148–e2167. [Google Scholar] [CrossRef]
- Liu, S.; Yan, Z.; Huang, Y.; Zheng, W.; Deng, Y.; Zou, Y.; Xie, H. A case of White–Sutton syndrome arising from a maternally-inherited mutation in POGZ. Psychiatr. Genet. 2021, 31, 135–139. [Google Scholar] [CrossRef]
- Trimarchi, G.; Caraffi, S.; Radio, F.; Barresi, S.; Contrò, G.; Pizzi, S.; Maini, I.; Pollazzon, M.; Fusco, C.; Sassi, S.; et al. Adducted Thumb and Peripheral Polyneuropathy: Diagnostic Supports in Suspecting White–Sutton Syndrome: Case Report and Review of the Literature. Genes 2021, 12, 950. [Google Scholar] [CrossRef]
- Donnarumma, B.; Riccio, M.P.; Terrone, G.; Palma, M.; Strisciuglio, P.; Scala, I. Expanding the neurological and behavioral phenotype of White-Sutton syndrome: A case report. Ital. J. Pediatr. 2021, 47, 148. [Google Scholar] [CrossRef]
- Garde, A.; Cornaton, J.; Sorlin, A.; Moutton, S.; Nicolas, C.; Juif, C.; Geneviève, D.; Perrin, L.; Khau-Van-Kien, P.; Smol, T.; et al. Neuropsychological study in 19 French patients with White-Sutton syndrome and POGZ mutations. Clin. Genet. 2021, 99, 407–417. [Google Scholar] [CrossRef]
- Dal, S.; Hopper, B.; du Chattel, M.V.R.; Goel, H. A case of White–Sutton syndrome with previously described loss-of-function variant in DDE domain of POGZ (p.Arg1211*) and Kartagener syndrome. Am. J. Med. Genet. Part A 2021, 185, 1006–1007. [Google Scholar] [CrossRef]
- Ye, Y.; Cho, M.T.; Retterer, K.; Alexander, N.; Ben-Omran, T.; Al-Mureikhi, M.; Cristian, I.; Wheeler, P.G.; Crain, C.; Zand, D.; et al. De novo POGZ mutations are associated with neurodevelopmental disorders and microcephaly. Mol. Case Stud. 2015, 1, a000455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316. [Google Scholar] [CrossRef]
- Wright, C.M.; Guter, S.J.; Cook, E.H. Case Report: Association of Comorbid Psychiatric Disorders and Sigmoid Prolapse with de novo POGZ Mutation. J. Autism Dev. Disord. 2021, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Murch, O.; Jain, V.; Benneche, A.; Metcalfe, K.; Hobson, E.; Prescott, K.; Chandler, K.; Ghali, N.; Carmichael, J.; Foulds, N.C.; et al. Further delineation of the clinical spectrum of White–Sutton syndrome: 12 new individuals and a review of the literature. Eur. J. Hum. Genet. 2021, 30, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.M.; Kamphausen, S.B.; Zenker, M.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Assia Batzir, N.; White, J.; Sutton, V.R. White-Sutton Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK573972/ (accessed on 19 December 2021).
- Zhao, W.; Tan, J.; Zhu, T.; Ou, J.; Li, Y.; Shen, L.; Wu, H.; Han, L.; Liu, Y.; Jia, X.; et al. Rare inherited missense variants of POGZ associate with autism risk and disrupt neuronal development. J. Genet. Genom. 2019, 46, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Nakazawa, T.; Nagayasu, K.; Gotoda-Nishimura, N.; Kasai, A.; Hayata-Takano, A.; Shintani, N.; Yamamori, H.; Yasuda, Y.; Hashimoto, R.; et al. De novo POGZ mutations in sporadic autism disrupt the DNA-binding activity of POGZ. J. Mol. Psychiatry 2016, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Chylack, L.T.; Fu, L.; Mancini, R.; Martin-Rehrmann, M.D.; Saunders, A.J.; Konopka, G.; Tian, D.; Hedley-Whyte, E.T.; Folkerth, R.D.; Goldstein, L.E. Lens epithelium-derived growth factor (LEDGF/p75) expression in fetal and adult human brain. Exp. Eye Res. 2004, 79, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeeusen, K.; Christ, F.; Hendrix, J.; Rain, J.-C.; Emiliani, S.; Benarous, R.; Debyser, Z.; Gijsbers, R.; de Rijck, J. Lens Epithelium-derived Growth Factor/p75 Interacts with the Transposase-derived DDE Domain of PogZ. J. Biol. Chem. 2009, 284, 11467–11477. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. DYSMORPHIC FACIAL FEATURES | CLINICAL SCORES |
|---|---|
| Broad/high forehead/bitemporal narrowing | Scoring from 1 to 4 (mild-to-severe facial dysmorphism)
|
| Hypertelorism | |
| Downslanting or upslanting palpebral fissures | |
| Epicanthus | |
| Ptosis | |
| High-arched/sparse eyebrows | |
| Broad nasal tip | |
| Depressed, flat nasal bridge | |
| Pear-shaped nose | |
| Midface hypoplasia/retrusion | |
| Short philtrum | |
| Downturned corners of mouth (triangular/tented) | |
| Upper lip (cupid’s bow) | |
| Thin vermillion/thin upper lip | |
| Everted upper/lower lip | |
| Open mouth | |
| Macrostomia | |
| Protrusion of the tongue/macroglossia | |
| High-arched palate | |
| Bifid uvula | |
| Mandibula (prognathia or micro-retrognathia) | |
| Pointed chin | |
| Low-set ears | |
| Posteriorly rotated ears | |
| Over-folded/abnormally folded helices | |
| 2. ABNORMALITY OF THE EYE | |
| Strabismus | 1 scoring point/symptom |
| Myopia | |
| Hypermetropia | |
| Anisometropia | |
| Astigmatism | |
| Iris coloboma | |
| Optic nerve atrophy or hypoplasia | |
| Rod-cone dystrophy | |
| Cortical visual impairment | |
| Abnormal electroretinogram | |
| Abnormal visual evoked potentials | |
| 3. ABNORMALITY OF THE NERVOUS SYSTEM | |
| Global developmental delay | Developmental delay and intellectual disability were scored from score 1 (+, mild) to score 4 (++++, severe) based on the MEAN of the scoring points given for gross motor (A), speech delay (B) and IQ-level (C) (Details seen in Supplementary Table S1):
|
| Gross motor developmental delay | |
| Age at walking | |
| Fine motor developmental delay | |
| Speech delay/No speech | |
| Age at talking | |
| Receptive language disorder | |
| Expressive language disorder | |
| Intellectual disability (IQ, if applicable) | |
| Aplasia/hypoplasia of the corpus callosum | Additional scores:
No score for antiepileptics |
| Cerebral atrophy | |
| Polymicrogyria/simplified gyral pattern | |
| Brainstem hypoplasia | |
| Cerebellar dysplasia/hypoplasia | |
| Periventricular white matter lesion | |
| Delayed myelination | |
| Optic chiasma dysplasia | |
| Dandy-Walker malformation/variant | |
| Ventriculomegaly | |
| Other central nervous system (CNS) abnormality | |
| Sensorineural hearing loss (bilateral/unilateral) | |
| Seizures | |
| EEG abnormality | |
| Hypoglycemic seizures | |
| Febrile seizures | |
| Antiepileptics (mono therapy/combined) | |
| 4. BEHAVIORAL ABNORMALITIES | |
| Autism spectrum disorder | 1 scoring point/behavioral abnormality |
| (Self-)injurious behavior | |
| Anxiety | |
| Attention deficit hyperactivity disorder | |
| Limited social interactions | |
| Low frustration tolerance (tantrums) | |
| 5. ABNORMALITY OF THE MUSCULATURE | |
| Hypotonia (facial, axial, appendicular, generalized, others) | 0 scoring point: no hypotonia/not reported 1 scoring point: if any type of hypotonia was reported |
| 6. NORMALITY OF THE CARDIOVASCULAR SYSTEM | |
| Congenital heart defect | 1 scoring point/cardiovascular defect |
| Atrial septal defect | |
| Persistent ductus arteriosus | |
| 7. ABNORMALITY OF THE SKELETAL SYSTEM | |
| Brachycephaly | 1 scoring point/skeletal abnormality |
| Microcephaly | |
| Plagiocephaly | |
| Head circumference in cm (percentile/-SD) | |
| Cleft palate | |
| Short neck | |
| Brachydactyly/Small hands | |
| Syndactyly | |
| Broad fingers and toes | |
| Clinodactyly | |
| Joint laxity | |
| Scoliosis | |
| Contractures | |
| Short stature | |
| Skeletal anomalies of the lower extremities | |
| 8. ABNORMALITY OF THE DIGESTIVE SYSTEM | |
| Feeding difficulties: dysphagia, swallowing difficulty | 1 scoring point/gastrointestinal abnormality |
| Tube feeding/Gastrostomy tube | |
| Gastroesophageal reflux | |
| Constipation | |
| Cyclic vomiting | |
| Failure to thrive | |
| Overweight/Obesity | |
| Diaphragmatic hernia | |
| Other hernias | |
| Intestinal malrotation, intussusception | |
| Rectal prolapse | |
| 9. PERINATAL MEDICAL HISTORY | |
| Prenatal or postnatal complications and findings (high nuchal translucency, low Apgar scores, microcephaly, etc.) | 0 scoring point: no prenatal/perinatal problem or not reported 1 scoring point: if any type of problem was reported |
| 10. GENITO-URINARY TRACT ABNORMALITY | |
| Duplicated renal collecting system | 1 scoring point/genito-urinary abnormality |
| Ureteropelvic junction obstruction | |
| Renal dysplasia | |
| Cryptorchidism | |
| Hypoplastic scrotum | |
| Hypoplastic testes | |
| Micropenis | |
| Phimosis | |
| Primary amenorrhea | |
| 11. MISCELLANEOUS | |
| Sleep disturbance (obstructive sleep apnea) | 1 scoring point/abnormality |
| Frequent respiratory infections | |
| Recurrent otitis media | |
| Others | |
| CUMULATIVE CLINICAL SCORE: | SUM of the scores given to organ system/category 1–11 |
| SEVERITY SCORES | CUMULATIVE CLINICAL SCORES IN: | ||
|---|---|---|---|
| Our Patients | Published Cases with Detailed Phenotypes | Published Cases with Less Detailed Phenotypes | |
| 1 | 1–10 | <9 | 1–3 |
| 2 | 11–20 | 9–14 | 4–6 |
| 3 | 21–30 | 15–19 | 7–10 |
| 4 | ≥31 | ≥20 | ≥11 |
| Patient ID | Age at Last Follow-Up/Age at the Diagnosis /Gender | Variant in POGZ Gene | ACMG Classification ** and ClinVar Submissions/ Frequency in Gnomad | De Novo | Ethnicity | Cumulative Clinical Scores | Severity Score |
|---|---|---|---|---|---|---|---|
| L01 | 2 ys/11 months/male | c.2873_2874delCA; p.Ala958Valfs*6 | Path (PVS1, PM2, PM6)/0 | de novo | Caucasian | 53 | 4 |
| L02 | 6.5 ys/4 ys /female | c.2763del; p.Thr922Leufs*6 | Path (PVS1, PM2, PM6, PP3)/0 | de novo | Caucasian | 31 | 4 |
| G01 | 35 ys/35 ys /female | c.1522C>T; p.Arg508 * | Path (PVS1, PM2, PP3) ClinVar +/0 | unknown | Caucasian | 9 | 1 |
| G02 | 5 ys/5 ys /male | c.1522C>T; p.Arg508 * | Path (PVS1, PM2, PP3) ClinVar +/0 | maternal | Caucasian | 7 | 1 |
| W01 | 11 ys/9 ys /female | c.2190T>G; p.Tyr730 * | Path (PVS1, PM2, PM6, PP3)/0 | de novo | Caucasian | 16 | 2 |
| S01 | 5 ys/4.2 ys | c.3259C>T; p.Arg1087* | Path (PVS1, PM2, PM6, PP3, PP5) ClinVar + + /0 | de novo | Caucasian | 13 | 2 |
| D01 | 17 ys/17 ys /male | c.2258G>A; p.Cys753Tyr | VUS (PM2, PP3, PM6)/0 | de novo | Caucasian | 11 | 2 |
| R01 | 8 ys/7 ys /male | c.600dupT; p.Gly201Trpfs*114 | Path (PVS1, PM2, PM6, PP3)/0 | de novo | Caucasian | 22 | 3 |
| R02 | 3 ys/2.5 ys /male | c.2103delT; p.Pro701fs*64 | Path (PVS1, PM2, PM6)/0 | de novo | Caucasian | 18 | 2 |
| US01 | 14 ys/14 ys /male | c.1180_1181delAT; p.Met394Valfs*9 | Path (PVS1, PM2, PM6, PP3, PP5) ClinVar +/0 | de novo | Caucasian | 13 | 2 |
| US02 * | 1 month/1 month/female | c.2545G>T; p.Gly849 * | Path (PVS1, PM2, PM6, PP3)/0 | de novo | Caucasian | 3 | 1 |
| US03 | 2 ys 5 months/ 2 months/male | c.3196A>T; p.Lys1066 * | Path (PVS1, PM2, PP3)/0 | unknown | Caucasian | 12 | 2 |
| NL01 | 7 ys/7 ys | Deletion 1q21.3 encompassing the whole POGZ gene | de novo | Caucasian | 16 | 2 |
| BINARY COMPARISON | NO OF CASES | MEAN AUC | AUC SD | P VALUE FOR AUC |
|---|---|---|---|---|
| Healthy vs. Mild | 79 vs. 21 | 0.90 | 0.04 | <0.001 |
| Healthy vs. Severe | 79 vs. 27 | 0.96 | 0.02 | <0.001 |
| Mild vs. Severe | 21 vs. 27 | 0.74 | 0.06 | 0.067 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, D.; Verheyen, S.; Wigby, K.M.; Borovikov, A.; Sharkov, A.; Slegesky, V.; Larson, A.; Fagerberg, C.; Brasch-Andersen, C.; Kibæk, M.; et al. Genotype-Phenotype Comparison in POGZ-Related Neurodevelopmental Disorders by Using Clinical Scoring. Genes 2022, 13, 154. https://doi.org/10.3390/genes13010154
Nagy D, Verheyen S, Wigby KM, Borovikov A, Sharkov A, Slegesky V, Larson A, Fagerberg C, Brasch-Andersen C, Kibæk M, et al. Genotype-Phenotype Comparison in POGZ-Related Neurodevelopmental Disorders by Using Clinical Scoring. Genes. 2022; 13(1):154. https://doi.org/10.3390/genes13010154
Chicago/Turabian StyleNagy, Dóra, Sarah Verheyen, Kristen M. Wigby, Artem Borovikov, Artem Sharkov, Valerie Slegesky, Austin Larson, Christina Fagerberg, Charlotte Brasch-Andersen, Maria Kibæk, and et al. 2022. "Genotype-Phenotype Comparison in POGZ-Related Neurodevelopmental Disorders by Using Clinical Scoring" Genes 13, no. 1: 154. https://doi.org/10.3390/genes13010154