Transcriptomic Analysis of the Influence of Methanol Assimilation on the Gene Expression in the Recombinant Pichia pastoris Producing Hirudin Variant 3


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Construction of Recombinant P. pastoris
2.3. Fermentation of P. pastoris GS115/pPIC9K-hv3
2.4. Antithrombin Activity Analysis
2.5. cDNA Library Construction and Illumina Sequencing
2.6. De Novo Assembly and Functional Annotation Analysis of Illumina Sequencing
3. Results
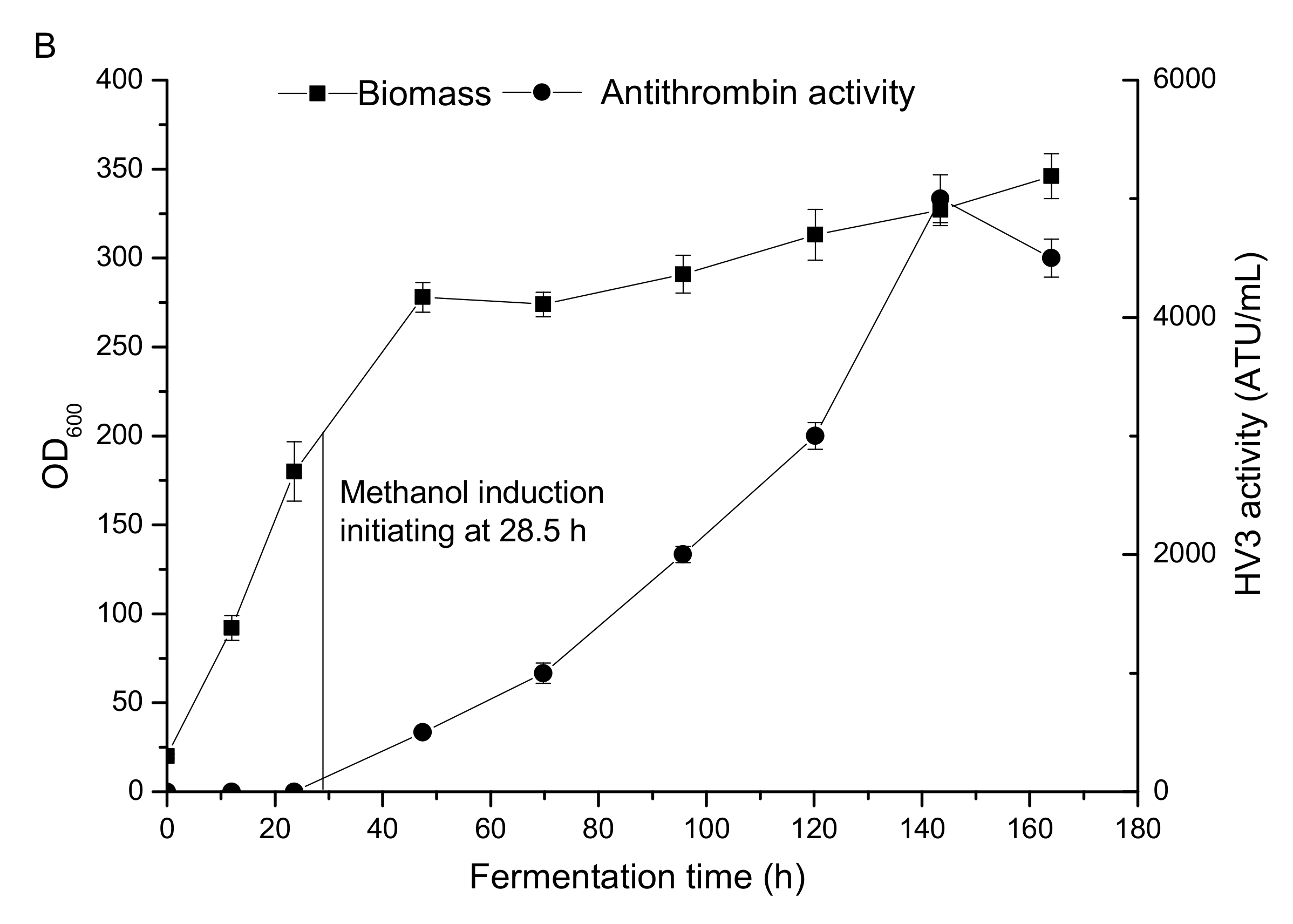
3.1. HV3 Fermentation by P. pastoris GS115/pPIC9K-hv3 in 5L Fermenter
3.2. Overall Evaluation of RNA-Seq Data
3.3. Overview of Transcriptomic Analysis
3.4. GO (Gene Ontology) Enrichment Analysis of DEGs
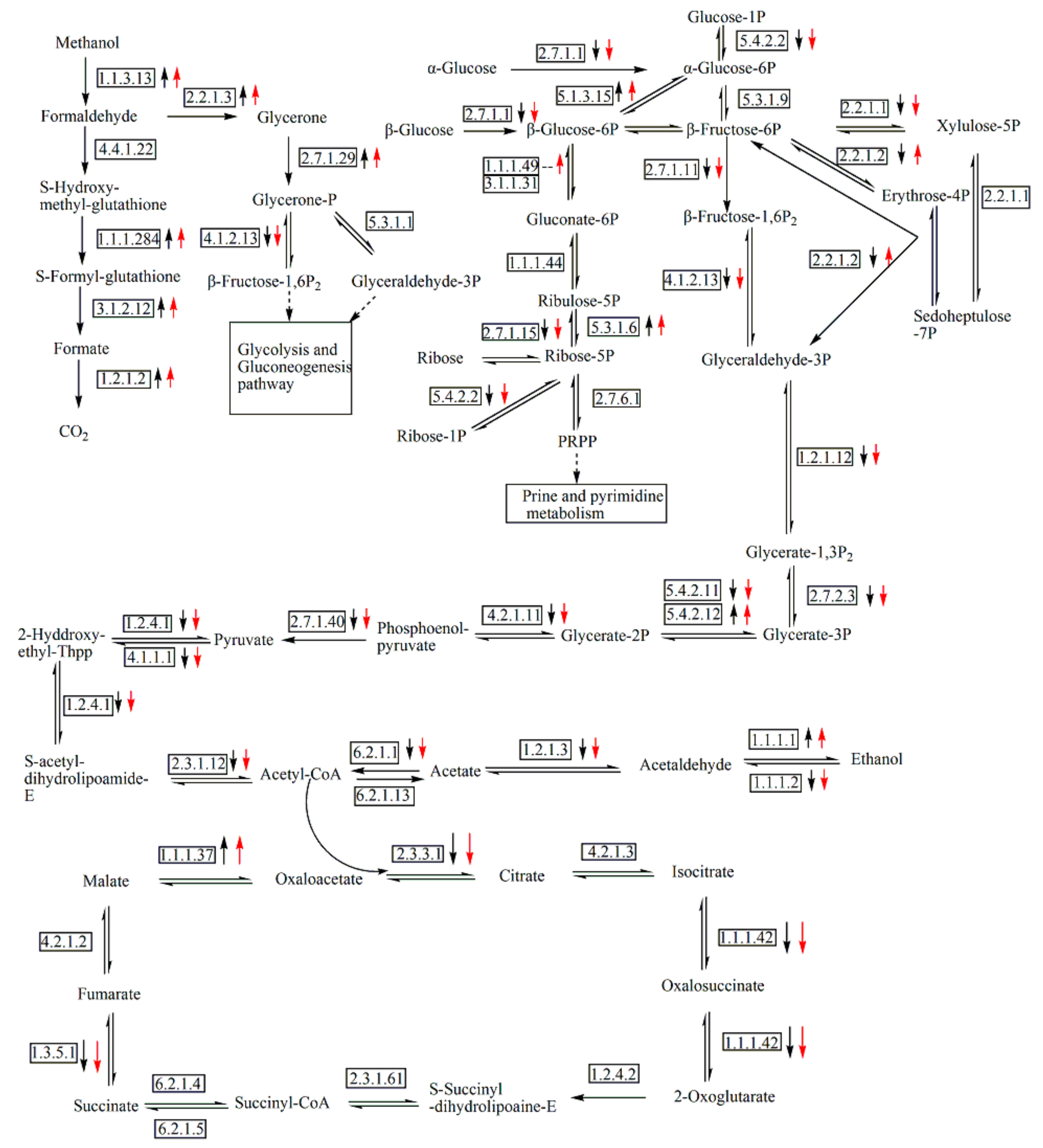
3.5. Analysis of DEGs Involved in Central Carbon Metabolic Pathway
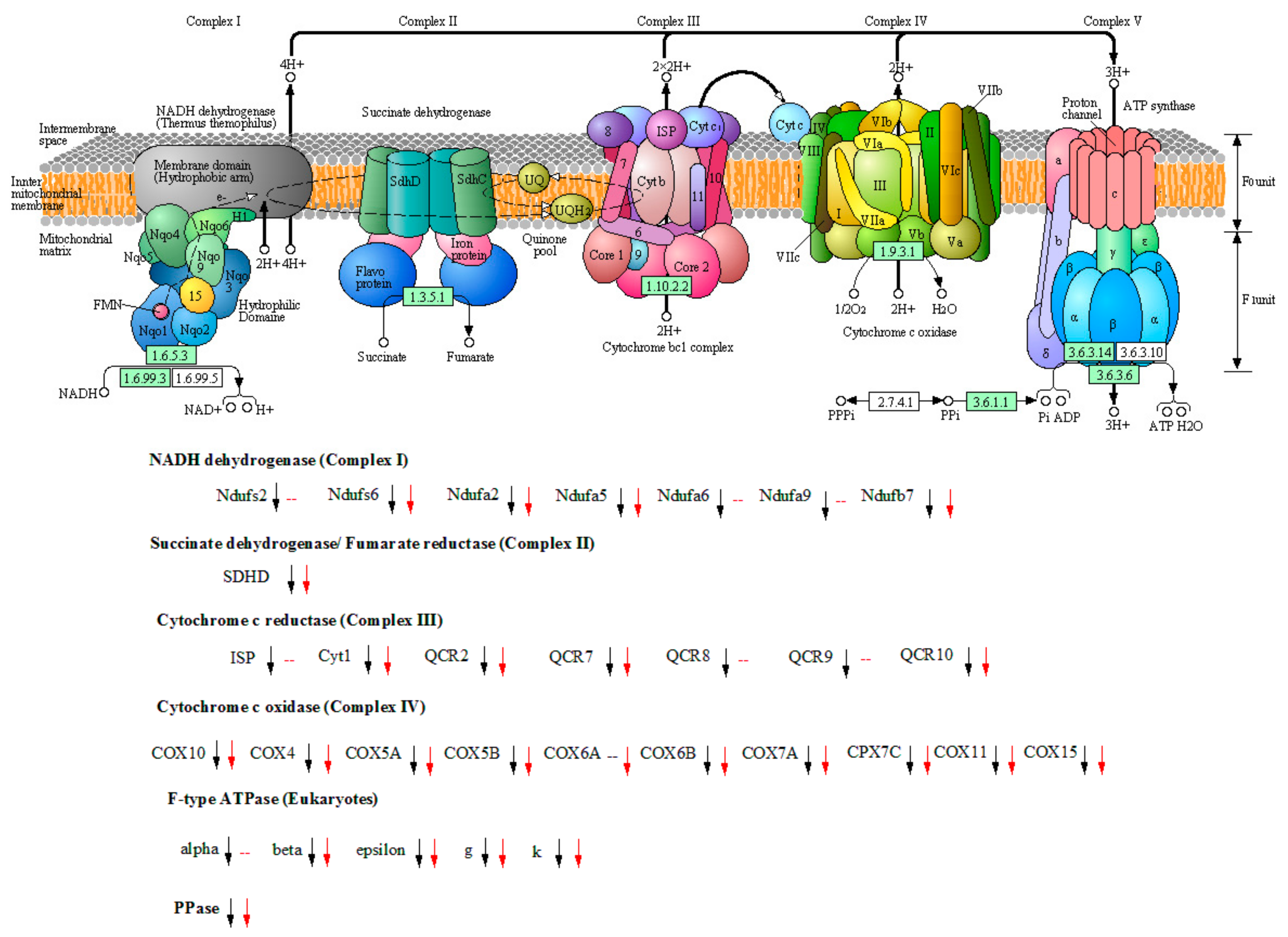
3.6. Carbon Source Switching from Glycerol to Methanol Resulted in a Significant Slowdown of Energy Metabolism
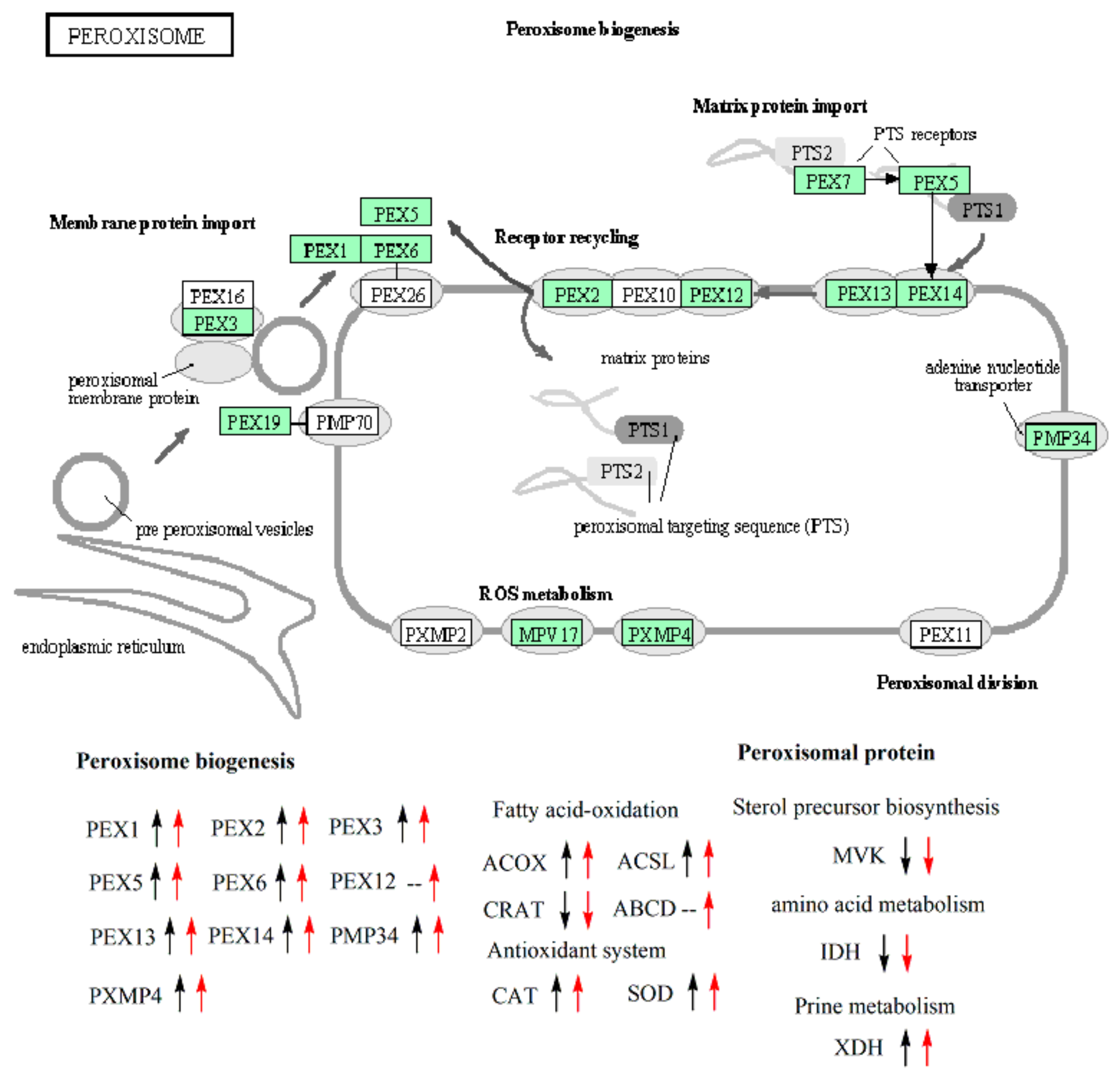
3.7. Expression Analysis of Genes Involved in the Biogenesis of Peroxisome and Peroxisomal Proteins
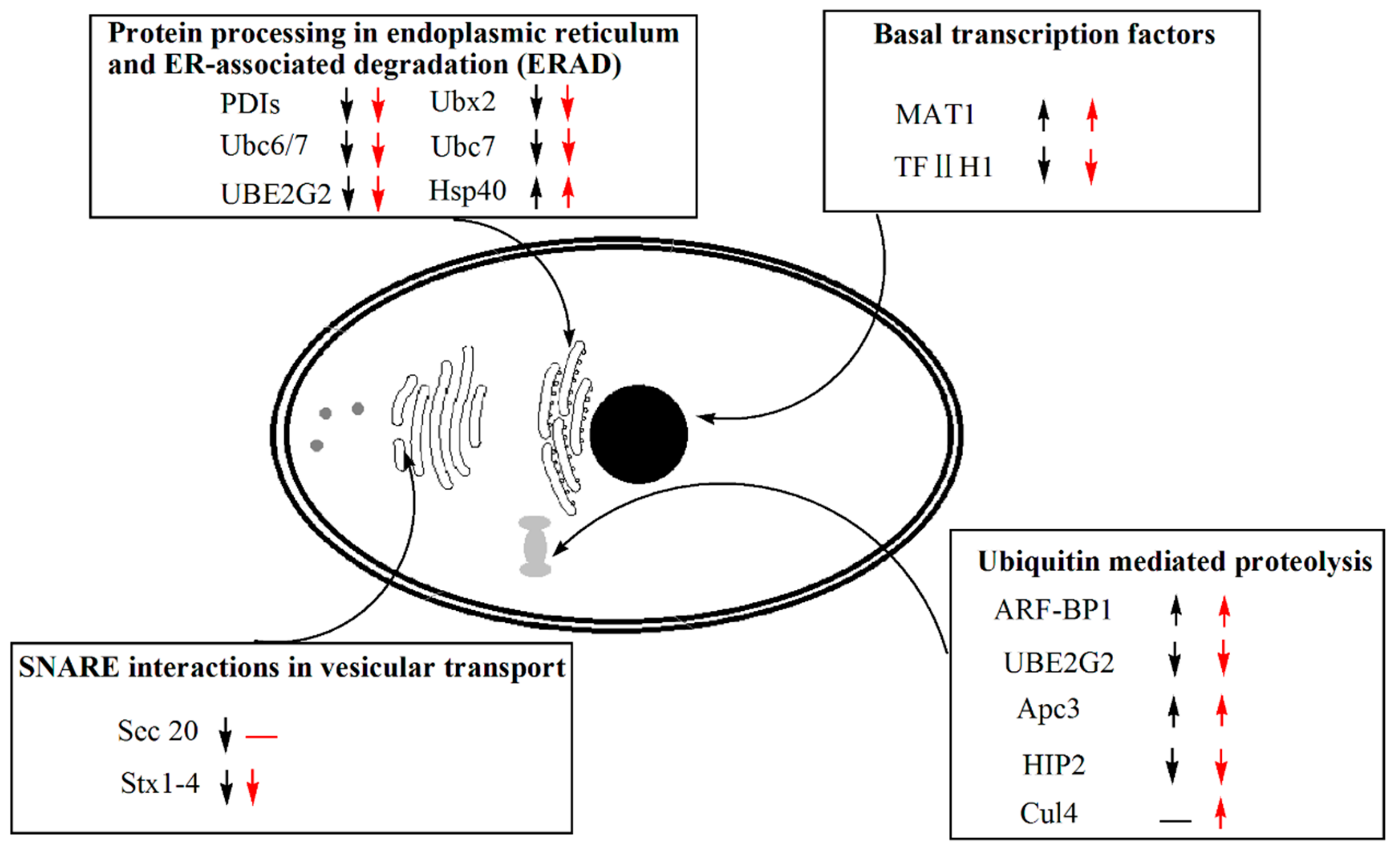
3.8. Transcriptomic Analysis of Genes Participating in the Protein Production and Degradation Processes
3.9. Overall Analysis of Genes Involved in Amino Acid Biosynthesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elliott, J.M.; Sawyer, R.T. Leech Biology and Behaviour-Review. J. Anim. Ecol. 1986, 56, 720. [Google Scholar] [CrossRef]
- Sket, B.; Trontelj, P. Global diversity of leeches (Hirudinea) in freshwater. Hydrobiologia 2008, 595, 129–137. [Google Scholar] [CrossRef]
- Fogden, S.C.L.; Proctor, J. Notes on the feeding of land leeches (Haemadipsa zeylanica Moore and H. picta Moore) in Gunung Mulu National Park, Sarawak. Biotropica 1985, 17, 172–174. [Google Scholar] [CrossRef]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [PubMed]
- Kvist, S.; Min, G.S.; Siddall, M.E. Diversity and selective pressures of anticoagulants in three medicinal leeches (Hirudinida: Hirudinidae, Macrobdellidae). Ecol. Evol. 2013, 3, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Lemke, S.; Müller, C.; Lipke, E.; Uhl, G.; Hildebrandt, J.-P. May salivary gland secretory proteins from hematophagous leeches (Hirudo verbana) reach pharmacologically relevant concentrations in the vertebrate host? PLoS ONE 2013, 8, e73809. [Google Scholar] [CrossRef] [PubMed]
- Lent, C.; Fliegner, K.; Freedman, E.; Dickinson, M. Ingestive behaviour and physiology of the medicinal leech. J. Exp. Biol. 1988, 137, 513–527. [Google Scholar]
- Tessler, M.; Marancik, D.; Champagne, D.; Dove, A.; Camus, A.; Siddall, M.E.; Kvist, S. Marine leech anticoagulant diversity and evolution. J. Parasitol. 2018, 104, 210–220. [Google Scholar] [CrossRef]
- Whitaker, I.; Izadi, D.; Oliver, D.; Monteath, G.; Butler, P. Hirudo medicinalis and the plastic surgeon. Br. J. Plast. Surg. 2004, 57, 348–353. [Google Scholar] [CrossRef]
- Shammas, R.L.; Cornejo, A.; Poveromo, L.P.; Glener, A.D.; Hollenbeck, S.T. Outcomes of leech therapy for venous congestion after reconstructive surgery. J. Am. Coll Surg. 2016, 223, S100. [Google Scholar] [CrossRef]
- Rajic, A.J.; Deleyiannis, F.W.-B. Determining the duration of leech therapy in the treatment of acute venous congestion in prefabricated free flaps. Plast. Reconstr. Surg. 2016, 137, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Huntington, J.A. Natural inhibitors of thrombin. Thromb. Haemost. 2014, 111, 583–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.-Y. The functional domain of hirudin, a thrombin-specific inhibitor. FEBS Lett. 1983, 164, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Dodt, J.; Müller, H.-P.; Seemüller, U.; Chang, J.-Y. The complete amino acid sequence of hirudin, a thrombin specific inhibitor: Application of colour carboxymethylation. FEBS Lett. 1984, 165, 180–184. [Google Scholar] [CrossRef]
- Harvey, R.; Degryse, E.; Stefani, L.; Schamber, F.; Cazenave, J.; Courtney, M.; Tolstoshev, P.; Lecocq, J. Cloning and expression of a cDNA coding for the anticoagulant hirudin from the bloodsucking leech, Hirudo medicinalis. Proc. Natl. Acad. Sci. USA 1986, 83, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Johannes, D.; Machleidt, W.; Seemüller, U.; Maschler, R.; Fritz, H. Isolation and characterization of hirudin isoinhibitors and sequence analysis of hirudin PA. Biol. Chem. Hoppe Seyler 1986, 367, 803–812. [Google Scholar]
- Johnson, P.H.; Sze, P.; Winant, R.; Payne, P.W.; Lazar, J.B. Biochemistry and genetic engineering of hirudin. Semin. Thromb. Hemost. 1989, 15, 302–315. [Google Scholar] [CrossRef]
- Cuicui, H.; Xuerui, Z.; Jia, Q.; Ping, Z.; Shuhua, T. Robust preparative-scale extracellular production of hirudin in Escherichia coli and its purification and characterization. J. Ind. Microbiol. Biotechnol. 2012, 39, 1487–1494. [Google Scholar]
- Kim, M.-D.; Rhee, S.-K.; Seo, J.-H. Enhanced production of anticoagulant hirudin in recombinant Saccharomyces cerevisiae by chromosomal δ-integration. J. Biotechnol. 2001, 85, 41–48. [Google Scholar] [CrossRef]
- Xiao, A.; Zhou, X.; Zhou, L.; Zhang, Y. Improvement of cell viability and hirudin production by ascorbic acid in Pichia pastoris fermentation. Appl. Microbiol. Biotechnol. 2006, 72, 837–844. [Google Scholar] [CrossRef]
- Tan, S.; Wu, W.; Li, X.; Cui, L.; Li, B.; Ruan, Q. Enhanced secretion of adhesive recognition sequence containing hirudin III mutein in E. coli. Mol. Biotechnol. 2007, 36, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Asgari, S.; Mirzahoseini, H.; Karimipour, M.; Rahimi, H.; Ebrahim-Habibi, A. Rational design of stable and functional hirudin III mutants with lower antigenicity. Biologicals 2015, 43, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Wu, W.; Liu, J.; Kong, Y.; Pu, Y.; Yuan, R. Efficient expression and secretion of recombinant hirudin III in E. coli using the L-asparaginase II signal sequence. Protein Expr. Purif. 2002, 25, 430–436. [Google Scholar] [CrossRef]
- Juturu, V.; Wu, J.C. Heterologous protein expression in Pichia pastoris: Latest research progress and applications. ChemBioChem 2018, 19, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Rydel, T.J.; Tulinsky, A.; Bode, W.; Huber, R. Refined structure of the hirudin-thrombin complex. J. Mol. Biol. 1991, 221, 583–601. [Google Scholar] [CrossRef]
- Aw, R.; Barton, G.R.; Leak, D.J. Insights into the prevalence and underlying causes of clonal variation through transcriptomic analysis in Pichia pastoris. Appl. Microbiol. Biotechnol. 2017, 101, 5045–5058. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Guo, N.; Lin, F.; Wu, L.; Zhou, H. Screening of multi-copy mannanase recombinants of Pichia pastoris based on colony size. World J. Microbiol. Biotechnol. 2013, 30, 579–584. [Google Scholar] [CrossRef]
- Inan, M.; Meagher, M.M. Non-repressing carbon sources for alcohol oxidase (AOX1) promoter of Pichia pastoris. J. Biosci. Bioeng. 2001, 92, 585–589. [Google Scholar] [CrossRef]
- Müller, C.; Mescke, K.; Liebig, S.; Mahfoud, H.; Lemke, S.; Hildebrandt, J.-P. More than just one: Multiplicity of hirudins and hirudin-like factors in the medicinal leech, Hirudo medicinalis. Mol. Genet. Genom. 2016, 291, 227–240. [Google Scholar] [CrossRef]
- Mulder, K.C.; de Lima, L.A.; Aguiar, P.S.; Carneiro, F.C.; Franco, O.L.; Dias, S.C.; Parachin, N.S. Production of a modified peptide clavanin in Pichia pastoris: Cloning, expression, purification and in vitro activities. AMB Express 2015, 5, 46. [Google Scholar] [CrossRef]
- Rosenfeld, S.A.; Nadeau, D.; Tirado, J.; Hollis, G.F.; Knabb, R.M.; Jia, S. Production and purification of recombinant hirudin expressed in the methylotrophic yeast Pichia pastoris. Protein Expr. Purif. 1996, 8, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Clare, J.; Rayment, F.; Ballantine, S.; Sreekrishna, K.; Romanos, M. High-level expression of tetanus toxin fragment C in Pichia pastoris strains containing multiple tandem integrations of the gene. Nat. Biotechnol. 1991, 9, 455. [Google Scholar] [CrossRef]
- Grieβbach, U.; Stürzebecher, J.; Markwardt, F. Assay of hirudin in plasma using a chromogenic thrombin substrate. Thromb. Res. 1985, 37, 347–350. [Google Scholar] [CrossRef]
- Markwardt, F. Hirudin as an inhibitor of thrombin. In Method Enzymol; Elsevier: Amsterdam, The Netherlands, 1970; Volume 19, pp. 924–932. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level–The DESeq Package; EMBL: Heidelberg, Germany, 2012. [Google Scholar]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, 14. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Zahrl, R.J.; Pena, D.A.; Mattanovich, D.; Gasser, B. Systems biotechnology for protein production in Pichia pastoris. FEMS Yeast Res. 2017, 17, 15. [Google Scholar] [CrossRef]
- Laukens, B.; Wachter, C.D.; Callewaert, N. Engineering the Pichia pastoris N-glycosylation pathway using the GlycoSwitch technology. Methods Mol. Biol. 2015, 1321, 103–120. [Google Scholar] [PubMed]
- Puxbaum, V.; Mattanovich, D.; Gasser, B. Quo vadis? The challenges of recombinant protein folding and secretion in Pichia pastoris. Appl. Microbiol. Biotechnol. 2015, 99, 2925–2938. [Google Scholar] [CrossRef] [PubMed]
- Gasser, B.; Steiger, M.G.; Mattanovich, D. Methanol regulated yeast promoters: Production vehicles and toolbox for synthetic biology. Microb. Cell Fact. 2015, 14, 196. [Google Scholar] [CrossRef] [PubMed]
- Vassileva, A.; Chugh, D.A.; Swaminathan, S.; Khanna, N. Effect of copy number on the expression levels of hepatitis B surface antigen in the methylotrophic yeast Pichia pastoris. Protein Expr. Purif. 2001, 21, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Marx, H.; Mecklenbräuker, A.; Gasser, B.; Sauer, M.; Mattanovich, D. Directed gene copy number amplification in Pichia pastoris by vector integration into the ribosomal DNA locus. FEMS Yeast Res. 2009, 9, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-S.; Zhang, Y.-X. Decrease of proteolytic degradation of recombinant hirudin produced by Pichia pastoris by controlling the specific growth rate. Biotechnol. Lett. 2002, 24, 1449–1453. [Google Scholar] [CrossRef]
- Kim, S.; Warburton, S.; Boldogh, I.; Svensson, C.; Pon, L.; D’Anjou, M.; Stadheim, T.A.; Choi, B.K. Regulation of alcohol oxidase 1 (AOX1) promoter and peroxisome biogenesis in different fermentation processes in Pichia pastoris. J. Biotechnol. 2013, 166, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.L.; Zhang, Z.S. Engineering strategies for enhanced production of protein and bio-products in Pichia pastoris: A review. Biotechnol. Adv. 2018, 36, 182–195. [Google Scholar] [CrossRef]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 2009, 13, 525–537. [Google Scholar] [CrossRef]
- Krainer, F.W.; Dietzsch, C.; Hajek, T.; Herwig, C.; Spadiut, O.; Glieder, A. Recombinant protein expression in Pichia pastoris strains with an engineered methanol utilization pathway. Microb. Cell Fact. 2012, 11, 22. [Google Scholar] [CrossRef]
- Jordà, J.; Jouhten, P.; Cámara, E.; Maaheimo, H.; Albiol, J.; Ferrer, P. Metabolic flux profiling of recombinant protein secreting Pichia pastoris growing on glucose:methanol mixtures. Microb. Cell Fact. 2012, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Delic, M.; Valli, M.; Graf, A.; Pfeffer, M.; Mattanovich, D.; Gasser, B. The secretory pathway: Exploring yeast diversity. FEMS Microbiol. Rev. 2013, 37, 872–914. [Google Scholar] [CrossRef] [PubMed]
- Love, K.R.; Shah, K.A.; Whittaker, C.A.; Wu, J.; Bartlett, M.C.; Ma, D.; Leeson, R.L.; Priest, M.; Borowsky, J.; Young, S.K.; et al. Comparative genomics and transcriptomics of Pichia pastoris. BMC Genom. 2016, 17, 550. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.L.; Wang, B.; Pan, L.; Ye, Y.R.; He, M.H.; Han, S.Y.; Zheng, S.P.; Wang, X.N.; Lin, Y. Comprehensive structural annotation of Pichia pastoris transcriptome and the response to various carbon sources using deep paired-end RNA sequencing. BMC Genom. 2012, 13, 738. [Google Scholar] [CrossRef] [PubMed]
- Cereghino, J.L.; Cregg, J.M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 2000, 24, 45–66. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EC No. | Gene Name | Gene ID | Gene Description | Log2 (Fold Change)/PI1 vs. BI | padj/PI1 vs. BI | Log2 (Fold change)/PI2 vs. BI | padj/PI2 vs. BI |
|---|---|---|---|---|---|---|---|
| 6.2.1.1 | PAS_chr3_0403 | 8199535 | Acetyl-coA synthetase isoform | −4.2415 | 0 | −4.2022 | 0 |
| 2.7.1.1 | PAS_chr1-4_0561 | 8197692 | Hexokinase-2 | −2.4875 | 0 | −3.6289 | 0 |
| 1.2.1.3 | PAS_chr3_0987 | 8199733 | Mitochondrial aldehyde dehydrogenase | −2.2758 | 1.95 × 10−237 | −1.5456 | 5.74 × 10−131 |
| 2.7.2.3 | PAS_chr1-4_0292 | 8197742 | 3-phosphoglycerate kinase | −2.1073 | 0 | −2.8743 | 0 |
| 4.2.1.11 | PAS_chr3_0082 | 8199366 | Enolase | −1.9763 | 7.89 × 10−307 | −2.5303 | 0 |
| 1.1.1.2 | PAS_chr4_0576 | 8200841 | NADPH-dependent medium chain alcohol dehydrogenase | −1.9642 | 7.25 × 10−181 | −1.8923 | 7.45 × 10−190 |
| 4.1.2.13 | PAS_chr1-1_0072 | 8197200 | Fructose 1,6-bisphosphate aldolase | −1.9342 | 3.70 × 10−282 | −2.0982 | 0 |
| 1.2.1.12 | PAS_chr2-1_0437 | 8198905 | Glyceraldehyde-3-phosphate dehydrogenase | −1.9318 | 1.00 × 10−134 | −2.3868 | 4.91 × 10−185 |
| 5.4.2.11 | PAS_chr3_0826 | 8200319 | Tetrameric phosphoglycerate mutase | −1.8358 | 0 | −2.0814 | 0 |
| 5.4.2.2 | PAS_chr1-4_0264 | 8197873 | Phosphoglucomutase | −1.7789 | 0 | −2.0539 | 0 |
| 2.7.1.11 | PAS_chr2-1_0402 | 8198870 | Phosphofructokinase involved in glycolysis | −1.759 | 0 | −2.711 | 0 |
| 2.7.1.15 | PAS_chr3_0604 | 8199807 | Putative ribokinase | −1.7244 | 0 | −2.3145 | 0 |
| 4.1.1.1 | PAS_chr3_0188 | 8199939 | Major of three pyruvate decarboxylase isozymes | −1.5442 | 4.26 × 10−187 | −2.4152 | 0 |
| 1.2.4.1 | PAS_chr1-4_0593 | 8197723 | E1 β subunit of the pyruvate dehydrogenase (PDH) complex | −1.5393 | 2.17 × 10−131 | −1.676 | 3.23 × 10−157 |
| 2.7.1.11 | PAS_chr1-4_0047 | 8196884 | β subunit of heterooctameric phosphofructokinase | −1.4239 | 0 | −1.8729 | 0 |
| 2.7.1.40 | PAS_chr2-1_0769 | 8198046 | Pyruvate kinase | −1.3692 | 0 | −1.9631 | 0 |
| 2.2.1.1 | PAS_chr1-4_0150 | 8197134 | Transketolase | −1.3666 | 5.53 × 10−118 | −1.3961 | 2.01 × 10−134 |
| 5.4.2.11 | PAS_chr3_0693 | 8200393 | Tetrameric phosphoglycerate mutase | −1.0681 | 4.40 × 10−55 | −1.2688 | 2.30 × 10−74 |
| 2.2.1.2 | PAS_chr2-2_0337 | 8198237 | Transaldolase | −1.0501 | 1.28 × 10−44 | -- | -- |
| 2.3.1.12 | PAS_chr1-1_0050 | 8197567 | Dihydrolipoamide acetyltransferase component (E2) of pyruvate dehydrogenase complex | −1.028 | 1.03 × 10−187 | −1.2588 | 3.15 × 10−240 |
| 5.3.1.6 | PAS_chr4_0213 | 8200884 | Ribose-5-phosphate ketol-isomerase | 1.8505 | 0 | 2.1047 | 4.199 × 10−314 |
| 2.7.1.29 | PAS_chr3_0841 | 8200330 | Dihydroxyacetone kinase | 2.1754 | 0 | 2.3308 | 0 |
| 5.1.3.15 | PAS_chr1-4_0042 | 8196879 | Glucose-6-phosphate 1-epimerase | 2.2902 | 0 | 2.6352 | 0 |
| 5.4.2.12 | PAS_chr2-2_0177 | 8198274 | Putative protein of unknown function | 2.419 | 0 | 2.267 | 0 |
| 1.1.1.284 | PAS_chr3_1028 | 8199772 | S-(hydroxymethyl)glutathione dehydrogenase | 3.401 | 0 | 3.714 | 0 |
| 1.1.1.1 | PAS_chr3_1028 | 8199772 | S-(hydroxymethyl)glutathione dehydrogenase | 3.401 | 0 | 3.714 | 0 |
| 3.1.2.12 | PAS_chr3_0867 | 8200354 | Esterase function as an S-formylglutathione hydrolase | 3.9897 | 0 | 4.2466 | 0 |
| 1.1.3.13 | PAS_chr4_0821 | 8201223 | Alcohol oxidase | 6.5268 | 0 | 6.6696 | 0 |
| 5.3.1.6 | PAS_chr4_0212 | 8200883 | Ribose-5-phosphate ketol-isomerase | 6.5546 | 0 | 6.9045 | 0 |
| 2.2.1.3 | PAS_chr3_0832 | 8199663 | Transketolase, dihydroxyacetone synthase | 6.7656 | 0 | 6.9536 | 0 |
| 1.2.1.2 | PAS_chr3_0932 | 8200284 | NAD (+)-dependent formate dehydrogenase | 7.1213 | 0 | 7.5608 | 0 |
| 2.2.1.3 | PAS_chr3_0834 | 8200324 | Transketolase | 7.8929 | 0 | 8.1561 | 0 |
| 1.1.1.49 | PAS_chr2-1_0308 | 8198996 | Glucose-6-phosphate dehydrogenase (G6PD) | -- | -- | 1.1109 | 2.86 × 10−271 |
| 2.2.1.2 | PAS_chr2-2_0338 | 8198238 | Transaldolase | -- | -- | 6.8356 | 0 |
| 1.1.1.42 | PAS_chr2-1_0580 | 8198933 | Cytosolic NADP-specific isocitrate dehydrogenase | −2.9882 | 4.16 × 10−277 | −3.1716 | 2.877 × 10−311 |
| 1.2.4.1 | PAS_chr1-4_0593 | 8197723 | E1 β subunit of the pyruvate dehydrogenase (PDH) complex | −1.5393 | 2.17 × 10−131 | −1.676 | 3.23 × 10−157 |
| 1.3.5.1 | PAS_chr3_0424 | 8199556 | Membrane anchor subunit of succinate dehydrogenase | −1.3199 | 3.86 × 10−139 | −1.4013 | 1.19 × 10−148 |
| 2.3.3.1 | PAS_chr1-1_0475 | 8197246 | Citrate synthase | −1.3184 | 1.47 × 10−121 | −1.3199 | 4.41 × 10−131 |
| 1.1.1.42 | PAS_chr1-1_0233 | 8196735 | Mitochondrial NADP-specific isocitrate dehydrogenase | −1.0698 | 4.44 × 10−202 | −1.0284 | 4.63 × 10−179 |
| 2.3.1.12 | PAS_chr1-1_0050 | 8197567 | Dihydrolipoamide acetyltransferase component (E2) of pyruvate dehydrogenase complex | −1.028 | 1.03 × 10−187 | −1.2588 | 3.15 × 10−240 |
| 1.1.1.37 | PAS_chr4_0815 | 8201217 | Mitochondrial malate dehydrogenase | 1.381 | 0 | 1.7676 | 0 |
| 1.2.4.1 | PAS_chr2-2_0294 | 8198194 | E1 α subunit of the pyruvate dehydrogenase (PDH) complex | -- | -- | −1.006 | 1.59 × 10−228 |
| Protein Name | Gene Name | Gene ID | Gene Description | Log2 (Fold Change)/PI1 vs. BI | padj/PI1 vs. BI | Log2 (Fold Change)/PI2 vs. BI | padj/PI2 vs. BI |
|---|---|---|---|---|---|---|---|
| Ndufb7 | PAS_chr1-1_0172 | 8196676 | Hypothetical protein | −1.0432 | 1.07 × 10−89 | −1.4134 | 1.01 × 10−108 |
| Ndufa2 | PAS_chr1-4_0575 | 8197446 | NADH-ubiquinone oxidoreductase | −1.2501 | 4.90 × 10−108 | −1.2525 | 6.41 × 10−106 |
| Ndufa5 | PAS_chr1-4_0371 | 8197883 | Hypothetical protein | −1.2085 | 1.92 × 10−191 | −1.1128 | 1.15 × 10−158 |
| Ndufs6 | PAS_chr2-2_0235 | 8198642 | Hypothetical protein | −1.1401 | 1.73 × 10−156 | −1.1829 | 6.66 × 10−160 |
| Ndufa9 | PAS_chr3_1188 | 8199532 | Hypothetical protein | −1.0381 | 5.51 × 10−229 | -- | -- |
| Ndufs2 | PAS_chr3_0808 | 8200498 | Hypothetical protein | −1.0048 | 1.02 × 10−157 | -- | -- |
| Ndufa6 | PAS_chr4_0535 | 8201285 | Hypothetical protein | −1.0031 | 6.90 × 10−124 | -- | -- |
| SDHD | PAS_chr3_0424 | 8199556 | Membrane anchor subunit of succinate dehydrogenase | −1.3199 | 3.86 × 10−139 | −1.4013 | 1.19 × 10−148 |
| ISP | PAS_chr2-1_0850 | 8198393 | Hypothetical protein | −1.2036 | 0 | -- | -- |
| Cyt1 | PAS_chr3_0997 | 8199742 | Cytochrome c1 | −1.2324 | 3.89 × 10−174 | −1.1014 | 8.88 × 10−171 |
| QCR2 | PAS_chr2-2_0430 | 8199133 | Subunit 2 of the ubiquinol Cytochrome-c reductase complex | −1.1952 | 2.78 × 10−246 | −1.0056 | 3.22 × 10−204 |
| QCR7 | PAS_chr1-1_0322 | 8196587 | Subunit 7 of the ubiquinol Cytochrome-c reductase complex | −1.199 | 3.64 × 10−298 | −1.0103 | 7.86 × 10−205 |
| QCR8 | PAS_chr1-4_0313 | 8197763 | Subunit 8 of ubiquinol Cytochrome-c reductase complex | −1.0855 | 1.41 × 10−265 | -- | -- |
| QCR9 | PAS_chr4_0520 | 8201463 | Hypothetical protein | −1.125 | 2.36 × 10−58 | -- | -- |
| QCR10 | PAS_chr2-1_0717 | 8198335 | Hypothetical protein | −1.4053 | 6.25 × 10−212 | −1.3829 | 1.80 × 10−136 |
| COX10 | PAS_chr1-3_0194 | 8196833 | Heme A: farnesyltransferase | −1.2751 | 7.51 × 10−119 | −1.0158 | 3.44 × 10−78 |
| COX7A | PAS_chr2-2_0265 | 8198670 | Hypothetical protein | −1.5686 | 5.23 × 10−35 | −1.7976 | 6.61 × 10−42 |
| COX7C | PAS_chr2-2_0266 | 8198671 | Subunit VIII of cytochrome c oxidase | −1.1987 | 3.73 × 10−43 | −1.0889 | 3.07 × 10−25 |
| COX11 | PAS_chr2-1_0226 | 8198685 | Mitochondrial inner membrane protein required for delivery of copper to the Cox1p subunit of cytochrome C | −1.4389 | 4.15 × 10−163 | −1.3231 | 5.35 × 10−138 |
| COX7A | PAS_chr2-1_0746 | 8198951 | Hypothetical protein | −1.378 | 3.59 × 10−114 | −1.4629 | 1.67 × 10−104 |
| COX5B | PAS_chr2-1_0361 | 8199049 | Subunit IV of cytochrome c oxidase | −1.2454 | 1.34 × 10−99 | −1.415 | 2.08 × 10−124 |
| COX4 | PAS_chr3_0615 | 8199817 | Subunit Va of cytochrome c oxidase | −1.2259 | 0 | −1.2926 | 0 |
| COX5A | PAS_chr3_0824 | 8200317 | Subunit VI of cytochrome c oxidase | −1.4422 | 0 | −1.6052 | 0 |
| COX6B | PAS_chr4_0422 | 8200705 | Subunit VIb of cytochrome c oxidase | −1.9104 | 0 | −2.2113 | 0 |
| COX15 | PAS_chr4_0449 | 8201398 | Protein required for the hydroxylation of heme O to form heme A | −1.4388 | 1.40 × 10−318 | −2.1416 | 0 |
| COX6A | PAS_chr2-1_0363 | 8198106 | Subunit VIa of cytochrome c oxidase | -- | -- | −1.0228 | 4.76 × 10−53 |
| epsilon | PAS_chr2-1_0612 | 8198937 | Hypothetical protein | −1.2169 | 2.50 × 10−212 | −1.1389 | 7.07 × 10−138 |
| k | PAS_chr3_0161 | 8199436 | Hypothetical protein | −1.3219 | 1.50 × 10−50 | −1.3717 | 3.29 × 10−37 |
| β | PAS_chr2-2_0165 | 8198741 | β subunit of the F1 sector of mitochondrial F1F0 ATP synthase | −1.2684 | 1.44 × 10−170 | −1.2277 | 3.37 × 10−182 |
| α | PAS_chr3_0576 | 8199781 | α subunit of the F1 sector of mitochondrial F1F0 ATP synthase | −1.029 | 9.73 × 10−114 | -- | - |
| g | PAS_chr3_0819 | 8200313 | Subunit g of the mitochondrial F1F0 ATP synthase | −1.1936 | 1.58 × 10−125 | −1.1555 | 1.52 × 10−124 |
| Ppase /EC 3.6.1.1 | PAS_chr1-3_0070 | 8197380 | Mitochondrial inorganic pyrophosphatase | −1.2811 | 5.87 × 10−273 | −1.2728 | 7.19 × 10−257 |
| Protein Name | Gene Name | Gene ID | Gene Description | Log2 (Fold Change)/PI1 vs. BI | padj/PI1 vs. BI | Log2 (Fold Change)/PI2 vs. BI | padj/PI2 vs. BI |
|---|---|---|---|---|---|---|---|
| PEX1 | PAS_chr3_1045 | 8200007 | Peroxisome biosynthesis protein PAS1 | 2.6156 | 0 | 2.7808 | 0 |
| PEX2 | PAS_chr3_0043 | 8199329 | Peroxisomal integral membrane protein | 2.3827 | 0 | 2.8031 | 0 |
| PEX3 | PAS_chr3_1073 | 8200033 | Peroxisomal membrane protein (PMP) | 1.4397 | 3.017 × 10−310 | 1.92 | 0 |
| PEX5 | PAS_chr2-2_0186 | 8198761 | Peroxisomal membrane signal receptor for the C-terminal tripeptide signal sequence (PTS1) | 2.949 | 0 | 3.5365 | 0 |
| PEX6 | PAS_chr1-4_0133 | 8197117 | AAA-peroxin that heterodimerizes with AAA-peroxin Pex1p | 2.0075 | 0 | 2.1995 | 0 |
| PEX13 | PAS_chr2-2_0207 | 8198615 | Integral peroxisomal membrane protein required for the translocation of peroxisomal matrix proteins | 3.7578 | 0 | 4.7576 | 0 |
| PEX14 | PAS_chr4_0794 | 8200572 | Peroxisomal membrane peroxin that is a central component of the peroxisomal protein import machinery | 2.2643 | 0 | 2.8084 | 0 |
| PMP34 | PAS_chr3_0099 | 8199380 | Mitochondrial NAD+ transporter, involved in the transport of NAD+ into the mitochondria | 4.9561 | 0 | 5.2536 | 0 |
| PXMP4 | PAS_chr1-1_0352 | 8196615 | Hypothetical protein | 1.1228 | 3.11 × 10−18 | 1.2063 | 1.85 × 10−49 |
| ACOX | PAS_chr1-4_0538 | 8197432 | Fatty-acyl coenzyme A oxidase | 2.3146 | 0 | 2.5487 | 0 |
| ACSL | PAS_chr4_0352 | 8200793 | Long chain fatty acyl-CoA synthetase | 2.9342 | 0 | 3.3382 | 0 |
| ACSL | PAS_chr2-1_0785 | 8198062 | Long chain fatty acyl-CoA synthetase | 1.092 | 0 | 1.0212 | 4.13 × 10−262 |
| CRAT | PAS_chr1-4_0074 | 8197291 | Outer mitochondrial carnitine acetyltransferase, minor ethanol-inducible enzyme | −1.0294 | 1.17 × 10−225 | −1.1661 | 8.13 × 10−269 |
| CAT | PAS_chr2-2_0131 | 8198267 | Catalase A | 2.1646 | 0 | 2.509 | 0 |
| SOD | PAS_chr4_0786 | 8200564 | Cytosolic superoxide dismutase | 1.8615 | 0 | 2.353 | 0 |
| SOD | PAS_chr4_0788 | 8200566 | Mitochondrial ribosomal protein of the small subunit | 1.0854 | 1.35 × 10−45 | -- | -- |
| MVK | PAS_chr1-3_0187 | 8197654 | Mevalonate kinase | −1.0019 | 6.82 × 10−64 | −1.1063 | 7.71 × 10−74 |
| IDH | PAS_chr1-1_0233 | 8196735 | Mitochondrial NADP-specific isocitrate dehydrogenase | −1.0698 | 4.44 × 10−202 | −1.0284 | 4.63 × 10−179 |
| IDH | PAS_chr2-1_0580 | 8198933 | Cytosolic NADP-specific isocitrate dehydrogenase | −2.9882 | 4.16 × 10−277 | −3.1716 | 2.87 × 10−311 |
| XDH | PAS_chr2-2_0112 | 8199216 | Hypothetical protein - xanthine dehydrogenase | 1.3445 | 0 | 1.36 | 0 |
| ABCD | PAS_chr2-2_0272 | 8198676 | Subunit of a heterodimeric peroxisomal ATP-binding cassette transporter complex | -- | -- | 1.227 | 1.19 × 10−160 |
| PEX12 | PAS_chr4_0759 | 8200652 | C3HC4-type RING-finger peroxisomal membrane peroxin | -- | -- | 1.7009 | 0 |
| Protein Name | Gene Name | Gene ID | Gene Description | Log2 (Fold Change)/PI1 vs. BI | padj/PI1 vs. BI | Log2 (Fold Change)/PI2 vs. BI | padj/PI2 vs. BI |
|---|---|---|---|---|---|---|---|
| MAT1 | PAS_chr4_0204 | 8201110 | Subunit of TFIIH and nucleotide excision repair factor 3 complexes | 3.3693 | 0 | 4.0208 | 0 |
| TFIIH1 | PAS_chr4_0745 | 8200638 | Subunit of TFIIH and nucleotide excision repair factor 3 complexes | −1.6733 | 6.87 × 10−286 | −2.1074 | 0 |
| ARF-BP1 | PAS_chr3_0044 | 8200180 | E3 ubiquitin ligase of the hect-domain class | 1.479 | 2.52 × 10−178 | 1.94 | 0 |
| UBE2G2 | PAS_chr3_0924 | 8200276 | Ubiquitin conjugating enzyme, involved in the ER-associated protein degradation pathway | −1.6354 | 5.35 × 10−176 | −1.7258 | 1.56 × 10−195 |
| Apc3 | PAS_chr1-3_0148 | 8197615 | Subunit of the Anaphase-Promoting Complex/Cyclosome (APC/C) | 1.1055 | 2.11 × 10−39 | 1.2557 | 1.02 × 10−59 |
| HIP2 | PAS_chr4_0429 | 8200711 | Ubiquitin-conjugating enzyme that mediates selective degradation of short-lived and abnormal protein | −1.0662 | 1.72 × 10−135 | −1.035 | 1.75 × 10−123 |
| Ubc6/7 | PAS_chr3_0924 | 8200276 | Ubiquitin conjugating enzyme, involved in the ER-associated protein degradation pathway | −1.6354 | 5.35 × 10−176 | −1.7258 | 1.56 × 10−195 |
| Ubc7 | PAS_chr3_0924 | 8200276 | Ubiquitin conjugating enzyme, involved in the ER-associated protein degradation pathway | −1.6354 | 5.35 × 10−176 | −1.7258 | 1.56 × 10−195 |
| Ubx2 | PAS_chr1-1_0084 | 8197210 | Protein involved in ER-associated protein degradation | −1.1051 | 3.95 × 10−82 | −1.146 | 5.05 × 10−84 |
| PDIs | PAS_chr1-1_0160 | 8196664 | Protein disulfide isomerase, multifunctional protein resident in the endoplasmic reticulum lumen | −1.3541 | 2.3 × 10−295 | −1.5559 | 0 |
| Hsp40 | PAS_chr2-2_0066 | 8199171 | Protein chaperone involved in regulation of the HSP90 and HSP70 functions | 1.3014 | 1.248 × 10−319 | 1.1231 | 3.2 × 10−221 |
| Sec20 | PAS_chr1-4_0462 | 8197050 | Membrane glycoprotein v-SNARE | −1.0588 | 1.18 × 10−57 | -- | -- |
| Stx1-4 | PAS_chr1-4_0294 | 8197744 | Plasma membrane t-SNARE involved in fusion of secretory vesicles at the plasma membrane | −1.1446 | 2.29 × 10−200 | −1.3775 | 3.9 × 10−264 |
| Cul4 | PAS_chr1-4_0609 | 8197739 | Cullin, structural protein of SCF complexes | -- | -- | 1.0004 | 3.17 × 10−46 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Ma, J.; Xu, Z.; Wang, S.; Wang, N.; Shao, S.; Yang, W.; Huang, L.; Liu, Y. Transcriptomic Analysis of the Influence of Methanol Assimilation on the Gene Expression in the Recombinant Pichia pastoris Producing Hirudin Variant 3. Genes 2019, 10, 606. https://doi.org/10.3390/genes10080606
Li T, Ma J, Xu Z, Wang S, Wang N, Shao S, Yang W, Huang L, Liu Y. Transcriptomic Analysis of the Influence of Methanol Assimilation on the Gene Expression in the Recombinant Pichia pastoris Producing Hirudin Variant 3. Genes. 2019; 10(8):606. https://doi.org/10.3390/genes10080606
Chicago/Turabian StyleLi, Tao, Jieying Ma, Zehua Xu, Shuang Wang, Nan Wang, Shulin Shao, Wei Yang, Lin Huang, and Yihan Liu. 2019. "Transcriptomic Analysis of the Influence of Methanol Assimilation on the Gene Expression in the Recombinant Pichia pastoris Producing Hirudin Variant 3" Genes 10, no. 8: 606. https://doi.org/10.3390/genes10080606