
Establishing the Mutational Spectrum of Hungarian Patients with Familial Hypercholesterolemia

 , and
, and
Abstract
:1. Introduction
1.1. Structure and Function of ApoB
1.2. Structure and Function of LDLR
2. Patients and Methods
2.1. Patients
2.2. Methods
2.2.1. Isolation of Genomic DNA
2.2.2. Sequencing of Genes Related to Familial Hypercholesterolemia
2.2.3. MLPA
2.2.4. Variant Filtering and Interpretation
2.2.5. Breakpoint Determination
3. Results
3.1. APOB Alterations
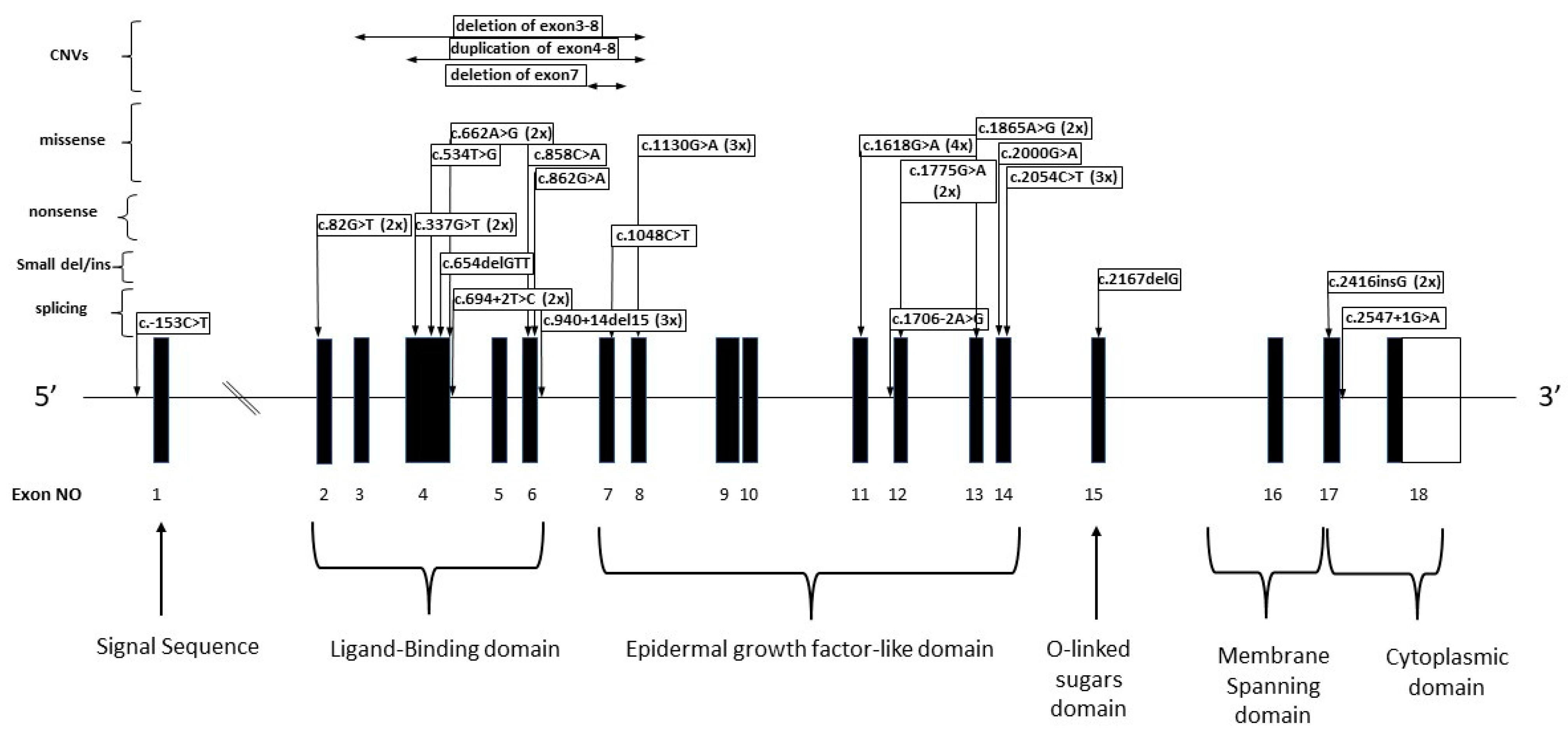
3.2. LDLR Alterations
3.2.1. Patients with Missense LDLR Alterations
3.2.2. Patients with Nonsense Variants
3.2.3. Patients with Splicing Alterations
3.2.4. Small Duplications/Deletions in the LDLR Gene
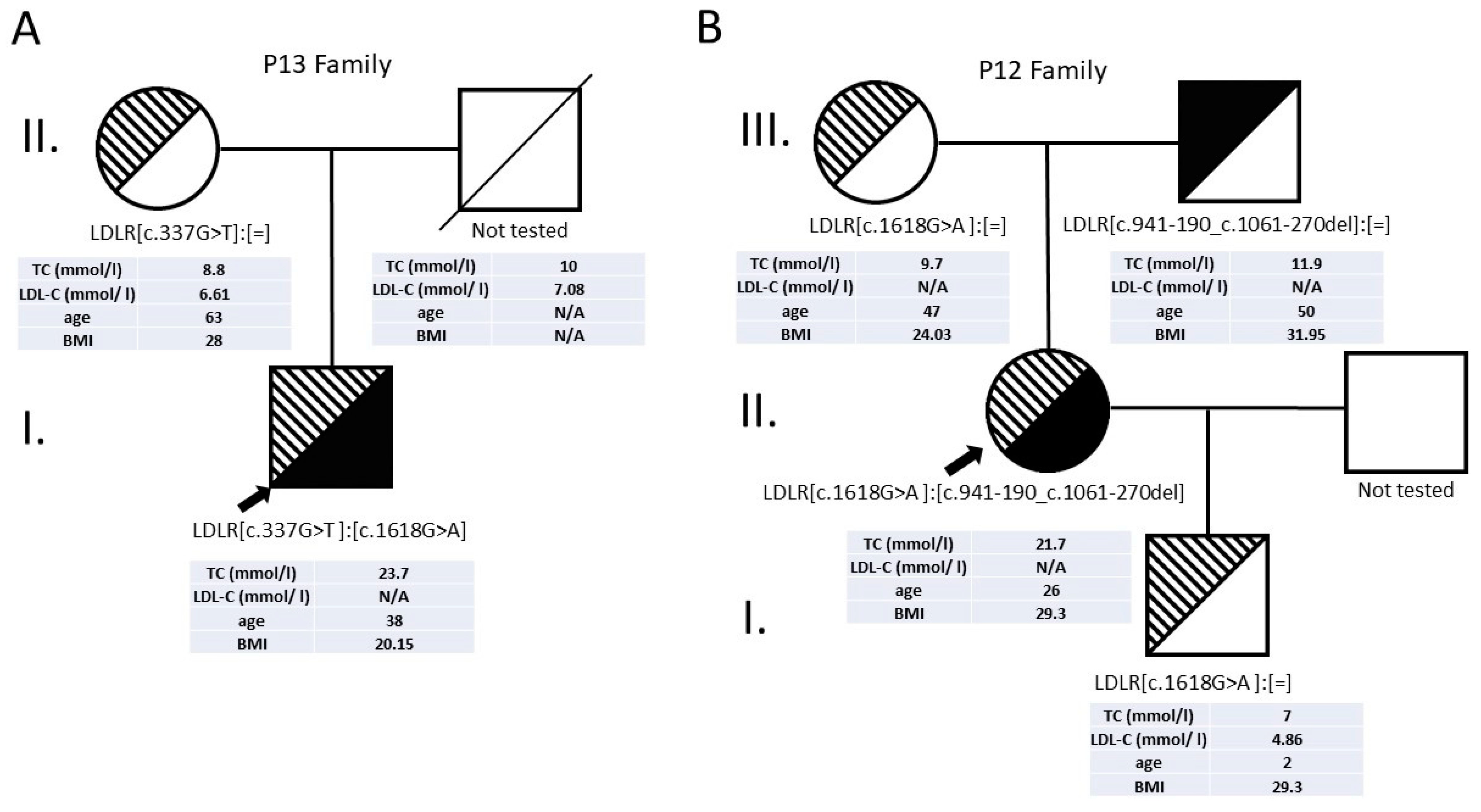
3.2.5. Patients with Compound Heterozygous LDLR Alterations
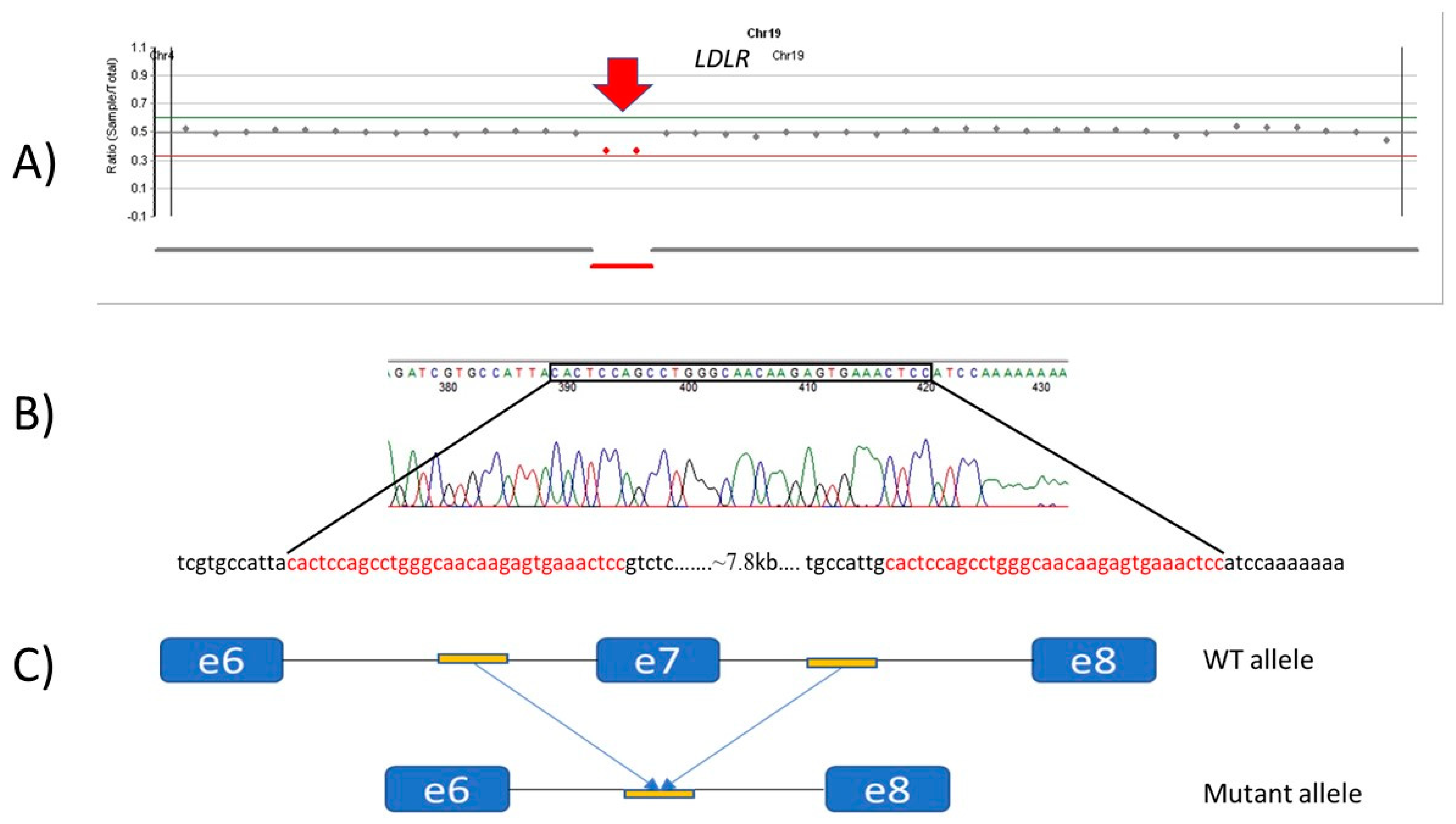
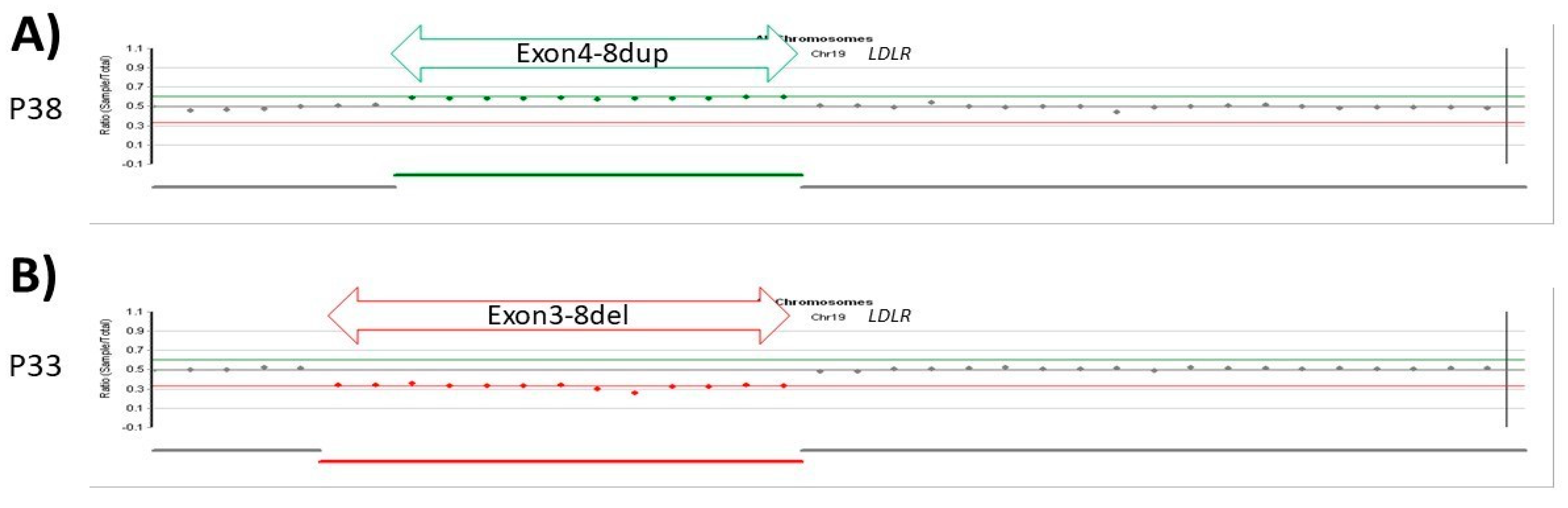
3.2.6. Patients with CNV LDLR Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paragh, G.; Harangi, M.; Karányi, Z.; Daróczy, B.; Németh, Á.; Fülöp, P. Identifying Patients with Familial Hypercholesterolemia Using Data Mining Methods in the Northern Great Plain Region of Hungary. Atherosclerosis 2018, 277, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial Hypercholesterolaemia Is Underdiagnosed and Undertreated in the General Population: Guidance for Clinicians to Prevent Coronary Heart Disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [Green Version]
- Cuchel, M.; Bruckert, E.; Ginsberg, H.N.; Raal, F.J.; Santos, R.D.; Hegele, R.A.; Kuivenhoven, J.A.; Nordestgaard, B.G.; Descamps, O.S.; Steinhagen-Thiessen, E.; et al. Homozygous Familial Hypercholesterolaemia: New Insights and Guidance for Clinicians to Improve Detection and Clinical Management. A Position Paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014, 35, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Gent, J.; Braakman, I. Low-Density Lipoprotein Receptor Structure and Folding. Cell. Mol. Life Sci. 2004, 61, 2461–2470. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Larrea-Sebal, A.; Alonso-Estrada, R.; Aguilo-Arce, J.; Ostolaza, H.; Palacios, L.; Martin, C. Mutation Type Classification and Pathogenicity Assignment of Sixteen Missense Variants Located in the EGF-Precursor Homology Domain of the LDLR. Sci. Rep. 2020, 10, 1727. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Einhorn, Y.; Einhorn, M.; Kamshov, A.; Lev, O.; Trabelsi, A.; Paz-Yaacov, N.; Gross, S.J. Gene-Specific Artificial Intelligence-Based Variant Classification Engine: Results of a Time-Capsule Experiment. 2019. Available online: https://assets.researchsquare.com/files/rs-2650/v1/94f75786-fde0-4719-b91d-91321b7cbac4.pdf?c=1631826043 (accessed on 19 December 2021). [CrossRef] [Green Version]
- Tybjærg-Hansen, A.; Humphries, S.E. Familial Defective Apolipoprotein B-100: A Single Mutation That Causes Hypercholesterolemia and Premature Coronary Artery Disease. Atherosclerosis 1992, 96, 91–107. [Google Scholar] [CrossRef]
- Francová, H.; Trbusek, M.; Zapletalová, P.; Kuhrová, V. New Promoter Mutations in the Low-Density Lipoprotein Receptor Gene Which Induce Familial Hypercholesterolaemia Phenotype: Molecular and Functional Analysis. J. Inherit. Metab. Dis. 2004, 27, 523–528. [Google Scholar] [CrossRef]
- Usifo, E.; Leigh, S.E.A.; Whittall, R.A.; Lench, N.; Taylor, A.; Yeats, C.; Orengo, C.A.; Martin, A.C.R.; Celli, J.; Humphries, S.E. Low-Density Lipoprotein Receptor Gene Familial Hypercholesterolemia Variant Database: Update and Pathological Assessment. Ann. Hum. Genet. 2012, 76, 387–401. [Google Scholar] [CrossRef]
- Hobbs, H.H.; Brown, M.S.; Goldstein, J.L. Molecular Genetics of the LDL Receptor Gene in Familial Hypercholesterolemia. Hum. Mutat. 1992, 1, 445–466. [Google Scholar] [CrossRef]
- Weiss, N.; Binder, G.; Keller, C. Mutations in the Low-Density-Lipoprotein Receptor Gene in German Patients with Familial Hypercholesterolaemia. J. Inherit. Metab. Dis. 2000, 23, 778–790. [Google Scholar] [CrossRef]
- Meiner, V.; Landsberger, D.; Berkman, N.; Reshef, A.; Segal, P.; Seftel, H.C.; van der Westhuyzen, D.R.; Jeenah, M.S.; Coetzee, G.A.; Leitersdorf, E. A Common Lithuanian Mutation Causing Familial Hypercholesterolemia in Ashkenazi Jews. Am. J. Hum. Genet. 1991, 49, 443–449. [Google Scholar]
- Bertolini, S.; Pisciotta, L.; Rabacchi, C.; Cefalù, A.B.; Noto, D.; Fasano, T.; Signori, A.; Fresa, R.; Averna, M.; Calandra, S. Spectrum of Mutations and Phenotypic Expression in Patients with Autosomal Dominant Hypercholesterolemia Identified in Italy. Atherosclerosis 2013, 227, 342–348. [Google Scholar] [CrossRef]
- Gudnason, V.; Sigurdsson, G.; Nissen, H.; Humphries, S.E. Common Founder Mutation in the LDL Receptor Gene Causing Familial Hypercholesterolaemia in the Icelandic Population. Hum. Mutat. 1997, 10, 36–44. [Google Scholar] [CrossRef]
- Maglio, C.; Mancina, R.M.; Motta, B.M.; Stef, M.; Pirazzi, C.; Palacios, L.; Askaryar, N.; Borén, J.; Wiklund, O.; Romeo, S. Genetic Diagnosis of Familial Hypercholesterolaemia by Targeted Next-Generation Sequencing. J. Intern. Med. 2014, 276, 396–403. [Google Scholar] [CrossRef]
- Ekström, U.; Abrahamson, M.; Wallmark, A.; Florén, C.H.; Nilsson-Ehle, P. Mutations in the Low-Density Lipoprotein Receptor Gene in Swedish Familial Hypercholesterolaemia Patients: Clinical Expression and Treatment Response. Eur. J. Clin. Investig. 1998, 28, 740–747. [Google Scholar] [CrossRef]
- Bochmann, H.; Geisel, J.; Herrmann, W.; Purcz, T.; Reuter, W.; Julius, U.; Metzler, W.; Bergmann, S.; Jaross, W.; Gehrisch, S. Eight Novel LDL Receptor Gene Mutations among Patients under LDL Apheresis in Dresden and Leipzig. Hum. Mutat. 2001, 17, 76–77. [Google Scholar] [CrossRef]
- Mehta, R.; Zubirán, R.; Martagón, A.J.; Vazquez-Cárdenas, A.; Segura-Kato, Y.; Tusié-Luna, M.T.; Aguilar-Salinas, C.A. The Panorama of Familial Hypercholesterolemia in Latin America: A Systematic Review. J. Lipid Res. 2016, 57, 2115–2129. [Google Scholar] [CrossRef] [Green Version]
- Miltiadous, G.; Elisaf, M.; Bairaktari, H.; Xenophontos, S.L.; Manoli, P.; Cariolou, M.A. Characterization and Geographic Distribution of the Low Density Lipoprotein Receptor (LDLR) Gene Mutations in Northwestern Greece. Hum. Mutat. 2001, 17, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Tichý, L.; Freiberger, T.; Zapletalová, P.; Soška, V.; Ravčuková, B.; Fajkusová, L. The Molecular Basis of Familial Hypercholesterolemia in the Czech Republic: Spectrum of LDLR Mutations and Genotype-Phenotype Correlations. Atherosclerosis 2012, 223, 401–408. [Google Scholar] [CrossRef]
- Chora, J.R.; Medeiros, A.M.; Alves, A.C.; Bourbon, M. Analysis of Publicly Available LDLR, APOB, and PCSK9 Variants Associated with Familial Hypercholesterolemia: Application of ACMG Guidelines and Implications for Familial Hypercholesterolemia Diagnosis. Genet. Med. 2018, 20, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajmal, M.; Ahmed, W.; Sadeque, A.; Ali, S.H.B.; Bokhari, S.H.; Ahmed, N.; Qamar, R. Identification of a Recurrent Insertion Mutation in the LDLR Gene in a Pakistani Family with Autosomal Dominant Hypercholesterolemia. Mol. Biol. Rep. 2010, 37, 3869–3875. [Google Scholar] [CrossRef]
- Bourbon, M.; Alves, A.C.; Medeiros, A.M.; Silva, S.; Soutar, A.K. Familial Hypercholesterolaemia in Portugal. Atherosclerosis 2008, 196, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebhardt, M.; Schmidt, H.; Doerk, T.; Tietge, U.; Haas, R.; Manns, M.P.; Schmidtke, J.; Stuhrmann, M. Mutation Analysis in 46 German Families with Familial Hypercholesterolemia: Identification of 8 New Mutations. Mutations in Brief No. 226. Online. Hum. Mutat. 1999, 13, 257. [Google Scholar] [CrossRef]
- Chmara, M.; Wasag, B.; Zuk, M.; Kubalska, J.; Wegrzyn, A.; Bednarska-Makaruk, M.; Pronicka, E.; Wehr, H.; Defesche, J.C.; Rynkiewicz, A.; et al. Molecular Characterization of Polish Patients with Familial Hypercholesterolemia: Novel and Recurrent LDLR Mutations. J. Appl. Genet. 2010, 51, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, S.W.; Defesche, J.C.; Umans-Eckenhausen, M.W.; Kastelein, J.P. The Molecular Basis of Familial Hypercholesterolemia in The Netherlands. Hum. Genet. 2001, 109, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Rimbert, A.; Vanhoye, X.; Coulibaly, D.; Marrec, M.; Pichelin, M.; Charrière, S.; Peretti, N.; Valéro, R.; Wargny, M.; Carrié, A.; et al. Phenotypic Differences Between Polygenic and Monogenic Hypobetalipoproteinemia. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e63–e71. [Google Scholar] [CrossRef]
- Sun, X.M.; Webb, J.C.; Gudnason, V.; Humphries, S.; Seed, M.; Thompson, G.R.; Knight, B.L.; Soutar, A.K. Characterization of Deletions in the LDL Receptor Gene in Patients with Familial Hypercholesterolemia in the United Kingdom. Arterioscler. Thromb. J. Vasc. Biol. 1992, 12, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.M.; Patel, D.D.; Knight, B.L.; Soutar, A.K. Influence of Genotype at the Low Density Lipoprotein (LDL) Receptor Gene Locus on the Clinical Phenotype and Response to Lipid-Lowering Drug Therapy in Heterozygous Familial Hypercholesterolaemia. The Familial Hypercholesterolaemia Regression Study Group. Atherosclerosis 1998, 136, 175–185. [Google Scholar] [CrossRef]
- Shawar, S.M.; Al-Drees, M.A.; Ramadan, A.R.; Ali, N.H.; Alfadhli, S.M. The Arabic Allele: A Single Base Pair Substitution Activates a 10-Base Downstream Cryptic Splice Acceptor Site in Exon 12 of LDLR and Severely Decreases LDLR Expression in Two Unrelated Arab Families with Familial Hypercholesterolemia. Atherosclerosis 2012, 220, 429–436. [Google Scholar] [CrossRef]
- Kalina, A.; Császár, A.; Czeizel, A.E.; Romics, L.; Szabóki, F.; Szalai, C.; Reiber, I.; Németh, A.; Stephenson, S.; Williams, R.R. Frequency of the R3500Q Mutation of the Apolipoprotein B-100 Gene in a Sample Screened Clinically for Familial Hypercholesterolemia in Hungary. Atherosclerosis 2001, 154, 247–251. [Google Scholar] [CrossRef]
- Alves, A.C.; Benito-Vicente, A.; Medeiros, A.M.; Reeves, K.; Martin, C.; Bourbon, M. Further Evidence of Novel APOB Mutations as a Cause of Familial Hypercholesterolaemia. Atherosclerosis 2018, 277, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Luirink, I.K.; Wiegman, A.; Kusters, D.M.; Hof, M.H.; Groothoff, J.W.; de Groot, E.; Kastelein, J.J.P.; Hutten, B.A. 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N. Engl. J. Med. 2019, 381, 1547–1556. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Exon | Variant at cDNA Level | Variant at Protein Level | Variant Type | ACMG Classification | TC (mmol/L) | LDL-C (mmol/L) | Age | Gender | BMI | Dutch Score | Xanthoma, Xanthelasma | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 26 | c.8213T>A | p.Ile2738Lys | missense | VUS | 8.1 | 5.72 | 69 | F | 22.5 | 11 | - | this study |
| 4 | 26 | c.10438A>G | p.Lys3480Glu | missense | VUS | 8.0 | 5.3 | 65 | F | 25.5 | 13 | - | this study |
| 30 | 26 | c.10580G>A | p.Arg3527Gln | missense | Likely Pathogenic | 7.66 | 5.51 | 60 | F | 29.9 | 11 | - | [8] |
| 37 | 26 | c.10580G>A | p.Arg3527Gln | missense | Likely Pathogenic | 8.3 | 6.0 | 51 | F | 30.1 | 14 | - | [8] |
| 1 | 26 | c.10580G>A | p.Arg3527Gln | missense | Likely Pathogenic | 6.9 | 4.6 | 78 | F | N/A | 10 | - | [8] |
| 39 | 26 | c.10580G>A | p.Arg3527Gln | missense | Likely Pathogenic | 8.1 | 5.2 | 61 | F | 23.5 | 4 | - | [8] |
| 29 | 29 | c.13242delG | p.Leu4415* | frameshift | VUS/Likely Pathogenic | 5.62 | 7.3 | 51 | M | 26.3 | 16 | - | SCV001357780 |
| Patient ID | Exon | Variant at cDNA Level | Variant at Protein Level | Variant Type | ACMG Classification | TC (mmol/L) | LDL-C (mmol/L) | Age | Gender | BMI | Dutch Score | Xanthoma, xanthelasma | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 43 | promoter | c.-153C>T | p.? | point | VUS/Likely Pathogenic | 9 | 6.13 | 21 | F | 22 | 13 | - | [9] |
| 10 | 2 | c.82G>T | p.Glu28* | nonsense | Pathogenic | 9.07 | 7.18 | 33 | F | 24.6 | 14 | - | [10] |
| 16 | 2 | c.82G>T | p.Glu28* | nonsense | Pathogenic | 13.3 | 10.23 | 53 | F | 34.7 | 18 | - | [10] |
| 35 | 4 | c.337G>T | p.Glu113* | nonsense | Pathogenic | 12.1 | 9.25 | 36 | M | 28.1 | 16 | - | [11] |
| 25 | 4 | c.534T>G | p.Asp178Glu | missense | Pathogenic | 4.2 | 2.4 | 48 | M | 27.8 | 11 | - | [12] |
| 41 | 4 | c.654_656delTGG | p.Gly219del | in-frame deletion | Pathogenic | 13 | n/a | 48 | M | 24.7 | 2 | - | [13] |
| 5 | 4 | c.662A>G | p.Asp221Gly | missense | Pathogenic | 5.6 | 3.8 | 66 | F | 25.2 | 2 | - | [14] |
| 20 | 4 | c.662A>G | p.Asp221Gly | missense | Pathogenic | 12 | 8.81 | 50 | F | 22 | 18 | - | [14] |
| 22 | intron 4 | c.694+2 T>C | p.? | splicing | Pathogenic | 9.4 | 9.4 | 47 | M | 34.2 | 17 | - | [15] |
| 32 | intron 4 | c.694+2 T>C | p.? | splicing | Pathogenic | 9 | 7.4 | 50 | M | 26.3 | 16 | - | [15] |
| 27 | 6 | c.858C>A | p.Ser286Arg | missense | Pathogenic | 9.2 | 6.93 | 34 | F | 22.9 | 13 | - | [14] |
| 8 | 6 | c.940_940+14del | p.? | splicing | Pathogenic | 10.4 | 7.9 | 30 | F | 25.8 | 14 | - | [16] |
| 11 | 6 | c.940_940+14del | p.? | splicing | Pathogenic | 10 | 10.5 | 14 | F | 22.4 | 17 | - | [16] |
| 23 | 6 | c.940_940+14del | p.? | splicing | Pathogenic | 11.4 | 8.7 | 53 | F | 22.2 | 17 | - | [16] |
| 9 | 7 | c.1048C>T | p.Arg350* | nonsense | Pathogenic | 11.9 | 9.75 | 52 | F | n/a | 16 | n/a | [14] |
| 36 | 8 | c.1130G>A | p.Cys377Tyr | missense | Pathogenic | 9.4 | 8.5 | 18 | M | n/a | 14 | - | [17] |
| 19 | 8 | c.1130G>A | p.Cys377Tyr | missense | Pathogenic | 8.4 | 6.2 | 18 | F | 19.6 | 12 | - | [17] |
| 24 | 8 | c.1130G>A | p.Cys377Tyr | missense | Pathogenic | 13.3 | 10.56 | 60 | F | 23 | 18 | - | [17] |
| 21 | 8 | c.1618G>A | p.Ala540Thr | missense | Pathogenic | n/a | n/a | 54 | F | n/a | n/a | n/a | [18] |
| 42 | 8 | c.1618G>A | p.Ala540Thr | missense | Pathogenic | 9.1 | 6.65 | 28 | M | 26.9 | 13 | - | [19] |
| 34 | intron 11 | c.1706-2A>G | p.? | splicing | Likely Pathogenic | 8.2 | 5.8 | 50 | F | 30.5 | 12 | - | this study |
| 26 | 12 | c.1775G>A | p.Gly592Glu | missense | Pathogenic | 10.3 | 7.7 | 45 | F | 19.8 | 13 | - | [20] |
| 31 | 12 | c.1775G>A | p.Gly592Glu | missense | Pathogenic | 10.2 | 7.62 | 53 | F | n/a | 13 | - | [20] |
| 3 | 13 | c.1865A>G | p.Asp622Gly | missense | Pathogenic | 9.6 | 7.3 | 67 | M | 24.3 | 8 | - | [21] |
| 6 | 13 | c.1865A>G | p.Asp622Gly | missense | Pathogenic | 10 | 7.9 | 60 | F | 27.8 | 11 | + | [21] |
| 2 | 14 | c.2000G>A | p.Cys667Tyr | missense | Pathogenic | 12.9 | 10.4 | 64 | F | 30.1 | 10 | - | [14] |
| 17 | 14 | c.2000G>A | p.Cys667Tyr | missense | Pathogenic | 8.1 | 5.4 | 40 | F | n/a | 12 | - | [14] |
| 15 | 14 | c.2054C>T | p.Pro685Leu | missense | Pathogenic | 9.6 | 7.6 | 22 | M | 21.1 | 14 | - | [22] |
| 40 | 14 | c.2054C>T | p.Pro685Leu | missense | Pathogenic | 9.6 | 7.14 | 56 | F | 24.2 | 19 | + | [22] |
| 18 | 17 | c.2416dupG | p.Val806fs*11 | frameshift | Pathogenic | 8.1 | 5.4 | 58 | F | n/a | 12 | - | [23] |
| 44 | 17 | c.2416dupG | p.Val806fs*11 | frameshift | Pathogenic | 10 | 3.1 | 58 | F | 22.6 | 9 | - | [23] |
| 14 | intron 17 | c.2547+1G>A | p.? | splicing | Pathogenic | 11.1 | 8.9 | 66 | M | 24.8 | 18 | - | [24] |
| Patient ID | Exon | Variant at cDNA Level | Variant at Protein Level | Variant Type | ACMG Classification | TC (mmol/L) | LDL-C (mmol/L) | Age | Gender | BMI | Dutch Score | Xanthoma, xanthelasma | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12 | 11 | c.1618G>A | p.Ala540Thr | missense | Pathogenic | 21.7 | n/a | 26 | F | 29.36 | 20 | + | [19] |
| 7 | c.941-190_c.1061-270del | ? | large deletion | Pathogenic | This study | ||||||||
| 13 | 4 | c.337G>T | p.Glu113* | nonsense | Pathogenic | 23.7 | n/a | 38 | M | 20.15 | 20 | + | [11] |
| 11 | c.1618G>A | p.Ala540Thr | missense | Pathogenic | [19] | ||||||||
| 28 | 6 | c.862G>A | p.Glu288Lys | missense | Pathogenic | 10.4 | 10.8 | 13 | M | 29.4 | 17 | - | [25] |
| 15 | c.2167delG | p.Glu723Arg fs*7 | frameshift | Pathogenic | [18] |
| Patient ID | Exon | Variant Type | ACMG Classification | TC (mmol/L) | LDL-C (mmol/L) | Age | Gender | BMI | Dutch Score | Xanthoma, xanthelasma | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 38 | exon 4–8 | duplication | Pathogenic | 16.6 | 9.8 | 69 | F | 25.7 | 24 | + | [26] |
| 33 | exon 3–8 | deletion | Pathogenic | n/a | n/a | 74 | F | 40.4 | 10 | - | [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madar, L.; Juhász, L.; Szűcs, Z.; Kerkovits, L.; Harangi, M.; Balogh, I. Establishing the Mutational Spectrum of Hungarian Patients with Familial Hypercholesterolemia. Genes 2022, 13, 153. https://doi.org/10.3390/genes13010153
Madar L, Juhász L, Szűcs Z, Kerkovits L, Harangi M, Balogh I. Establishing the Mutational Spectrum of Hungarian Patients with Familial Hypercholesterolemia. Genes. 2022; 13(1):153. https://doi.org/10.3390/genes13010153
Chicago/Turabian StyleMadar, László, Lilla Juhász, Zsuzsanna Szűcs, Lóránt Kerkovits, Mariann Harangi, and István Balogh. 2022. "Establishing the Mutational Spectrum of Hungarian Patients with Familial Hypercholesterolemia" Genes 13, no. 1: 153. https://doi.org/10.3390/genes13010153