



Genes 2023, 14(2), 428; https://doi.org/10.3390/genes14020428 - 08 Feb 2023
Cited by 1 | Viewed by 2072
Abstract
►
Show Figures
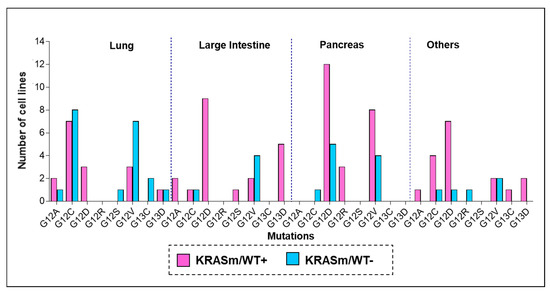
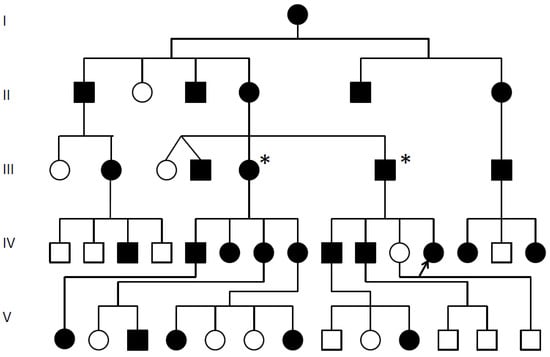
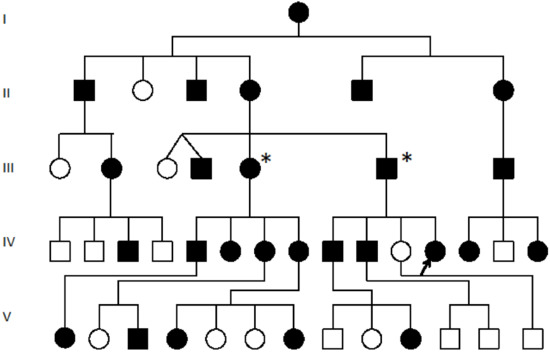
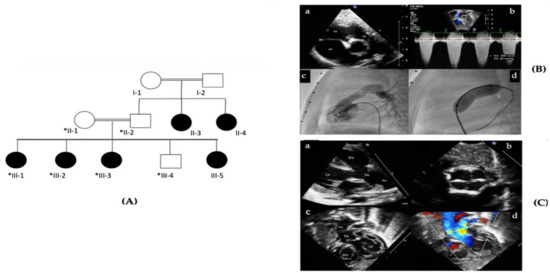
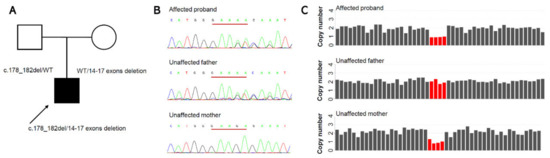
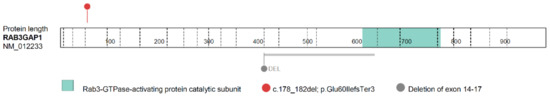
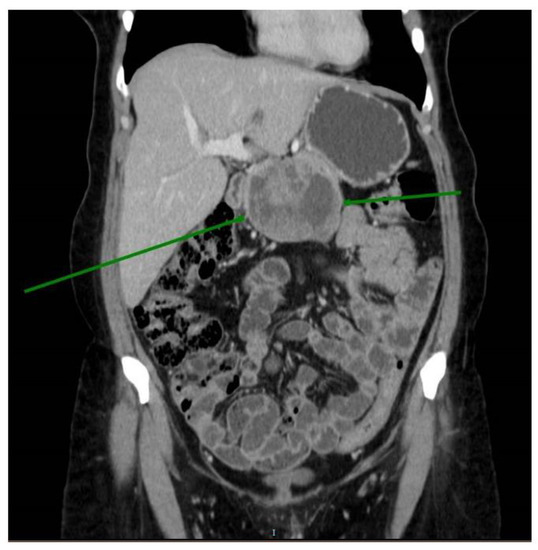
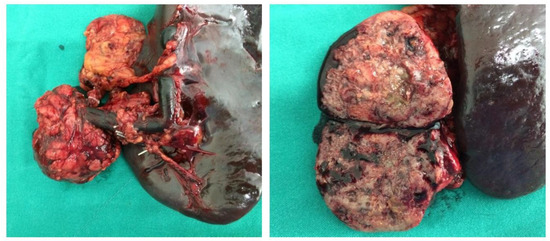
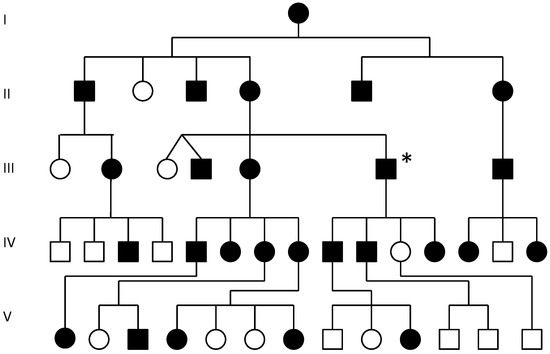
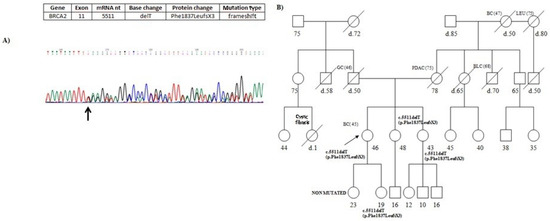
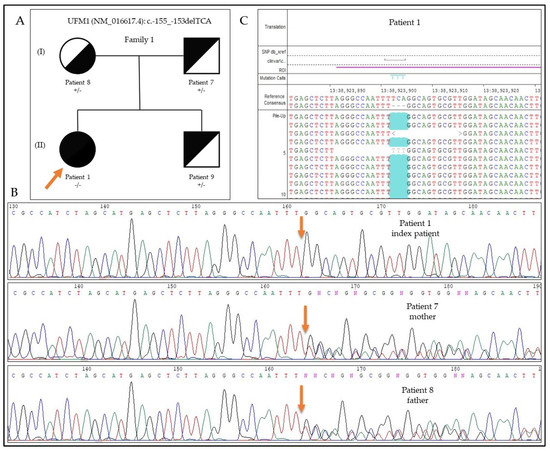
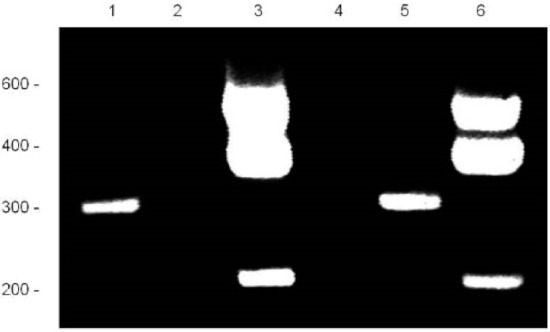
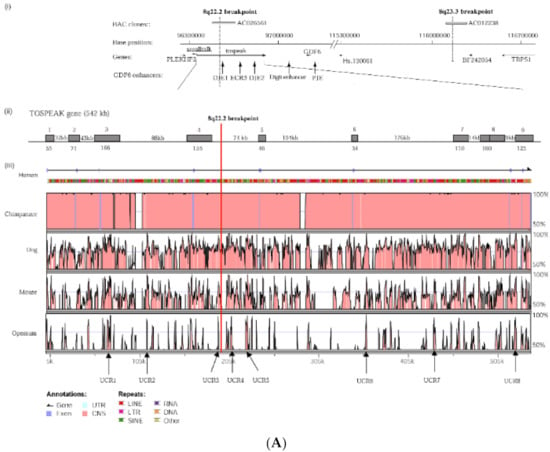
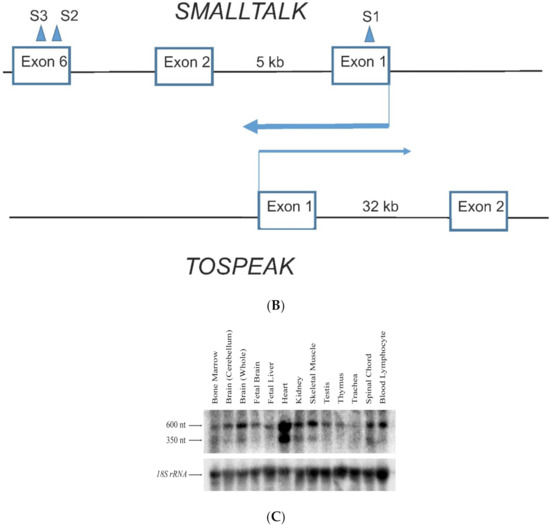
Hereditary cancer syndromes predispose to several types of cancer due to inherited pathogenic variants in susceptibility genes. We describe the case of a 57-year-old woman, diagnosed with breast cancer, and her family. The proband belongs to a family with a suspected tumor syndrome,
[...] Read more.
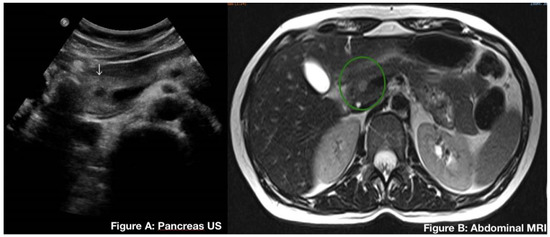
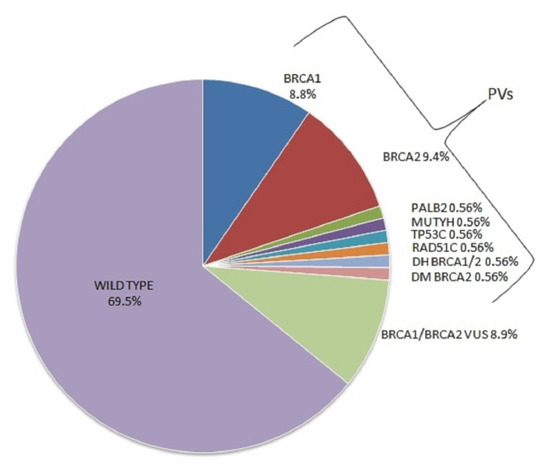
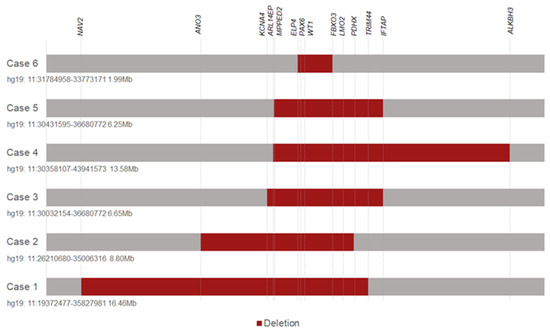
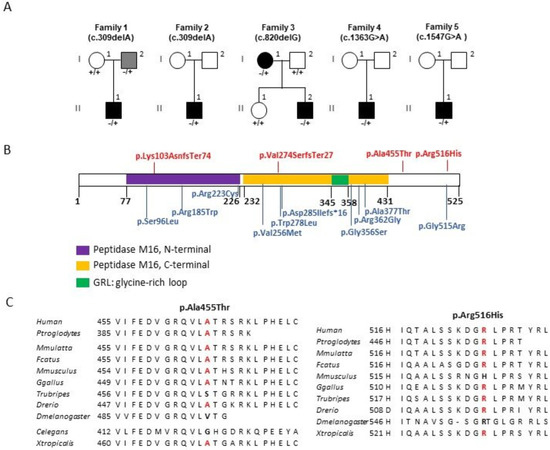
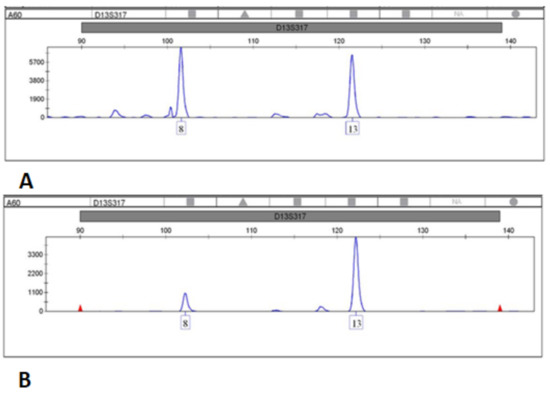
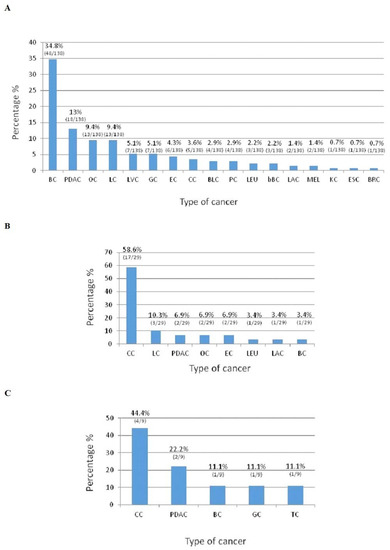
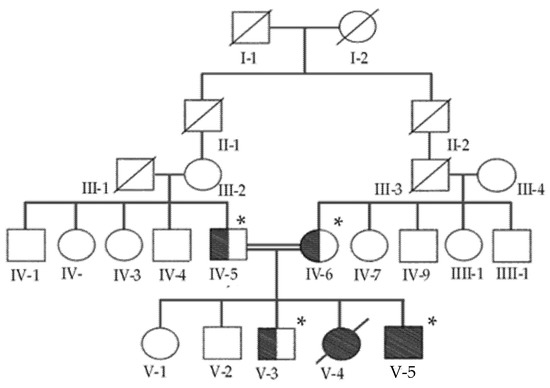
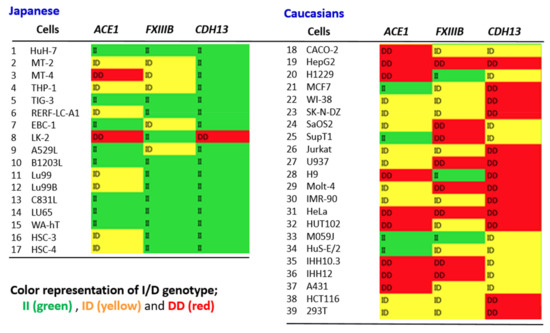
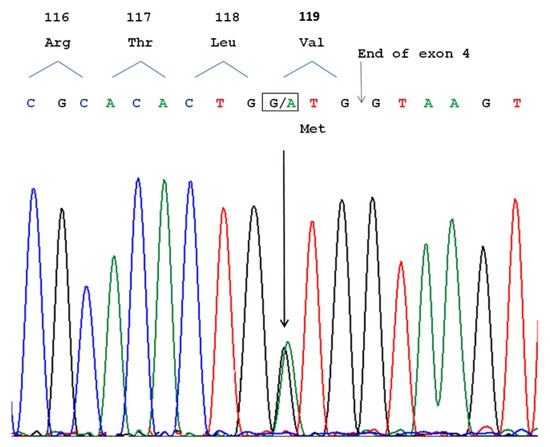
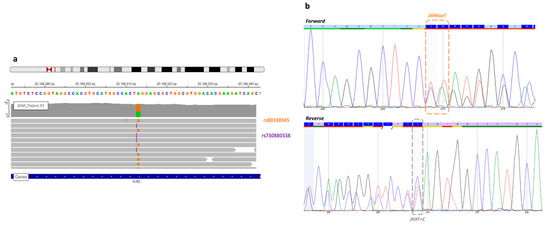
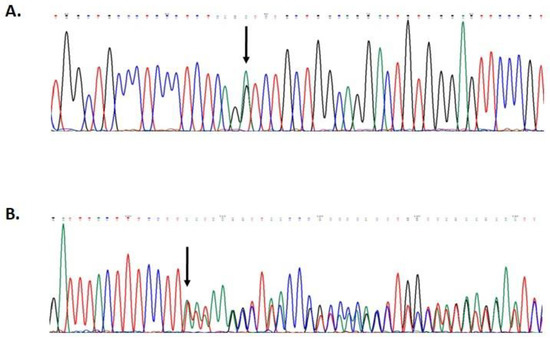
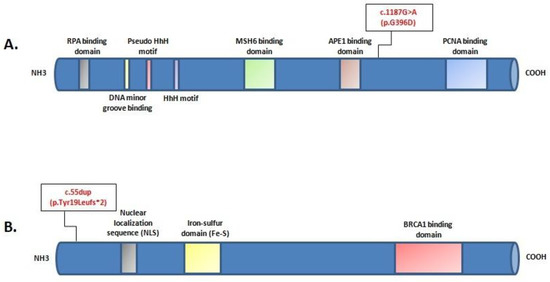
Hereditary cancer syndromes predispose to several types of cancer due to inherited pathogenic variants in susceptibility genes. We describe the case of a 57-year-old woman, diagnosed with breast cancer, and her family. The proband belongs to a family with a suspected tumor syndrome, due to other cancer cases in her family from the paternal and maternal sides. After oncogenetic counseling, she was subjected to mutational analysis with an NGS panel analyzing 27 genes. The genetic analysis showed two monoallelic mutations in low penetrance genes, c.1187G>A (p.G396D) in MUTYH and c.55dup (p.Tyr19Leufs*2) in BRIP1. One of the mutations was inherited from the maternal side and the other from the paternal side, suggesting two different cancer syndrome types in the family. MUTYH mutation was related to the onset of cancers on the paternal side, as confirmed by the occurrence of the same mutation in the proband’s cousin. BRIP1 mutation was found in the proband’s mother, indicating that it was related to the cancer cases observed on the maternal side, including breast cancer and sarcoma. Advances in NGS technologies have allowed the identification of mutations in families with hereditary cancers in genes other than those related to a specific suspected syndrome. A complete oncogenetic counseling, together with molecular tests that enable a simultaneous analysis of multiple genes, is essential for the identification of a correct tumor syndrome and for clinical decision-making in a patient and his/her family. The detection of mutations in multiple susceptibility genes allows the initiation of early risk-reducing measures for identified mutation carriers among family members and to include them in a proper surveillance program for specific syndromes. Moreover, it may enable an adapted treatment for the affected patient, permitting personalized therapeutic options.
Full article
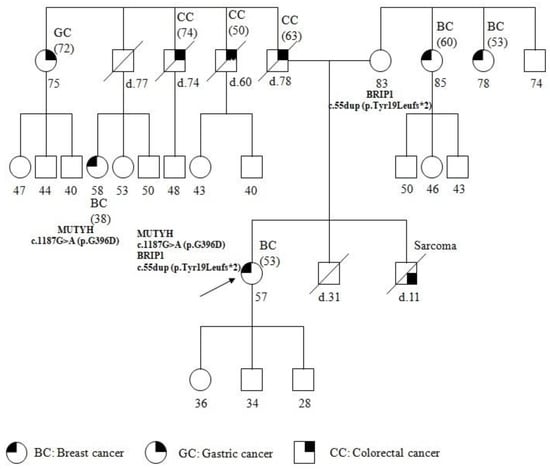
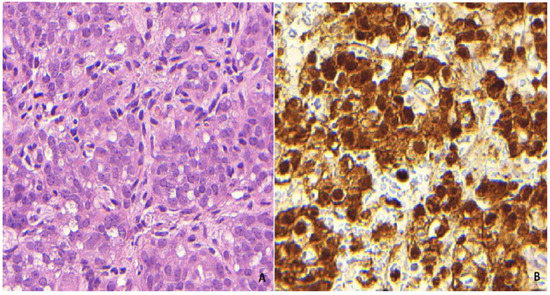
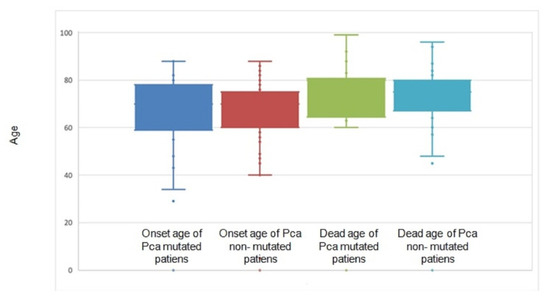
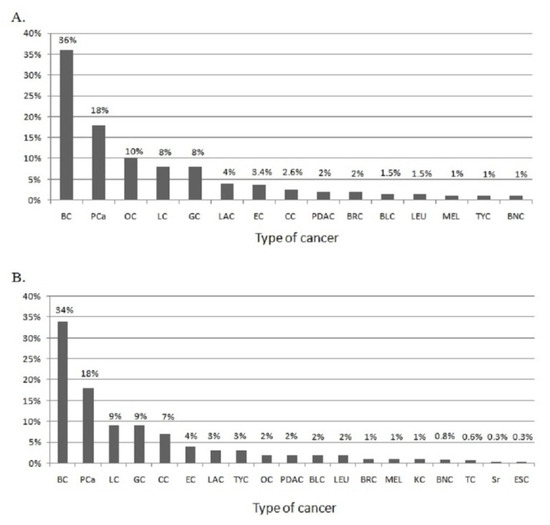
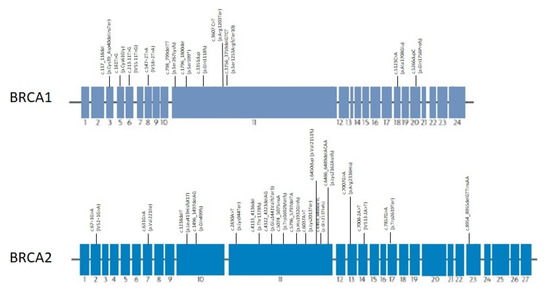
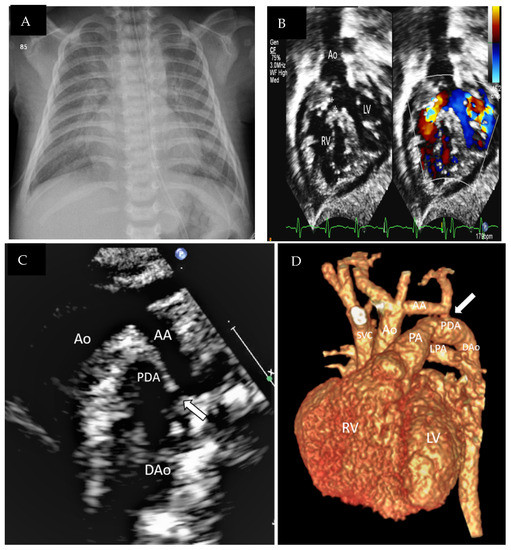
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}