Transcriptome Analysis of Acid-Responsive Genes and Pathways Involved in Polyamine Regulation in Iron Walnut


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Growth under Acidic Soil Conditions
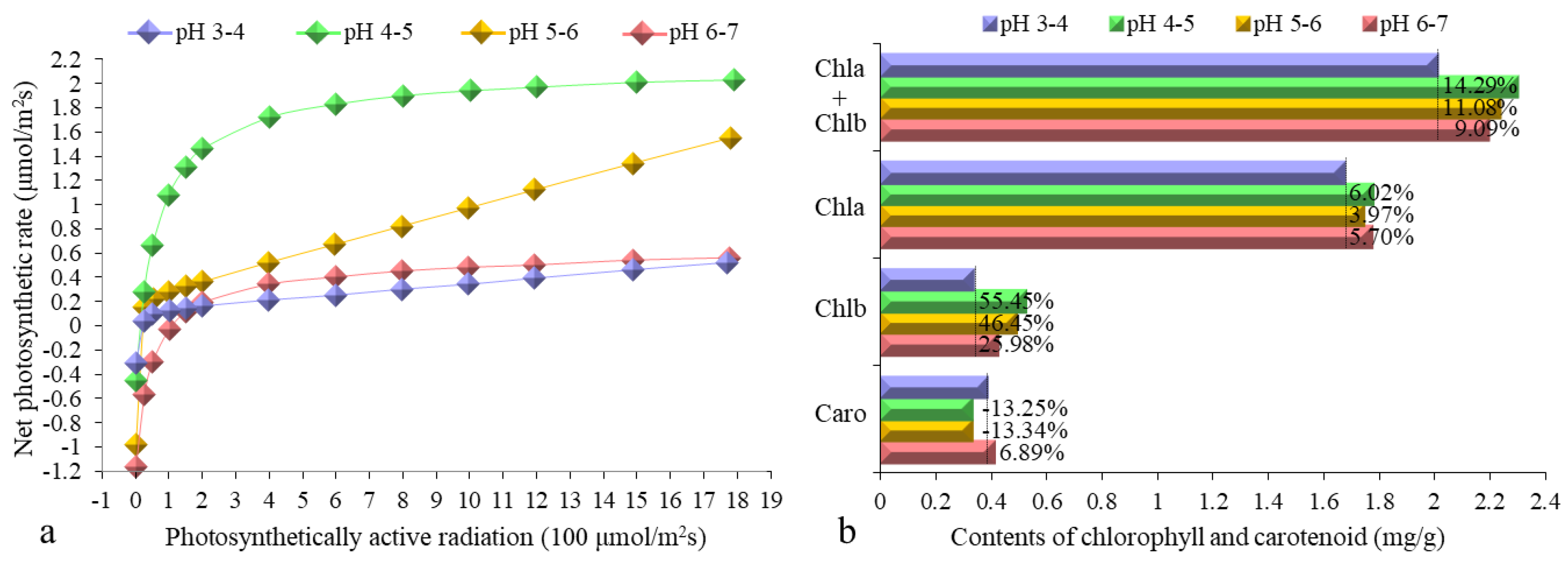
2.2. Contents of Photosynthetic Pigments and Pn-PAR Response Curves
2.3. RNA Isolation and Iron Walnut RNA-Seq
2.4. DEG Identification and Functional Annotation
2.5. Acid-Responsive Gene Identification and Enrichment Analysis
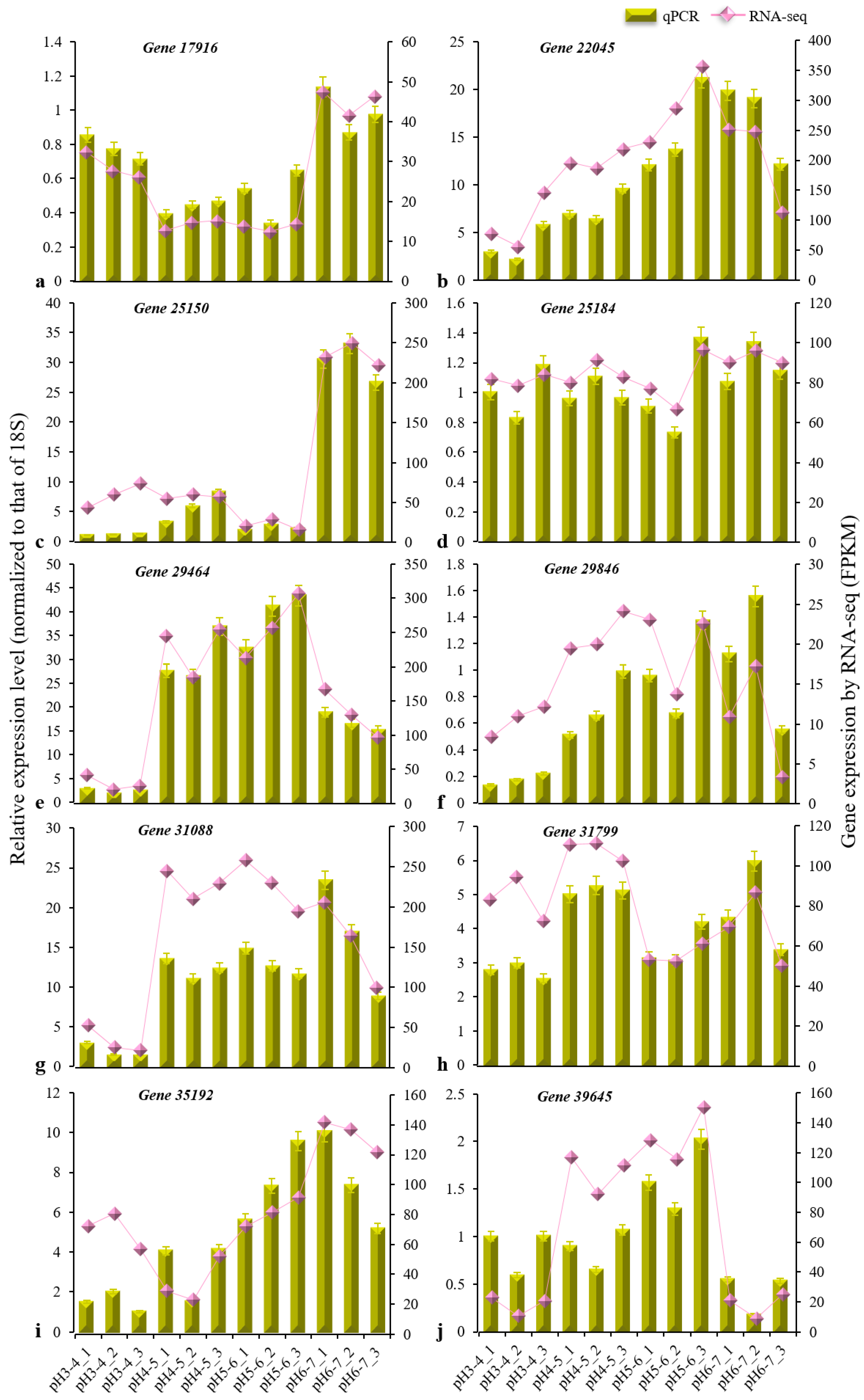
2.6. Quantitative Real-Time PCR (qRT-PCR) Validation
3. Results
3.1. Determination of Pn-PAR Response Curves and Photosynthetic Pigment Contents
3.2. Illumina Sequencing of Different cDNA Libraries
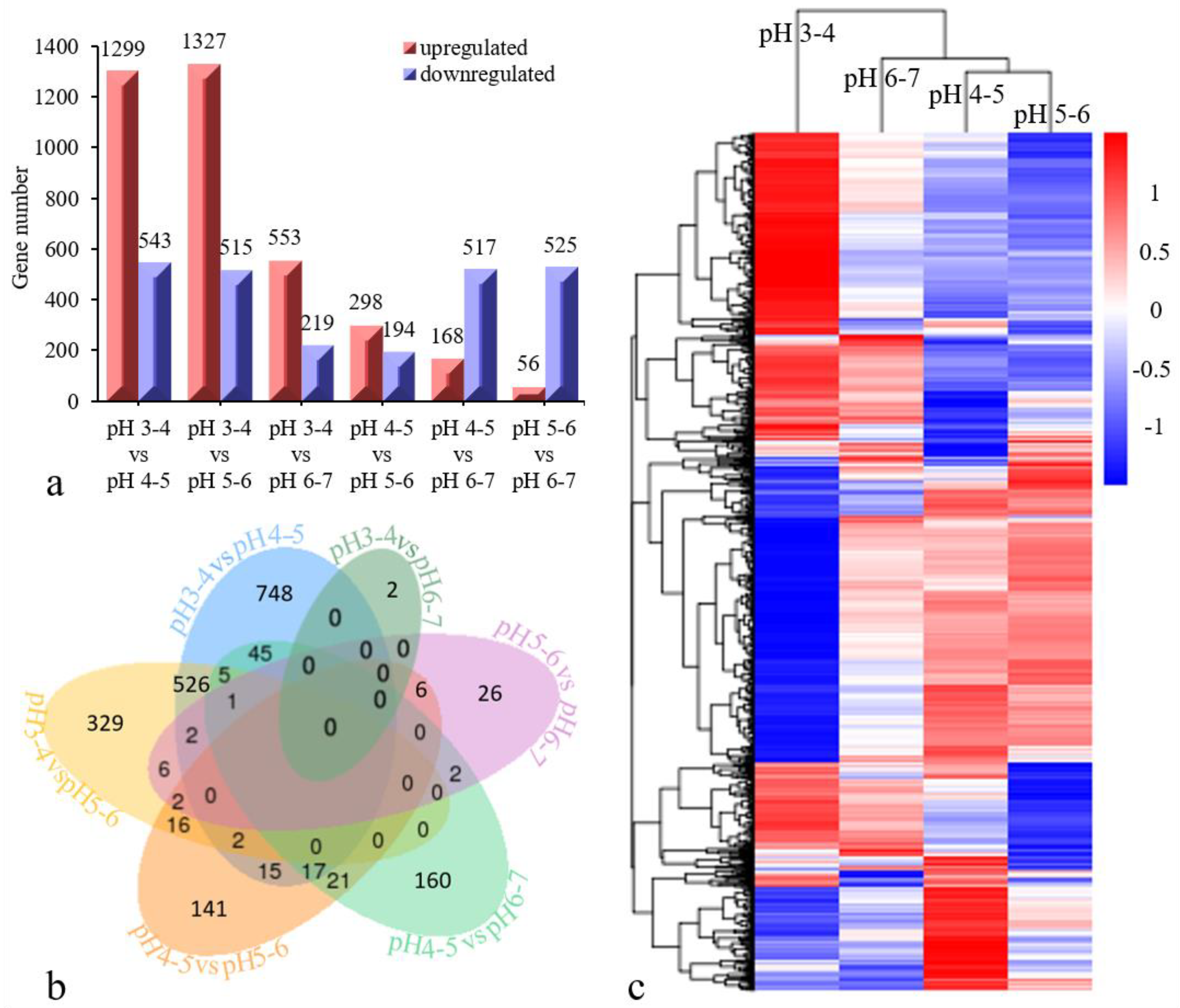
3.3. DEG Analysis and Validation of the Iron Walnut Sequencing Data
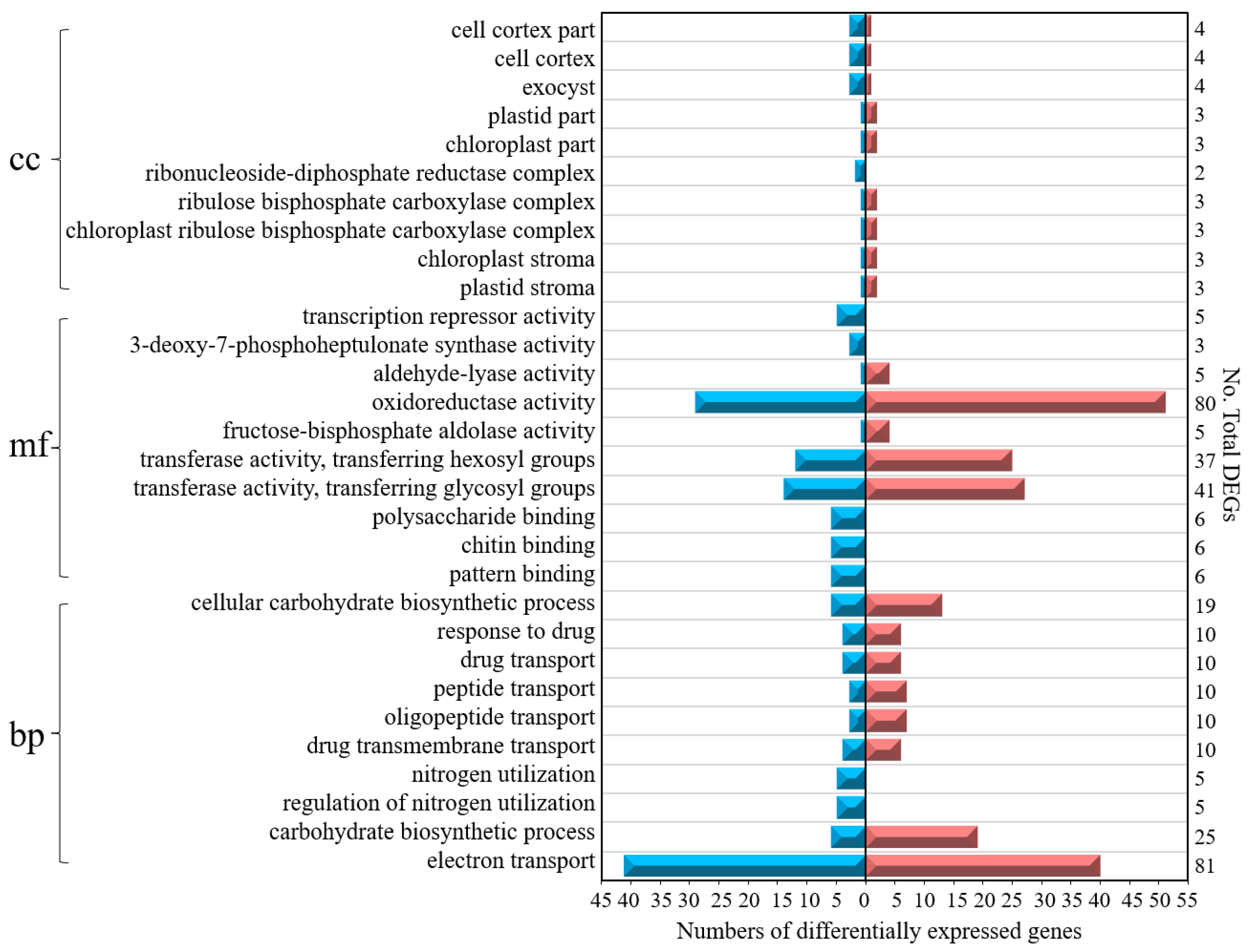
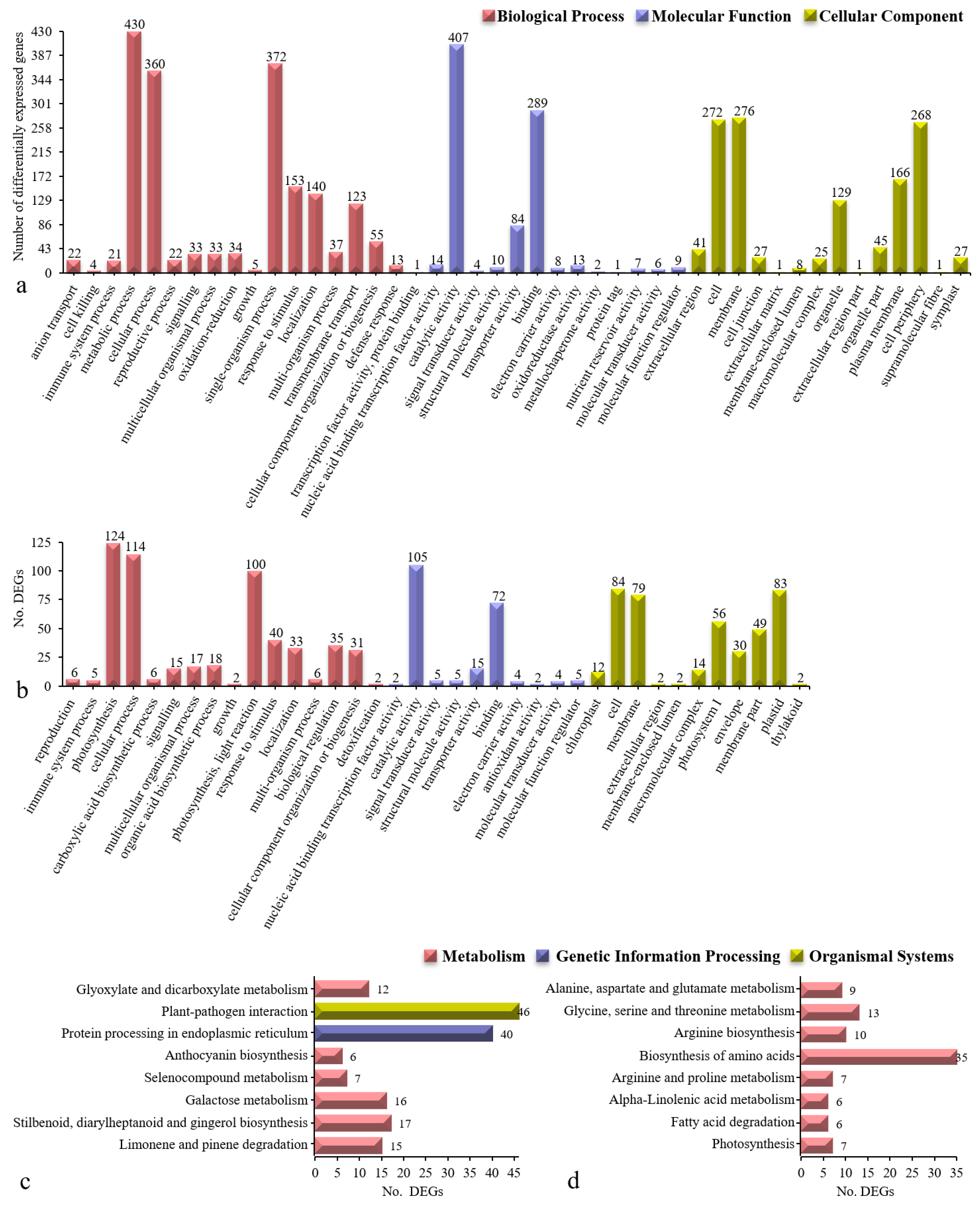
3.4. GO-and KEGG-Annotated DEGs
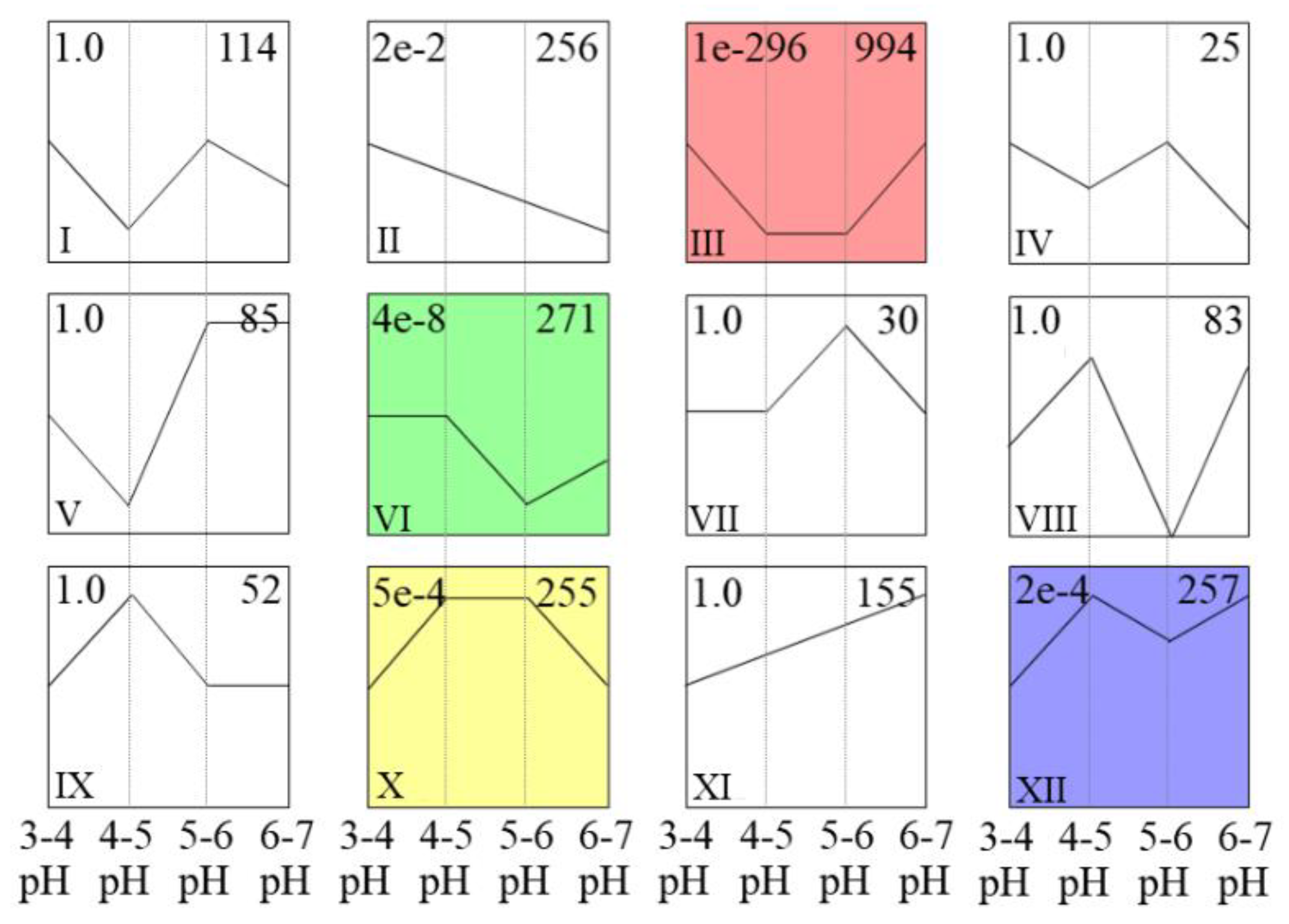
3.5. STEM Cluster Analysis of DEGs and Identification of Acid Resistance-Related Genes
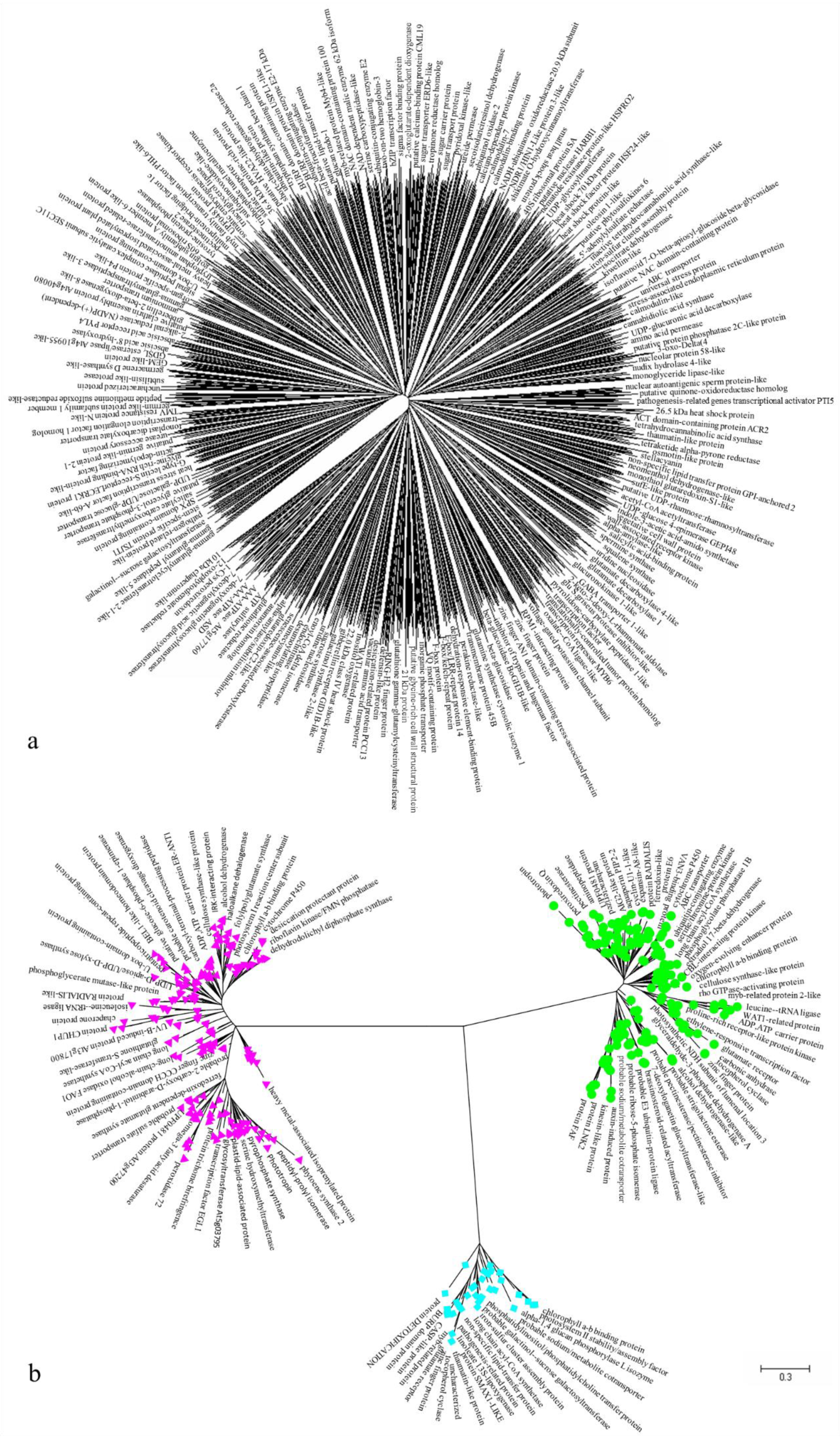
3.6. Molecular Phylogenetic Analysis of Acid Resistance-Related Genes
3.7. Gene Function and Pathway Enrichment Analyses of Acid Resistance-Related Genes
4. Discussion
4.1. Acid Resistance-Related Gene Identification
4.2. Analysis of Acid Resistance-Related Genes
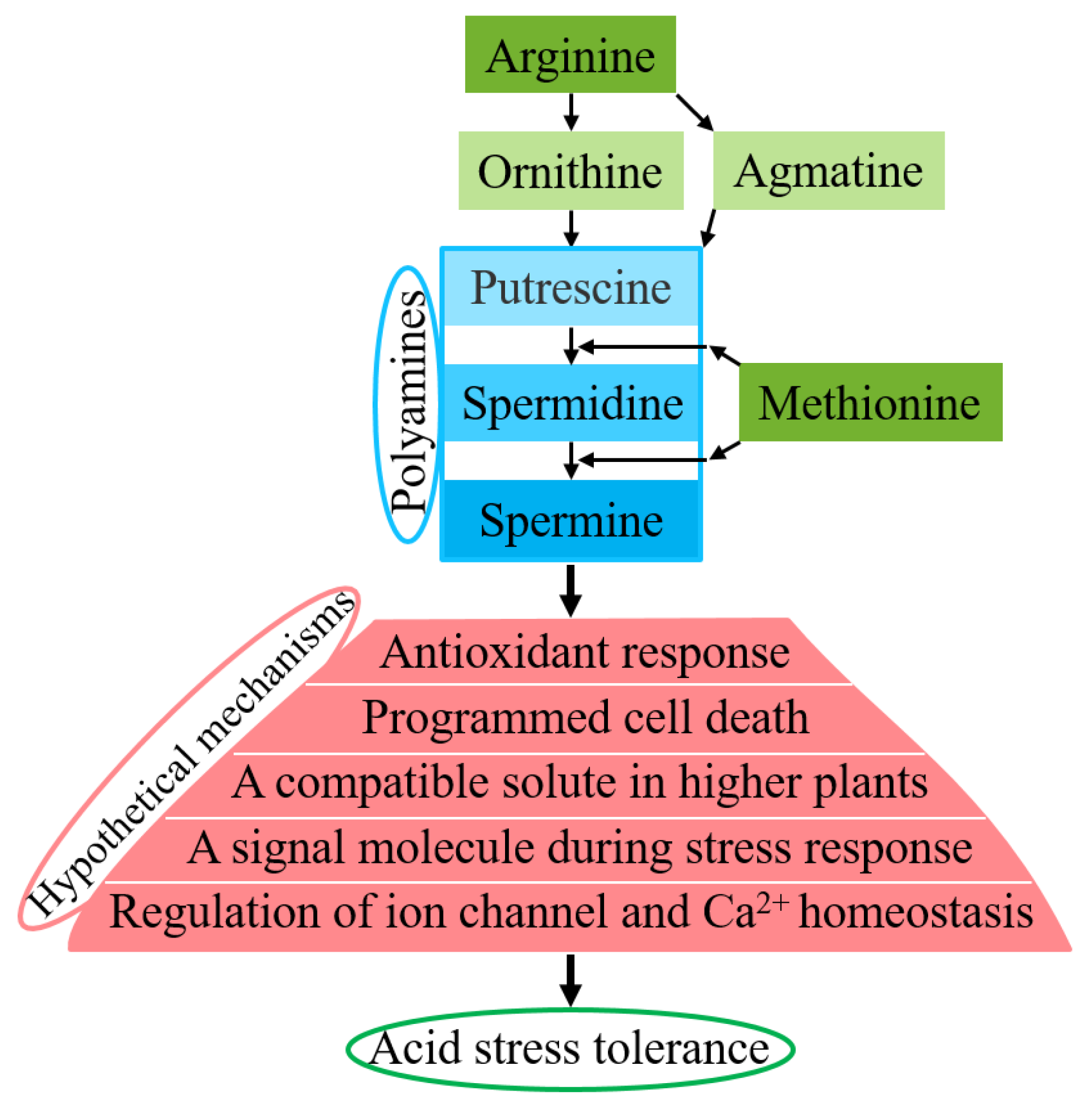
4.3. Hypothetical Regulatory Mechanisms of Acid Resistance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Surhone, L.M.; Tennoe, M.T.; Henssonow, S.F. Juglans Sigillata; Betascript Publishing: Beau Bassin, Mauritius, 2010; pp. 1–68. ISBN 13 978-613-3-34400-6. [Google Scholar]
- Kochian, L.V. Cellular mechanisms of aluminum toxicity and resistance in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1995, 46, 237–260. [Google Scholar] [CrossRef]
- Oberto, J.; Nabti, S.; Jooste, V.; Mignot, H.; Rouviere-Yaniv, J. The HU regulon is composed of genes responding to anaerobiosis, acid stress, high osmolarity and SOS induction. PLoS ONE 2009, 4, e4367. [Google Scholar] [CrossRef] [PubMed]
- Velikova, V.; Yordanov, I.; Edreva, A. Oxidative stress and some antioxidant systems in acid rain-treated bean plants: Protective role of exogenous polyamines. Plant Sci. 2000, 151, 59–66. [Google Scholar] [CrossRef]
- Garcíaquintáns, N.; Magni, C.; Mendoza, D.D.; López, P. The citrate transport system of Lococcuslactis subsp. Lactisbio var. diacetylactis is induced by acid stress. Appl. Environ. Microbiol. 1998, 64, 850–857. [Google Scholar]
- Duary, R.K.; Batish, V.K.; Grover, S. Expression of the atpD gene in probiotic Lactobacillus plantarum strains under in vitro acidic conditions using RT-qPCR. Res. Microbiol. 2010, 161, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Wemekamp Kamphuis, H.H.; Wouters, J.A.; de Leeuw, P.P.; Hain, T.; Chakraborty, T.; Abee, T. Identification of sigma factor sigma b-controlled genes and their impact on acid stress, high hydrostatic pressure, and freeze survival in Listeria monocytogenes egd-e. Appl. Environ. Microbiol. 2004, 70, 3457–3466. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Zheng, S.J.; He, Y.F.; Matsumoto, H. Aluminium resistance requires resistance to acid stress: A case study with spinach that exudes oxalate rapidly when exposed to Al stress. J. Exp. Bot. 2005, 56, 1197–1203. [Google Scholar] [CrossRef]
- Hussain, S.S.; Ali, M.; Ahmad, M.; Siddique, K.H. Polyamines: Natural and engineered abiotic and biotic stress tolerance in plants. Biotechnol. Adv. 2011, 29, 300–311. [Google Scholar] [CrossRef]
- Willekens, H.; Inze, D.; van Montagu, M.; van Camp, W. Catalases in plants. Mol. Breed. 1995, 1, 207–228. [Google Scholar] [CrossRef]
- Gupta, K.; Dey, A.; Gupta, B. Plant polyamines in abiotic stress responses. Acta Physiol. Plant 2013, 35, 2015–2036. [Google Scholar] [CrossRef]
- Kaur-Sawhney, R.; Galston, A.W. Physiological and biochemical studies on the anti-senescence properties of polyamines in plants. In Biochemistry and Physiology of Polyamines in Plants; Slocum, R.D., Floress, H.E., Eds.; CRC Press: Boca Raton, FL, USA, 1991; pp. 201–211. [Google Scholar]
- Berta, G.; Altamura, M.M.; Fusconi, A.; Cerruti, F.; Capitani, F.; Bagni, N. The plant cell wall is altered by inhibition of polyamine biosynthesis. New Phytol. 1997, 137, 569–577. [Google Scholar] [CrossRef]
- Borrell, A.; Carbonell, L.; Farra’s, R.; Puig-Parellada, P.; Tiburcio, A.F. Polyamines inhibit lipid peroxidation in senescing oat leaves. Physiol. Plant. 1997, 99, 385–390. [Google Scholar] [CrossRef]
- Amrani, A.E.; Couée, I.; Berthomé, R.; Ramel, F.; Gouesbet, G.; Sulmon, C. Involvement of polyamines in sucrose-induced tolerance to atrazine-mediated chemical stress in Arabidopsis thaliana. J. Plant Physiol. 2019, 238, 1–11. [Google Scholar] [CrossRef]
- Taie, H.A.A.; Seif El-Yazal, M.A.; Ahmed, S.M.A.; Rady, M.M. Polyamines modulate growth, antioxidant activity, and genomic DNA in heavy metal-stressed wheat plant. Environ. Sci. Pollut. Res. Int. 2019, 1, 1–13. [Google Scholar] [CrossRef]
- Zarza, X.; Shabala, L.; Fujita, M.; Shabala, S.; Haring, M.A.; Tiburcio, A.F.; Munnik, T. Extracellular Spermine Triggers a rapid intracellular phosphatidic acid response in Arabidopsis, involving PLD activation and stimulating ion flux. Front. Plant Sci. 2019, 10, 601. [Google Scholar] [CrossRef]
- Kahana, C. The antizyme family for regulating polyamines. J. Biol. Chem. 2018, 293, 18730–18735. [Google Scholar] [CrossRef] [Green Version]
- Porat, Z.; Wender, N.; Erez, O.; Kahana, C. Mechanism of polyamine tolerance in yeast: Novel regulators and insights. Cell. Mol. Life Sci. 2006, 62, 3106–3116. [Google Scholar] [CrossRef]
- Landau, G.; Ran, A.; Bercovich, Z.; Feldmesser, E.; Horn-Saban, S.; Korkotian, E.; Jacob-Hirsh, E.; Rechavi, G.; Ron, D.; Kahana, C. Expression profiling and biochemical analysis suggest stress response as a potential mechanism inhibiting proliferation of polyamine-depleted cells. J. Biol. Chem. 2012, 287, 35825–35837. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Z.; Zhang, S.; Yan, W.; Si, C.; Lee, M.H.; Li, Z. Inhibitory effects of the extracts of Juglans sigillata green husks on the proliferation, migration and survival of KYSE150 and EC9706 human esophageal cancer cell lines. Nutr. Cancer 2019, 71, 149–158. [Google Scholar] [CrossRef]
- Gao, P.; Liu, R.; Jin, Q.; Wang, X. Comparative study of chemical compositions and antioxidant capacities of oils obtained from two species of walnut: Juglans regia and Juglans sigillata. Food Chem. 2019, 279, 279–287. [Google Scholar] [CrossRef]
- Zhao, P.; Zhou, H.J.; Potter, D.; Hu, Y.H.; Feng, X.J.; Dang, M.; Feng, L.; Zulfiqar, S.; Liu, W.Z.; Zhao, G.F.; et al. Population genetics, phylogenomics and hybrid speciation of Juglans in China determined from whole chloroplast genomes, transcriptomes, and genotyping-by-sequencing (GBS). Mol. Phylogenet. Evol. 2018, 126, 250–265. [Google Scholar] [CrossRef]
- Bai, W.N.; Yan, P.C.; Zhang, B.W.; Woeste, K.E.; Lin, K.; Zhang, D.Y. Demographically idiosyncratic responses to climate change and rapid Pleistocene diversification of the walnut genus Juglans (Juglandaceae) revealed by whole-genome sequences. New Phytol. 2018, 217, 1726–1736. [Google Scholar] [CrossRef]
- Persic, M.; Mikulic-Petkovsek, M.; Halbwirth, H.; Solar, A.; Veberic, R.; Slatnar, A. Red walnut: Characterization of the phenolic profiles, activities and gene expression of selected enzymes related to the phenylpropanoid pathway in pellicle during walnut development. J. Agricul. Food Chem. 2018, 66, 2742–2748. [Google Scholar] [CrossRef]
- Li, Y.; Luo, X.; Wu, C.; Cao, S.; Zhou, Y.; Jie, B.; Cao, Y.; Meng, H.; Wu, G. Comparative transcriptome analysis of genes involved in anthocyanin biosynthesis in red and green walnut (Juglans regia L.). Molecules 2017, 23, 25. [Google Scholar] [CrossRef]
- Woodworth, R.H. Meiosis of microsporogenesis in the Juglandaceae. Am. J. Bot. 1930, 17, 863–869. [Google Scholar] [CrossRef]
- Martínez-García, P.J.; Crepeau, M.W.; Puiu, D.; Gonzalez-Ibeas, D.; Whalen, J.; Stevens, K.A.; Paul, R.; Butterfield, T.S.; Britton, M.T.; Reagan, R.L.; et al. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J. 2016, 87, 507–532. [Google Scholar] [CrossRef]
- Farquhar, G.D.; von Caemmerer, S.; Berry, J.A. A biochemical model of photosynthetic CO2 assimilation in leaves of C3 species. Planta 1980, 149, 78–90. [Google Scholar] [CrossRef]
- Romero, J.; Sanchez, M.A. Extraction of chlorophyll and carotenoids of whole protein concentrate obtained from green sweet potato by-products of harvest (Ipomoea batatas). Available online: http://aims.fao.org/serials/c_debd36ae (accessed on 9 August 2019).
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Ernst, J.; Barjoseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Li, X.; Pan, X.J.; Zhang, W.E.; Zhang, R.; Chen, J. Stability evaluation of reference genes for quantitative real-time PCR analysis in walnut (Juglans spp.). Plant Physiol. J. 2017, 53, 1795–1802. [Google Scholar]
- Wu, J.; Gu, Y.Q.; Hu, Y.; You, F.M.; Dandekar, A.M.; Leslie, C.A.; Aradhya, M.; Dvorak, J.; Luo, M.C. Characterizing the walnut genome through analyses of BAC end sequences. Plant Mol. Biol. 2012, 78, 95–107. [Google Scholar] [CrossRef]
- Xu, X.; Pan, S. Genome sequence and analysis of the tuber crop potato. In Proceedings of the 12th National Congress on Plant Genomics, San Diego, CA, USA, 4–6 December 2011; pp. 189–195. [Google Scholar]
- Hu, G.; Wei, B.; Wang, L.; Wang, L.; Kong, D.; Jin, Y.; Sun, Z. Analysis of gene expression profiles associated with glioma progression. Mol. Med. Rep. 2015, 12, 1884. [Google Scholar] [CrossRef]
- Forestan, C.; Cigliano, R.A.; Farinati, S.; Lunardon, A.; Sanseverino, W.; Varotto, S. Stress-induced and epigenetic-mediated maize transcriptome regulation study by means of transcriptome reannotation and differential expression analysis. Sci. Rep. 2016, 6, 30446. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Li, X.; Huang, Y.; Jia, L.; Li, W. Time series clustering of mRNA and LncRNA expression during osteogenic differentiation of periodontal ligament stem cells. PeerJ 2018, 6, e5214. [Google Scholar] [CrossRef]
- Uyttewaal, M.; Burian, A.; Alim, K.; Landrein, B.; Borowska-Wykret, D.; Dedieu, A.; Peaucelle, A.; Ludynia, M.; Traas, J.; Boudaoud, A. Mechanical stress acts via katanin to amplify differences in growth rate between adjacent cells in Arabidopsis. Cell 2012, 149, 439–451. [Google Scholar] [CrossRef]
- Piñero, M.C.; Porras, M.E.; López-Marín, J.; Sánchez-Guerrero, M.C.; Medrano, E.; Lorenzo, P.; del Amor, F.M. Differential nitrogen nutrition modifies polyamines and the amino-acid profile of sweet pepper under salinity stress. Front. Plant Sci. 2019, 10, 301. [Google Scholar] [CrossRef]
- Chen, M.J.; Tang, H.Y.; Chiang, M.L. Effects of heat, cold, acid and bile salt adaptations on the stress tolerance and protein expression of kefir-isolated probiotic Lactobacillus kefiranofaciens M1. Food Microbiol. 2017, 66, 20–27. [Google Scholar] [CrossRef]
- Räsänen, K.; Laurila, A.; Merilä, J. Geographic variation in acid stress tolerance of the moor frog, Rana arvalis. II. Adaptive maternal effects. Evolution 2003, 57, 352–362. [Google Scholar] [CrossRef]
- Majumdar, R.; Minocha, R.; Lebar, M.D.; Rajasekaran, K.; Long, S.; Carter-Wientjes, C.; Minocha, S.; Cary, J.W. Contribution of maize polyamine and amino acid metabolism toward resistance against Aspergillus flavus infection and aflatoxin production. Front. Plant Sci. 2019, 10, 692. [Google Scholar] [CrossRef]
- Shikanai, T. Central role of cyclic electron transport around photosystem I in the regulation of photosynthesis. Curr. Opin. Biotechnol. 2014, 26, 25–30. [Google Scholar] [CrossRef]
- Guan, N.; Du, B.; Li, J.; Shin, H.D.; Chen, R.R.; Du, G.; Chen, J.; Liu, L. Comparative genomics and transcriptomics analysis-guided metabolic engineering of Propionibacterium acidipropionici for improved propionic acid production. Biotechnol Bioeng. 2018, 115, 483–494. [Google Scholar] [CrossRef]
- Otani, T.; Kato, Y.; Shikanai, T. Specific substitutions of light-harvesting complex I proteins associated with photosystem I are required for super complex formation with chloroplast NADH dehydrogenase-like complex. Plant J. 2018, 94, 122–130. [Google Scholar] [CrossRef]
- Benes, K.; Krekule, J. Plant Growth Substances from the Viewpoint of Regulation of Biological Processes; Biologické Listy: Prague, Czech, 1979; ISSN 0366–0486. [Google Scholar]
- Yang, M.; Cao, X.; Wu, R.; Liu, B.; Ye, W.; Yue, X.; Wu, J. Comparative proteomic exploration of whey proteins in human and bovine colostrum and mature milk using iTRAQ-coupled LC-MS/MS. Int. J. Food Sci. Nutr. 2017, 68, 671–681. [Google Scholar] [CrossRef]
- Yin, X.M.; Yu, S.Q.; Jiang, H.; Liu, M.H. Effects of acid rain stress on Eleocarpus glabripetalus seedlings leaf chlorophyll fluorescence characteristics and growth. Chinese. J. Appl. Ecol. 2010, 21, 1374–1380. [Google Scholar]
- Liu, J.; Kitashiba, H.; Wang, J.; Ban, Y.; Moriguchi, T. Polyamines and their ability to provide environmental stress tolerance to plants. Plant Biotechnol. 2007, 24, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Grienenberger, E.; Besseau, S.; GeoVroy, P.; Debayle, D.; Heintz, D.; Lapierre, C.; Pollet, B.; Heitz, T.; Legrand, M. A BAHD acyltransferase is expressed in the tapetum of Arabidopsis anthers and is involved in the synthesis of hydroxycinnamoyl spermidines. Plant J. 2009, 58, 246–259. [Google Scholar] [CrossRef]
- Watson, M.B.; Malmberg, R.L. Regulation of Arabidopsis thaliana (L.) Heynh arginine decarboxilase by potassium deficiency stress. Plant Physiol. 1996, 111, 1077–1083. [Google Scholar] [CrossRef]
- Nam, K.H.; Lee, S.H.; Lee, J. Purification and characterization of arginine decarboxylase from soybean (Glycine max) hypocotyls. Plant Cell Physiol. 1997, 38, 1150–1155. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Q.S.; Wang, Y.; Lafyatis, R.; Stamm, S.; Andreadis, A. Tau exon 10, whose missplicing causes frontotemporal dementia, is regulated by an intricate interplay of cis elements and trans factors. J. Neurochem. 2004, 88, 1078–1090. [Google Scholar] [CrossRef] [Green Version]
- Camacho, J.A.; Rioseco-Camacho, N. The human and mouse SLC25A29 mitochondrial transporters rescue the deficient ornithine metabolism in fibroblasts of patients with the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr. Res. 2009, 66, 35–41. [Google Scholar] [CrossRef]
- Bitrián, M.; Zarza, X.; Altabella, T.; Tiburcio, A.F.; Alcázar, R. Polyamines under abiotic stress: Metabolic crossroads and hormonal cross talks in plants. Metabolites 2012, 2, 516–528. [Google Scholar] [CrossRef]
- Fuell, C.; Elliott, K.A.; Hanfrey, C.C.; Franceschetti, M.; Michael, A.J. Polyamine biosynthetic diversity in plants and algae. Plant Physiol. Biochem. 2010, 48, 513–520. [Google Scholar] [CrossRef]
- Gandre, S.; Bercovich, Z.; Kahana, C. Ornithine decarboxylase-antizyme is rapidly degraded through a mechanism that requires functional ubiquitin-dependent proteolytic activity. Eur. J. Biochem. 2002, 269, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Keren-Paz, A.; Bercovich, Z.; Kahana, C. Antizyme inhibitor: A defective ornithine decarboxylase or a physiological regulator of polyamine biosynthesis and cellular proliferation. Biochem. Soc. Trans. 2007, 35, 311–313. [Google Scholar] [CrossRef]
- Minguet, E.G.; Vera-Sirera, F.; Marina, A.; Carbonell, J.; Blázquez, M.A. Evolutionary diversification in polyamine biosynthesis. Mol. Biol. Evol. 2008, 25, 2119–2128. [Google Scholar] [CrossRef]
- Tabor, C.W.; Tabor, H. Polyamines. Annu. Rev. Biochem. 1984, 53, 749–790. [Google Scholar] [CrossRef]
- Takahashi, T.; Kakehi, J.I. Polyamines: Ubiquitous polycations with unique roles in growth and stress responses. Ann. Bot. 2010, 105, 1–6. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Reads | Q30 | GC | Mapped Genome | Unique Mapped Genome | Mapped Genes | Expressed Genes |
|---|---|---|---|---|---|---|---|
| pH 3–4_1 | 28,020,622 | 94.93% | 45.98% | 87.60% | 68.87% | 75.69% | 26,186 |
| pH 3–4_2 | 26,738,560 | 96.36% | 46.37% | 82.25% | 63.00% | 69.82% | 28,134 |
| pH 3–4_3 | 27,011,236 | 96.32% | 46.85% | 85.60% | 66.22% | 73.12% | 28,922 |
| pH 4–5_1 | 27,605,126 | 96.02% | 46.39% | 85.75% | 68.97% | 74.31% | 27,392 |
| pH 4–5_2 | 27,634,854 | 96.44% | 47.05% | 88.56% | 70.73% | 75.93% | 27,670 |
| pH 4–5_3 | 27,109,750 | 96.50% | 46.59% | 87.53% | 69.72% | 75.85% | 27,582 |
| pH 5–6_1 | 26,864,464 | 96.47% | 46.51% | 87.59% | 71.06% | 73.90% | 26,644 |
| pH 5–6_2 | 27,448,384 | 96.36% | 46.37% | 84.86% | 67.01% | 73.6% | 27,525 |
| pH 5–6_3 | 27,338,826 | 96.40% | 46.60% | 87.13% | 68.69% | 75.66% | 26,942 |
| pH 6–7_1 | 27,698,734 | 94.98% | 46.42% | 88.08% | 68.34% | 76.53% | 26,864 |
| pH 6–7_2 | 27,894,576 | 96.50% | 46.45% | 84.43% | 66.27% | 72.39% | 27,221 |
| pH 6–7_3 | 27,288,782 | 96.36% | 46.73% | 86.17% | 69.15% | 72.39% | 26,706 |
| Gene ID | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) | Amplicon Size (bp) |
|---|---|---|---|
| gene17916 | GAAGGCGAAGAAGAAGAAGA | CTGGCGGTAACTGTAACTC | 96 |
| gene22045 | GGCGTGAAGGAGTTGATT | ACAGTGTTAAGGTCGTATCG | 97 |
| gene25150 | AAGACGATGTTGATGATTCC | TTCCAGTATTAGCGGTAAGA | 100 |
| gene25184 | TGAGGCTGAAGAGTATGC | CGTAGATGGTTGGATGGT | 82 |
| gene29464 | CTGTGTTGTGGTAGAGGA | TCTTCATCGGCTGTGTAA | 81 |
| gene29846 | GAGAAGGCTATCACAAGAAG | CCAGTATGACAAGGAGTAATC | 113 |
| gene31088 | GATGCTGTGTTGCTGTTC | CCGCCATTATCTGCTTGA | 131 |
| gene31799 | GCTATAACTACGCTATATTCG | TTACTTCTGATTCTCCTATGT | 111 |
| gene35192 | GCTGGAAGTCATAGTAAGG | ATGGCTGCTAATCACAAG | 167 |
| gene39645 | GAGTGGGAATGAAGGAAGA | ATTGGCAGAGGAATTGGA | 72 |
| Pathway ID | Pathway | FDR < 0.05 | No. DEGs |
|---|---|---|---|
| ko01200 | Carbon metabolism | 5.70 × 10−7 | 50 |
| ko01100 | Metabolic pathways | 5.70 × 10−7 | 229 |
| ko01110 | Biosynthesis of secondary metabolites | 1.65 × 10−6 | 146 |
| ko00710 | Carbon fixation in photosynthetic organisms | 1.83 × 10−6 | 28 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 8.98 × 10−6 | 17 |
| ko00908 | Zeatin biosynthesis | 2.25 × 10−5 | 14 |
| ko00195 | Photosynthesis | 1.20 × 10−4 | 12 |
| ko00010 | Glycolysis/gluconeogenesis | 1.04 × 10−3 | 29 |
| ko03430 | Mismatch repair | 1.81 × 10−3 | 2 |
| ko03050 | Proteasome | 1.81 × 10−3 | 1 |
| ko00220 | Arginine biosynthesis | 2.13 × 10−3 | 10 |
| ko00903 | Limonene and pinene degradation | 4.64 × 10−3 | 14 |
| ko00910 | Nitrogen metabolism | 4.64 × 10−3 | 10 |
| ko00030 | Pentose phosphate pathway | 4.64 × 10−3 | 20 |
| ko01230 | Biosynthesis of amino acids | 5.00 × 10−3 | 35 |
| ko00071 | Fatty acid degradation | 5.88 × 10−3 | 6 |
| ko00511 | Other glycan degradation | 6.71 × 10−3 | 1 |
| ko00500 | Starch and sucrose metabolism | 7.97 × 10−3 | 40 |
| ko00670 | One carbon pool by folate | 8.93 × 10−3 | 7 |
| ko04075 | Plant hormone signal transduction | 1.07 × 10−2 | 43 |
| ko00260 | Glycine, serine and threonine metabolism | 1.22 × 10−2 | 13 |
| ko00250 | Alanine, aspartate and glutamate metabolism | 1.42 × 10−2 | 9 |
| Ko00330 | Arginine and proline metabolism | 1.42 × 10−2 | 7 |
| ko00051 | Fructose and mannose metabolism | 1.42 × 10−2 | 17 |
| ko03015 | mRNA surveillance pathway | 1.60 × 10−2 | 1 |
| ko03040 | Spliceosome | 1.60 × 10−2 | 2 |
| ko01040 | Biosynthesis of unsaturated fatty acids | 2.06 × 10−2 | 1 |
| ko00945 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 2.06 × 10−2 | 4 |
| ko00450 | Selenocompound metabolism | 2.15 × 10−2 | 7 |
| ko00740 | Riboflavin metabolism | 2.25 × 10−2 | 4 |
| ko04626 | Plant-pathogen interaction | 2.72 × 10−2 | 51 |
| ko00052 | Galactose metabolism | 2.90 × 10−2 | 12 |
| ko00943 | Isoflavonoid biosynthesis | 2.90 × 10−2 | 1 |
| ko00240 | Pyrimidine metabolism | 2.90 × 10−2 | 2 |
| ko00592 | α-linolenic acid metabolism | 3.33 × 10−2 | 6 |
| ko00196 | Photosynthesis-antenna proteins | 4.07 × 10−2 | 4 |
| ko00942 | Anthocyanin biosynthesis | 4.44 × 10−2 | 6 |
| ko04141 | Protein processing in endoplasmic reticulum | 4.44 × 10−2 | 40 |
| Gene ID | Descriptions | |
|---|---|---|
| GO Terms | KEGG Pathways | |
| gene42780 | spermidine hydroxycinnamoyl transferase-like | |
| ---- | ko01110, Biosynthesis of secondary metabolites | |
| ko01100, Metabolic pathways | ||
| ko00945, Stilbenoid, diarylheptanoid and gingerol biosynthesis | ||
| ko00941, Flavonoid biosynthesis | ||
| ko00940, Phenylpropanoid biosynthesis | ||
| gene28192 | spermine synthase-like | |
| GO:0003674, molecular function GO:0003824, catalytic activity | ko00410, β-Alanine metabolism | |
| ko00480, Glutathione metabolism | ||
| ko00270, Cysteine and methionine metabolism | ||
| ko00330, Arginine and proline metabolism | ||
| ko01100, Metabolic pathways | ||
| gene4089 | S-adenosylmethionine synthase 1-like | |
| ---- | ko01230, Biosynthesis of amino acids | |
| gene31260 | arginine decarboxylase-like | |
| ---- | ko00330, Arginine and proline metabolism | |
| gene24987 | serine/arginine-rich splicing factor RS40-like | |
| ---- | ko03040, Spliceosome | |
| gene9388 | mitochondrial arginine transporter BAC2-like | |
| GO:0016053, organic acid biosynthetic process | ko04714, Thermogenesis | |
| GO:0046394, carboxylic acid biosynthetic process | ||
| GO:0005488, binding | ||
| GO:0006810, transport | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, X.; Liu, J. Transcriptome Analysis of Acid-Responsive Genes and Pathways Involved in Polyamine Regulation in Iron Walnut. Genes 2019, 10, 605. https://doi.org/10.3390/genes10080605
Luo X, Liu J. Transcriptome Analysis of Acid-Responsive Genes and Pathways Involved in Polyamine Regulation in Iron Walnut. Genes. 2019; 10(8):605. https://doi.org/10.3390/genes10080605
Chicago/Turabian StyleLuo, Xiaomei, and Juncheng Liu. 2019. "Transcriptome Analysis of Acid-Responsive Genes and Pathways Involved in Polyamine Regulation in Iron Walnut" Genes 10, no. 8: 605. https://doi.org/10.3390/genes10080605