The Patchy Distribution of Restriction–Modification System Genes and the Conservation of Orphan Methyltransferases in Halobacteria

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Search Approach
2.2. Reference Phylogeny
2.3. Presence–absence Phylogeny
2.4. Horizontal Gene Transfer Detection
2.5. Data Analysis and Presentation
3. Results
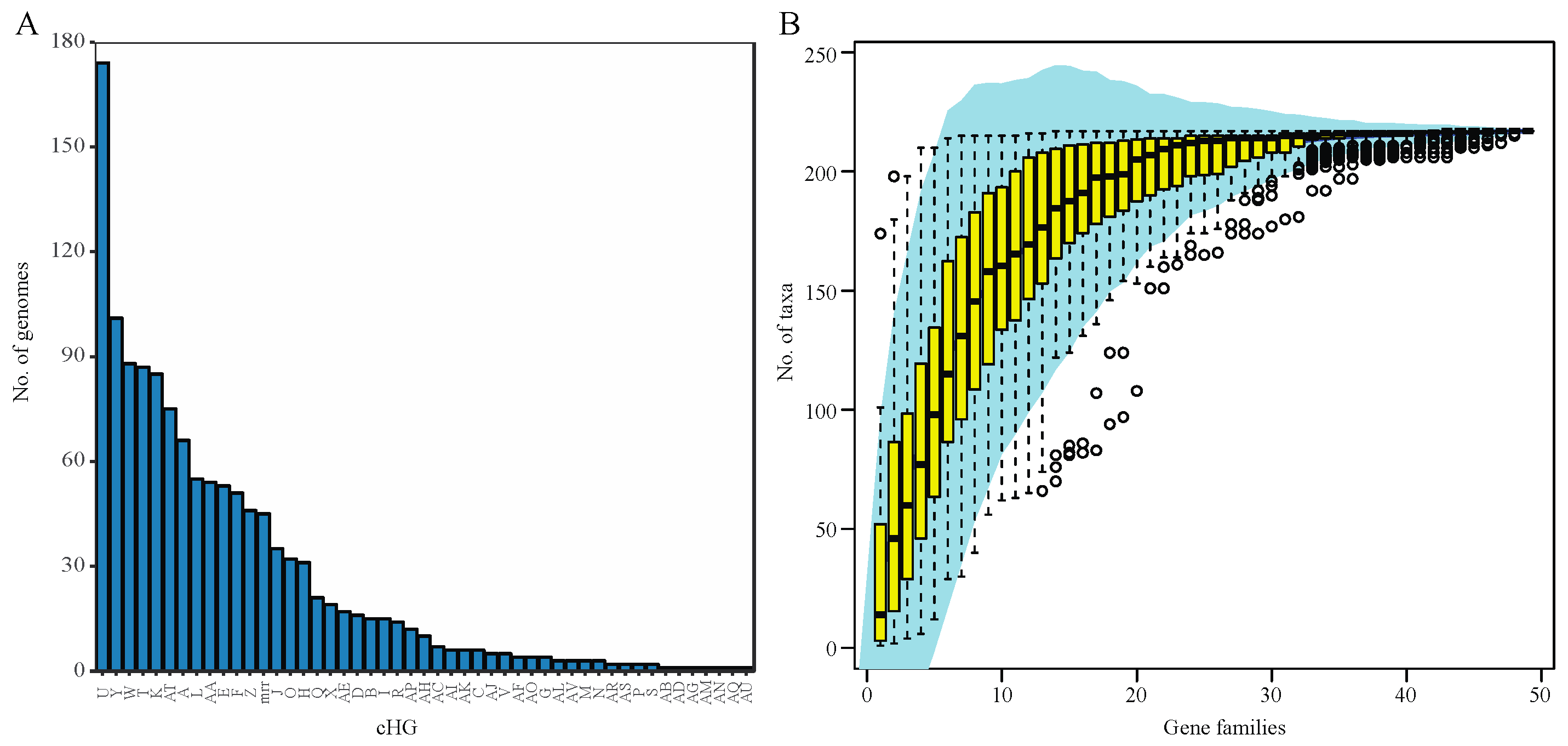
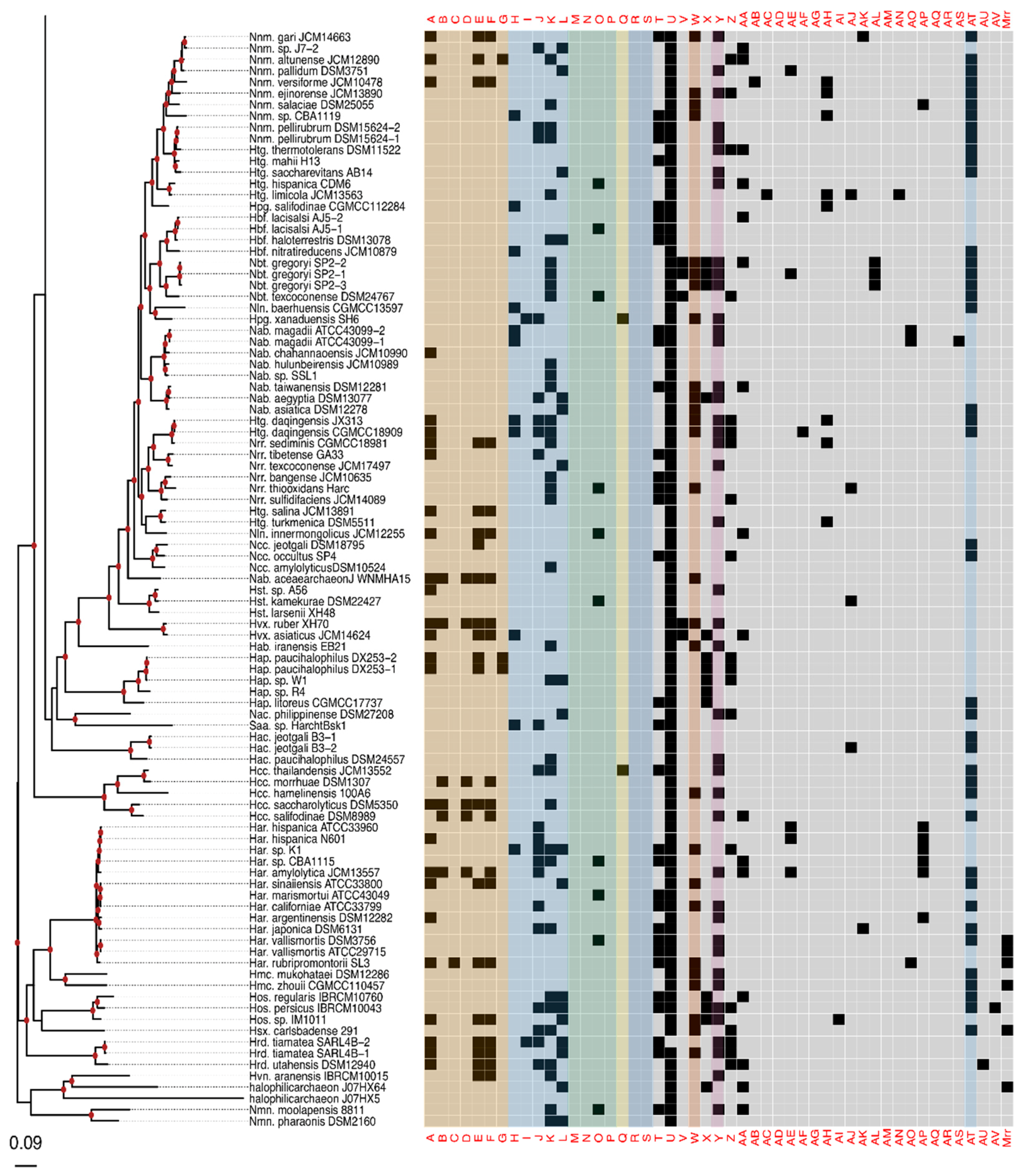
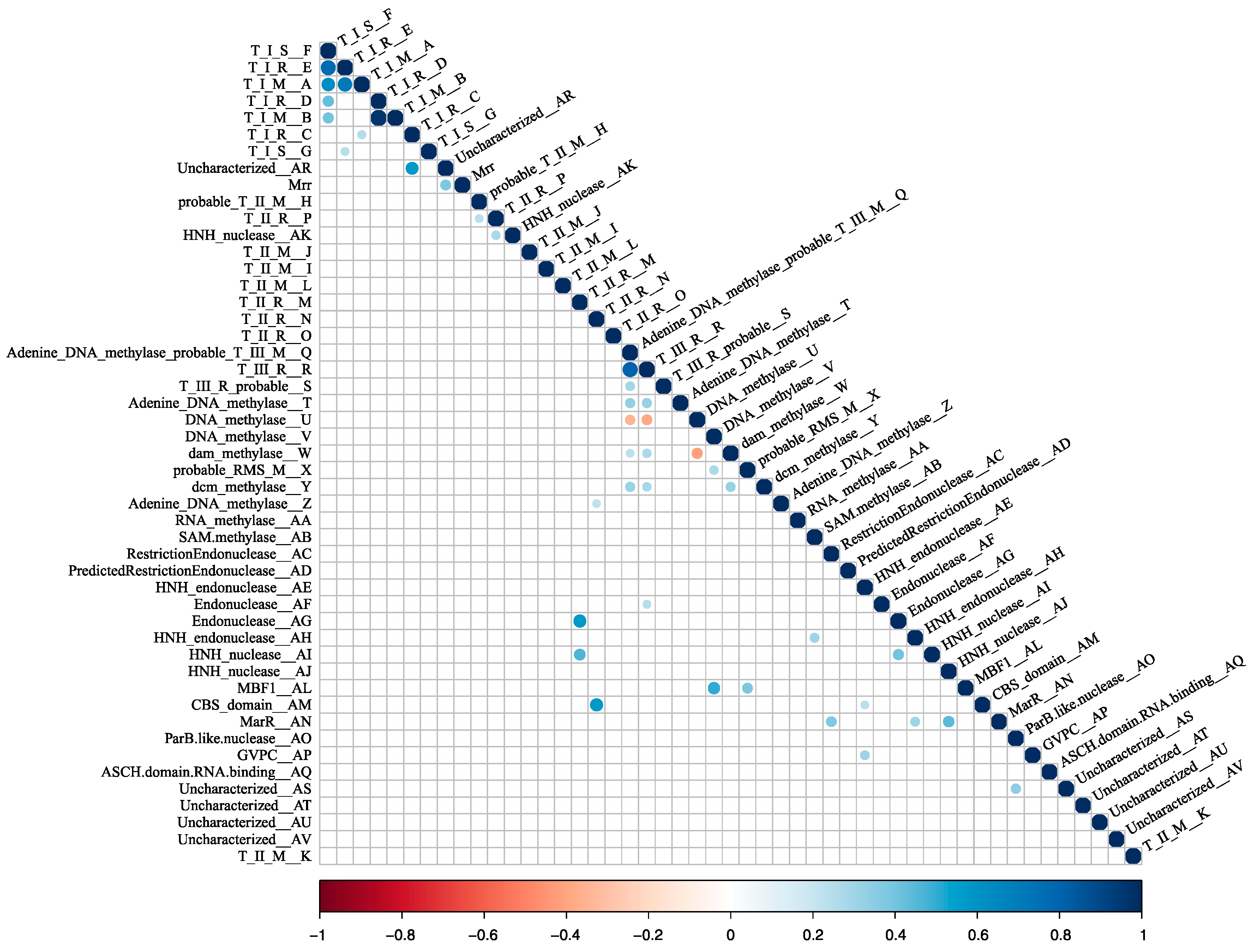
3.1. RM-System Gene Distribution
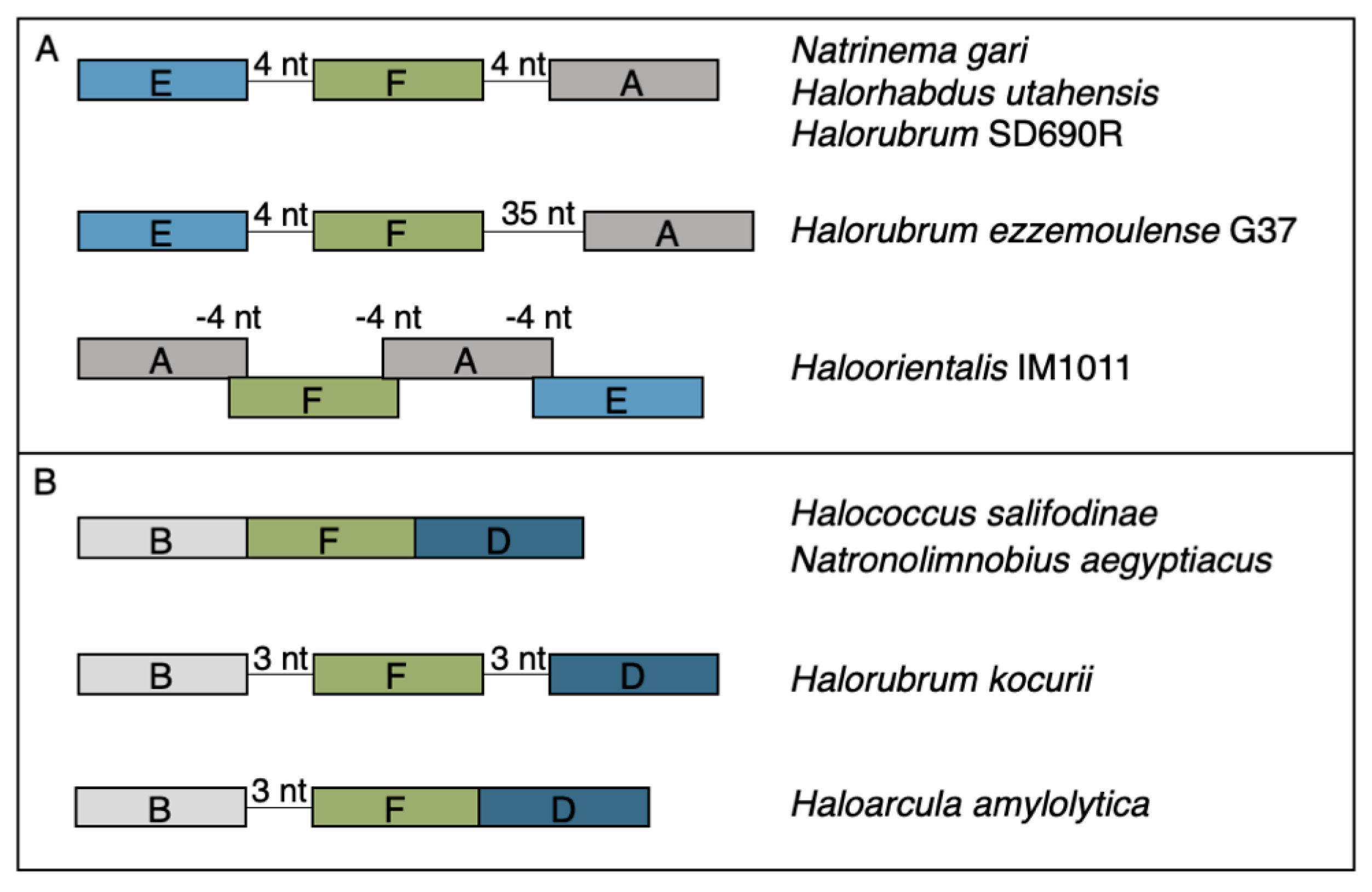
3.2. Horizontal Gene Transfer Explains Patchy Distribution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Korlach, J.; Turner, S.W. Going beyond five bases in DNA sequencing. Curr. Opin. Struct. Biol. 2012, 22, 251–261. [Google Scholar] [CrossRef]
- Bheemanaik, S.; Reddy, Y.V.R.; Rao, D.N. Structure, function and mechanism of exocyclic DNA methyltransferases. Biochem. J. 2006, 399, 177–190. [Google Scholar] [CrossRef]
- Malone, T.; Blumenthal, R.M.; Cheng, X. Structure-guided analysis reveals nine sequence motifs conserved among DNA mmino-methyl-transferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995, 253, 618–632. [Google Scholar] [CrossRef]
- Bujnicki, J.M.; Radlinska, M. Molecular evolution of DNA-(cytosine-N4) methyltransferases: evidence for their polyphyletic origin. Nucleic Acids Res. 1999, 27, 4501–4509. [Google Scholar] [CrossRef]
- Bujnicki, J.M. Sequence permutations in the molecular evolution of DNA methyltransferases. BMC Evol. Biol. 2002, 2, 3. [Google Scholar] [CrossRef]
- Tock, M.R.; Dryden, D.T. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005, 8, 466–472. [Google Scholar] [CrossRef]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Pleška, M.; Qian, L.; Okura, R.; Bergmiller, T.; Wakamoto, Y.; Kussell, E.; Guet, C.C. Bacterial autoimmunity due to a restriction-modification system. Curr. Biol. CB 2016, 26, 404–409. [Google Scholar] [CrossRef]
- Ohno, S.; Handa, N.; Watanabe-Matsui, M.; Takahashi, N.; Kobayashi, I. Maintenance forced by a restriction-modification system can be modulated by a region in its modification enzyme not essential for methyltransferase activity. J. Bacteriol. 2008, 190, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001, 29, 3742–3756. [Google Scholar] [CrossRef]
- Budroni, S.; Siena, E.; Hotopp, J.C.D.; Seib, K.L.; Serruto, D.; Nofroni, C.; Comanducci, M.; Riley, D.R.; Daugherty, S.C.; Angiuoli, S.V.; Covacci, A.; Pizza, M.; Rappuoli, R.; Moxon, E.R.; Tettelin, H.; Medini, D. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc. Natl. Acad. Sci. USA 2011, 108, 4494–4499. [Google Scholar] [CrossRef]
- Erwin, A.L.; Sandstedt, S.A.; Bonthuis, P.J.; Geelhood, J.L.; Nelson, K.L.; Unrath, W.C.T.; Diggle, M.A.; Theodore, M.J.; Pleatman, C.R.; Mothershed, E.A.; Sacchi, C.T.; Mayer, L.W.; Gilsdorf, J.R.; Smith, A.L. Analysis of genetic relatedness of Haemophilus influenzae isolates by multilocus sequence typing. J. Bacteriol. 2008, 190, 1473–1483. [Google Scholar] [CrossRef]
- Roer, L.; Aarestrup, F.M.; Hasman, H. The EcoKI type I restriction-modification system in Escherichia coli affects but Is not an absolute barrier for conjugation. J. Bacteriol. 2015, 197, 337–342. [Google Scholar] [CrossRef]
- McKane, M.; Milkman, R. Transduction, restriction and recombination patterns in Escherichia coli. Genetics 1995, 139, 35–43. [Google Scholar] [PubMed]
- Chang, S.; Cohen, S.N. In vivo site-specific genetic recombination promoted by the EcoRI restriction endonuclease. Proc. Natl. Acad. Sci. USA 1977, 74, 4811–4815. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.A.; Zhang, X.-S.; Levine, S.M.; Gill, S.R.; Falush, D.; Blaser, M.J. Natural transformation of Helicobacter pylori involves the Iintegration of short DNA fragments interrupted by gaps of variable size. PLoS Pathog. 2009, 5, e1000337. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.J. The relation of recombination to mutational advance. Mutat. Res. 1964, 1, 2–9. [Google Scholar] [CrossRef]
- Ershova, A.S.; Rusinov, I.S.; Spirin, S.A.; Karyagina, A.S.; Alexeevski, A.V. Role of restriction-modification systems in prokaryotic evolution and ecology. Biochemistry 2015, 80, 1373–1386. [Google Scholar] [CrossRef]
- Roberts, R.J.; Belfort, M.; Bestor, T.; Bhagwat, A.S.; Bickle, T.A.; Bitinaite, J.; Blumenthal, R.M.; Degtyarev, S.K.; Dryden, D.T.F.; Dybvig, K.; et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003, 31, 1805–1812. [Google Scholar] [CrossRef]
- Loenen, W.A.M.; Dryden, D.T.F.; Raleigh, E.A.; Wilson, G.G. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014, 42, 20–44. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Tang, Q.; Zhang, J.-Z.; Tian, L.-F.; Gao, P.; Yan, X.-X. Structural basis underlying complex assembly and conformational transition of the type I R-M system. Proc. Natl. Acad. Sci. USA 2017, 114, 11151–11156. [Google Scholar] [CrossRef] [PubMed]
- Pingoud, A.; Wilson, G.G.; Wende, W. Type II restriction endonucleases—A historical perspective and more. Nucleic Acids Res. 2014, 42, 7489–7527. [Google Scholar] [CrossRef]
- Morgan, R.D.; Bhatia, T.K.; Lovasco, L.; Davis, T.B. MmeI: A minimal Type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res. 2008, 36, 6558–6570. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.N.; Dryden, D.T.F.; Bheemanaik, S. Type III restriction-modification enzymes: A historical perspective. Nucleic Acids Res. 2014, 42, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Czapinska, H.; Kowalska, M.; Zagorskaitė, E.; Manakova, E.; Slyvka, A.; Xu, S.; Siksnys, V.; Sasnauskas, G.; Bochtler, M. Activity and structure of EcoKMcrA. Nucleic Acids Res. 2018, 46, 9829–9841. [Google Scholar] [CrossRef]
- Adhikari, S.; Curtis, P.D. DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiol. Rev. 2016, 40, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Messer, W.; Bellekes, U.; Lother, H. Effect of dam methylation on the activity of the E. coli replication origin, oriC. EMBO J. 1985, 4, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, H.; Han, J.S.; Hwang, D.S. Interaction of SeqA and Dam methylase on the hemimethylated origin of Escherichia coli chromosomal DNA replication. J. Biol. Chem. 1999, 274, 11463–11468. [Google Scholar] [CrossRef]
- Sanchez-Romero, M.A.; Busby, S.J.W.; Dyer, N.P.; Ott, S.; Millard, A.D.; Grainger, D.C. Dynamic distribution of SeqA protein across the chromosome of Escherichia coli K-12. MBio 2010, 1. [Google Scholar] [CrossRef]
- Welsh, K.M.; Lu, A.L.; Clark, S.; Modrich, P. Isolation and characterization of the Escherichia coli mutH gene product. J. Biol. Chem. 1987, 262, 15624–15629. [Google Scholar] [PubMed]
- Au, K.G.; Welsh, K.; Modrich, P. Initiation of methyl-directed mismatch repair. J. Biol. Chem. 1992, 267, 12142–12148. [Google Scholar]
- Putnam, C.D. Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair (Amst). 2016, 38, 32–41. [Google Scholar] [CrossRef]
- Zweiger, G.; Marczynski, G.; Shapiro, L. A Caulobacter DNA methyltransferase that functions only in the predivisional cell. J. Mol. Biol. 1994, 235, 472–485. [Google Scholar] [CrossRef]
- Domian, I.J.; Reisenauer, A.; Shapiro, L. Feedback control of a master bacterial cell-cycle regulator. Proc. Natl. Acad. Sci. USA 1999, 96, 6648–6653. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Kozdon, J.B.; McAdams, H.H.; Shapiro, L.; Collier, J. The functions of DNA methylation by CcrM in Caulobacter crescentus: A global approach. Nucleic Acids Res. 2014, 42, 3720–3735. [Google Scholar] [CrossRef]
- Seshasayee, A.S.N.; Singh, P.; Krishna, S. Context-dependent conservation of DNA methyltransferases in bacteria. Nucleic Acids Res. 2012, 40, 7066–7073. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The epigenomic landscape of prokaryotes. PLOS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed]
- Nölling, J.; de Vos, W.M. Identification of the CTAG-recognizing restriction-modification systems MthZI and MthFI from Methanobacterium thermoformicicum and characterization of the plasmid-encoded mthZIM gene. Nucleic Acids Res. 1992, 20, 5047–5052. [Google Scholar] [CrossRef] [PubMed]
- Grogan, D.W. Cytosine methylation by the SuaI restriction-modification system: Implications for genetic fidelity in a hyperthermophilic archaeon. J. Bacterio. 2003, 185, 4657–4661. [Google Scholar] [CrossRef]
- Ishikawa, K.; Watanabe, M.; Kuroita, T.; Uchiyama, I.; Bujnicki, J.M.; Kawakami, B.; Tanokura, M.; Kobayashi, I. Discovery of a novel restriction endonuclease by genome comparison and application of a wheat-germ-based cell-free translation assay: PabI (5’-GTA/C) from the hyperthermophilic archaeon Pyrococcus abyssi. Nucleic Acids Res. 2005, 33, e112. [Google Scholar] [CrossRef]
- Watanabe, M.; Yuzawa, H.; Handa, N.; Kobayashi, I. Hyperthermophilic DNA methyltransferase M.PabI from the archaeon Pyrococcus abyssi. Appl. Environ. Microbiol. 2006, 72, 5367–5375. [Google Scholar] [CrossRef] [PubMed]
- Couturier, M.; Lindås, A.-C. The DNA methylome of the hyperthermoacidophilic crenarchaeon Sulfolobus acidocaldarius. Front. Microbiol. 2018, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Chimileski, S.; Dolas, K.; Naor, A.; Gophna, U.; Papke, R.T. Extracellular DNA metabolism in Haloferax volcanii. Front. Microbiol. 2014, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Zerulla, K.; Chimileski, S.; Näther, D.; Gophna, U.; Papke, R.T.; Soppa, J. DNA as a phosphate storage polymer and the alternative advantages of polyploidy for growth or survival. PLoS ONE 2014, 9, e94819. [Google Scholar] [CrossRef]
- Ouellette, M.; Gogarten, J.; Lajoie, J.; Makkay, A.; Papke, R. Characterizing the DNA methyltransferases of Haloferax volcanii via bioinformatics, gene deletion, and SMRT sequencing. Genes 2018, 9, 129. [Google Scholar] [CrossRef]
- Ouellette, M.; Jackson, L.; Chimileski, S.; Papke, R.T. Genome-wide DNA methylation analysis of Haloferax volcanii H26 and identification of DNA methyltransferase related PD-(D/E)XK nuclease family protein HVO_A0006. Front. Microbiol. 2015, 6, 251. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, gr-186072. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Macelis, D. REBASE—Restriction enzymes and methylases. Nucleic Acids Res. 2001, 29, 268–269. [Google Scholar] [CrossRef]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- HMMER. Available online: http://hmmer.org/ (accessed on 18 March 2019).
- Analyses for RMS paper. Available online: https://github.com/Gogarten-Lab/rms_analysis.
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Archaeal clusters of orthologous genes (arCOGs): An update and application for analysis of shared features between Thermococcales, Methanococcales, and Methanobacteriales. Life (Basel, Switzerland) 2015, 5, 818–840. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695. [Google Scholar]
- Caspi, R.; Altman, T.; Dale, J.M.; Dreher, K.; Fulcher, C.A.; Gilham, F.; Kaipa, P.; Karthikeyan, A.S.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; Paley, S.; Popescu, L.; Pujar, A.; Shearer, A.G.; Zhang, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2010, 38, D473–D479. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Le, S.V. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.S.; Wu, Y.-C.; Alm, E.J.; Kellis, M. Improved gene tree error correction in the presence of horizontal gene transfer. Bioinformatics 2015, 31, 1211–1218. [Google Scholar] [CrossRef]
- Bansal, M.S.; Kellis, M.; Kordi, M.; Kundu, S. RANGER-DTL 2.0: Rigorous reconstruction of gene-family evolution by duplication, transfer and loss. Bioinformatics 2018, 34, 3214–3216. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. GGTREE: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Yu, G.; Lam, T.T.-Y.; Zhu, H.; Guan, Y. Two methods formapping and visualizing associated data on phylogeny using ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, 2016; ISBN 3319242776. [Google Scholar]
- Hmisc - Main - Vanderbilt Biostatistics Wiki Available online:. Available online: http://biostat.mc.vanderbilt.edu/wiki/Main/Hmisc (accessed on 18 March 2019).
- Schliep, K.P. phangorn: phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef]
- Salichos, L.; Rokas, A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 2013, 497, 327–331. [Google Scholar] [CrossRef]
- Drost, H. Philentropy: Information Theory and Distance Quantification with R. J. Open Source Softw. 2018, 3. [Google Scholar] [CrossRef]
- Gogarten, J.P. Perl script to measure frequency and distribution of CTAG and GATC motifs in DNA Available online:. Available online: https://github.com/Gogarten-Lab/CTAG-GATC-frequencies (accessed on 12 January 2019).
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2016, 45, D158–D169. [Google Scholar]
- Soucy, S.M.; Fullmer, M.S.; Papke, R.T.; Gogarten, J.P. Inteins as indicators of gene flow in the halobacteria. Front. Microbiol. 2014, 5, 1–14. [Google Scholar] [CrossRef]
- Gupta, R.S.; Naushad, S.; Fabros, R.; Adeolu, M. A phylogenomic reappraisal of family-level divisions within the class Halobacteria: Proposal to divide the order Halobacteriales into the families Halobacteriaceae, Haloarculaceae fam. nov., and Halococcaceae fam. nov., and the order Haloferacales into th. Antonie Van Leeuwenhoek 2016, 109, 565–587. [Google Scholar] [CrossRef]
- Gupta, R.S.; Naushad, S.; Baker, S. Phylogenomic analyses and molecular signatures for the class Halobacteria and its two major clades: A proposal for division of the class Halobacteria into an emended order Halobacteriales and two new orders, Haloferacales ord. nov. and Natrialbales ord. n. Int. J. Syst. Evol. Microbiol. 2015, 65, 1050–1069. [Google Scholar] [CrossRef] [PubMed]
- Allers, T.; Barak, S.; Liddell, S.; Wardell, K.; Mevarech, M. Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl. Environ. Microbiol. 2010, 76, 1759–1769. [Google Scholar] [CrossRef]
- Holmes, M.L.; Nuttall, S.D.; Dyall-Smith, M.L. Construction and use of halobacterial shuttle vectors and further studies on Haloferax DNA gyrase. J. Bacteriol. 1991, 173, 3807–3813. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Ma, J.H.; Warren, K.; Tsang, R.S.W.; Low, D.E.; Jamieson, F.B.; Alexander, D.C.; Hao, W. Homologous recombination drives both sequence diversity and gene content variation in Neisseria meningitidis. Genome Biol. Evol. 2013, 5, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Namba-Fukuyo, H.; Shibata, T.F.; Nishiyama, T.; Shigenobu, S.; Suzuki, Y.; Sugano, S.; Hasebe, M.; Kobayashi, I. Methylome diversification through changes in DNA methyltransferase sequence specificity. PLoS Genet. 2014, 10, e1004272. [Google Scholar] [CrossRef]
- Furuta, Y.; Kobayashi, I. Mobility of DNA sequence recognition domains in DNA methyltransferases suggests epigenetics-driven adaptive evolution. Mob. Genet. Elements 2012, 2, 292–296. [Google Scholar] [CrossRef]
- Furuta, Y.; Kawai, M.; Uchiyama, I.; Kobayashi, I. Domain movement within a gene: A novel evolutionary mechanism for protein diversification. PLoS ONE 2011, 6, e18819. [Google Scholar] [CrossRef]
- Gophna, U.; Brodt, A. CRISPR/Cas systems in archaea. Mob. Genet. Elements 2012, 2, 63–64. [Google Scholar] [CrossRef]
- Fullmer, M.S.; Soucy, S.M.; Swithers, K.S.; Makkay, A.M.; Wheeler, R.; Ventosa, A.; Gogarten, J.P.; Papke, R.T. Population and genomic analysis of the genus Halorubrum. Front. Microbiol. 2014, 5, 140. [Google Scholar] [CrossRef] [PubMed]
- Shalev, Y.; Soucy, S.; Papke, R.; Gogarten, J.; Eichler, J.; Gophna, U. Comparative analysis of surface layer glycoproteins and genes involved in protein glycosylation in the genus Haloferax. Genes 2018, 9, 172. [Google Scholar] [CrossRef]
- Kandiba, L.; Aitio, O.; Helin, J.; Guan, Z.; Permi, P.; Bamford, D.H.; Eichler, J.; Roine, E. Diversity in prokaryotic glycosylation: an archaeal-derived N-linked glycan contains legionaminic acid. Mol. Microbiol. 2012, 84, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.-B.; Rodriguez-Brito, B.; Pašić, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009, 7, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-L.; Nuttall, S.; Ngui, K.; Fisher, C.; Lopez, P.; Dyall-Smith, M. HF2: A double-stranded DNA tailed haloarchaeal virus with a mosaic genome. Mol. Microbiol. 2002, 44, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Olendzenski, L.; Gogarten, J.P. Evolution of genes and organisms: The tree/web of life in light of horizontal gene transfer. Ann. N. Y. Acad. Sci. 2009, 1178, 137–145. [Google Scholar] [CrossRef]
- Naor, A.; Altman-Price, N.; Soucy, S.M.; Green, A.G.; Mitiagin, Y.; Turgeman-Grott, I.; Davidovich, N.; Gogarten, J.P.; Gophna, U. Impact of a homing intein on recombination frequency and organismal fitness. Proc. Natl. Acad. Sci. USA 2016, 113, E4654-61. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P.C. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Mrázek, J.; Campbell, A.M. Compositional biases of bacterial genomes and evolutionary implications. J. Bacteriol. 1997, 179, 3899–3913. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.; Paulus, F.; Otten, L. IS870 requires a 5’-CTAG-3’ target sequence to generate the stop codon for its large ORF1. J. Bacteriol. 1993, 175, 3151–3160. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Schevitz, R.W.; Zhang, R.-G.; Lawson, C.L.; Joachimiak, A.; Marmorstein, R.Q.; Luisi, B.F.; Sigler, P.B. Crystal structure of trp represser/operator complex at atomic resolution. Nature 1988, 335, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Campbell, A.M.; Karlin, S. Over- and under-representation of short oligonucleotides in DNA sequences. Proc. Natl. Acad. Sci. USA 1992, 89, 1358–1362. [Google Scholar] [CrossRef]
- Bath, C.; Cukalac, T.; Porter, K.; Dyall-Smith, M.L. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology 2006, 350, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.; Tang, S.-L.; Chen, C.-P.; Chiang, P.-W.; Hong, M.-J.; Dyall-Smith, M. PH1: An archaeovirus of Haloarcula hispanica related to SH1 and HHIV-2. Archaea 2013, 2013, 456318. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alpha Code | Numerical Code | Annotated arCOG Function $$ | arCOG Number |
|---|---|---|---|
| A | cHG_021 | T_I_M | arCOG02632 |
| B | cHG_024 | T_I_M | arCOG05282 |
| C | cHG_018 | T_I_R | arCOG00880 |
| D | cHG_034 | T_I_R | arCOG00879 |
| E | cHG_045 | T_I_R | arCOG00878 |
| F | cHG_006 | T_I_S | arCOG02626 |
| G | cHG_025 | T_I_S | arCOG02628 |
| H | cHG_036 | probable_T_II_M | arCOG00890 |
| I | cHG_001 | T_II_M | arCOG02635 |
| J | cHG_003 | T_II_M | arCOG02634 |
| K | cHG_011 | T_II_M | arCOG04814 |
| L | cHG_033 | T_II_M | arCOG03521 |
| M | cHG_007 | T_II_R | arCOG11279 |
| N | cHG_013 | T_II_R | arCOG11717 |
| O | cHG_023 | T_II_R | arCOG03779 |
| P | cHG_029 | T_II_R | arCOG08993 |
| Q | cHG_042 | Adenine_DNA_methylase_probable_T_III_M | arCOG00108 |
| R | cHG_008 | T_III_R | arCOG06887 |
| S | cHG_009 | T_III_R_probable | arCOG07494 |
| T | cHG_014 | Adenine_DNA_methylase | arCOG00889 |
| U | cHG_022 | DNA_methylase | arCOG00115 |
| V | cHG_027 | DNA_methylase | arCOG00129 |
| W | cHG_031 | dam_methylase | arCOG03416 |
| X | cHG_035 | probable_RMS_M | arCOG08990 |
| Y | cHG_044 | dcm_methylase | arCOG04157 |
| Z | cHG_048 | Adenine_DNA_methylase | arCOG02636 |
| AA | cHG_010 | RNA_methylase | arCOG00910 |
| AB | cHG_040 | SAM-methylase | arCOG01792 |
| AC | cHG_012 | RestrictionEndonuclease | arCOG05724 |
| AD | cHG_038 | PredictedRestrictionEndonuclease | arCOG06431 |
| AE | cHG_015 | HNH_endonuclease | arCOG07787 |
| AF | cHG_019 | Endonuclease | arCOG02782 |
| AG | cHG_020 | Endonuclease | arCOG02781 |
| AH | cHG_004 | HNH_endonuclease | arCOG09398 |
| AI | cHG_037 | HNH_nuclease | arCOG05223 |
| AJ | cHG_039 | HNH_nuclease | arCOG03898 |
| AK | cHG_041 | HNH_nuclease | arCOG08099 |
| AL | cHG_046 | MBF1 | arCOG01863 |
| AM | cHG_028 | CBS_domain | arCOG00608 |
| AN | cHG_005 | MarR | arCOG03182 |
| AO | cHG_030 | ParB-like nuclease | arCOG01875 |
| AP | cHG_016 | GVPC | arCOG06392 |
| AQ | cHG_002 | ASCH domain RNA binding | arCOG01734 |
| AR | cHG_017 | Uncharacterized | arCOG10082 |
| AS | cHG_026 | Uncharacterized | arCOG13171 |
| AT | cHG_032 | Uncharacterized | arCOG08946 |
| AU | cHG_043 | Uncharacterized | arCOG08856 |
| AV | cHG_047 | Uncharacterized | arCOG04588 |
| Alpha (Numeric) cHG | No. of Taxa | No. of Transfers a | Function b | Predicted Recognition Sites c | Frequency e |
|---|---|---|---|---|---|
| I (001) | 16 | 9 | T_II_M | GAAGGC | 31% |
| GGRCA | 31% | ||||
| J (003) | 38 | 21 | T_II_M | CANCATC | 53% |
| TAGGAG | 21% | ||||
| AH (004) | 12 | 4 | HNH_endonuclease | GGCGCC | 89% |
| GATC | 11% | ||||
| F (006) | 61 | 44 | T_I_S | GGAYNNNNNNTGG | 24% |
| CAGNNNNNNTGCT | 16% | ||||
| R (008) | 14 | 0 | T_III_R | NA d | 100% |
| AA (010) | 55 | 15 | RNA_methylase | ATTAAT | 33% |
| K (011) | 137 | 97 | T_II_M | GCAAGG | 49% |
| GKAAYG | 28% | ||||
| AC (012) | 8 | 5 | Restriction Endonuclease | GCGAA | 29% |
| CAACNNNNNTC | 29% | ||||
| CTGGAG | 29% | ||||
| T (014) | 130 | 93 | Adenine_DNA_methylase | GCAGG | 45% |
| AAGCTT | 32% | ||||
| AE (015) | 21 | 13 | HNH_endonuclease | GGCGCC | 70% |
| YSCNS | 15% | ||||
| AP (016) | 12 | 6 | GVPC | CANCATC | 83% |
| C (018) | 7 | 4 | T_I_R | AACNNNNNNGTGC | 73% |
| CTANNNNNNRTTC | 27% | ||||
| AF (019) | 4 | 3 | Endonuclease | NAd | 100% |
| A (021) | 88 | 58 | T_I_M | GGAYNNNNNNTGG | 37% |
| GTCANNNNNNRTCA | 12% | ||||
| CTCGAG | 9% | ||||
| U (022) | 290 | 120 | DNA_methylase | CTAG | 59% |
| CATTC | 14% | ||||
| CCCGGG | 7% | ||||
| O (023) | 37 | 28 | T_II_R | NAd | 100% |
| B (024) | 16 | 8 | T_I_M | GAGNNNNNNVTGAC | 75% |
| GACNNNNNNRTAC | 19% | ||||
| G (025) | 4 | 2 | T_I_S | GAGNNNNRTAA | 75% |
| GAGNNNNNTAC | 25% | ||||
| V (027) | 5 | 1 | DNA_methylase | CATTC | 100% |
| AO (030) | 4 | 2 | ParB-like_nuclease | GATC | 75% |
| CTAG | 25% | ||||
| W (031) | 153 | 70 | dam_methylase | GATC | 70% |
| AB/SAAM | 22% | ||||
| AT (032) | 116 | 60 | Uncharacterized | GCAAGG | 43% |
| GKAAYG | 26% | ||||
| GGTTAG | 14% | ||||
| L (033) | 66 | 38 | T_II_M-033 | CAARCA | 40% |
| CTGAAG | 36% | ||||
| D (034) | 16 | 11 | T_I_R-034 | GCANNNNNRTTA | 69% |
| GGCANNNNNNTTC | 19% | ||||
| X (035) | 19 | 9 | probable_RMS_M | GGGAC | 83% |
| H (036) | 38 | 24 | probable_T_II_M | CCWGG | 42% |
| CCSGG | 18% | ||||
| GTAC | 16% | ||||
| AI (037) | 6 | 4 | HNH_nuclease | NA d | 100% |
| AJ (039) | 5 | 4 | HNH_nuclease | GGCGCC | 100% |
| AK (041) | 6 | 4 | HNH_nuclease | NA d | 100% |
| Q (042) | 21 | 8 | Adenine_DNA_methylase probable_T_III_M | RGTAAT | 71% |
| NA d | 19% | ||||
| Y (044) | 179 | 110 | dcm_methylase | CGGCCG | 24% |
| GTCGAC | 13% | ||||
| ACGT | 11% | ||||
| E (045) | 58 | 42 | T_I_R | CCCNNNNNRTTGY | 63% |
| GCANNNNNRTTA | 28% | ||||
| Z (048) | 54 | 35 | Adenine_DNA_methylase | CCRGAG | 36% |
| GTMKAC | 30% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fullmer, M.S.; Ouellette, M.; Louyakis, A.S.; Papke, R.T.; Gogarten, J.P. The Patchy Distribution of Restriction–Modification System Genes and the Conservation of Orphan Methyltransferases in Halobacteria. Genes 2019, 10, 233. https://doi.org/10.3390/genes10030233
Fullmer MS, Ouellette M, Louyakis AS, Papke RT, Gogarten JP. The Patchy Distribution of Restriction–Modification System Genes and the Conservation of Orphan Methyltransferases in Halobacteria. Genes. 2019; 10(3):233. https://doi.org/10.3390/genes10030233
Chicago/Turabian StyleFullmer, Matthew S., Matthew Ouellette, Artemis S. Louyakis, R. Thane Papke, and Johann Peter Gogarten. 2019. "The Patchy Distribution of Restriction–Modification System Genes and the Conservation of Orphan Methyltransferases in Halobacteria" Genes 10, no. 3: 233. https://doi.org/10.3390/genes10030233