Structure-Specific Endonucleases and the Resolution of Chromosome Underreplication


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Intrinsic Safeguards Against Chromosomal Underreplication
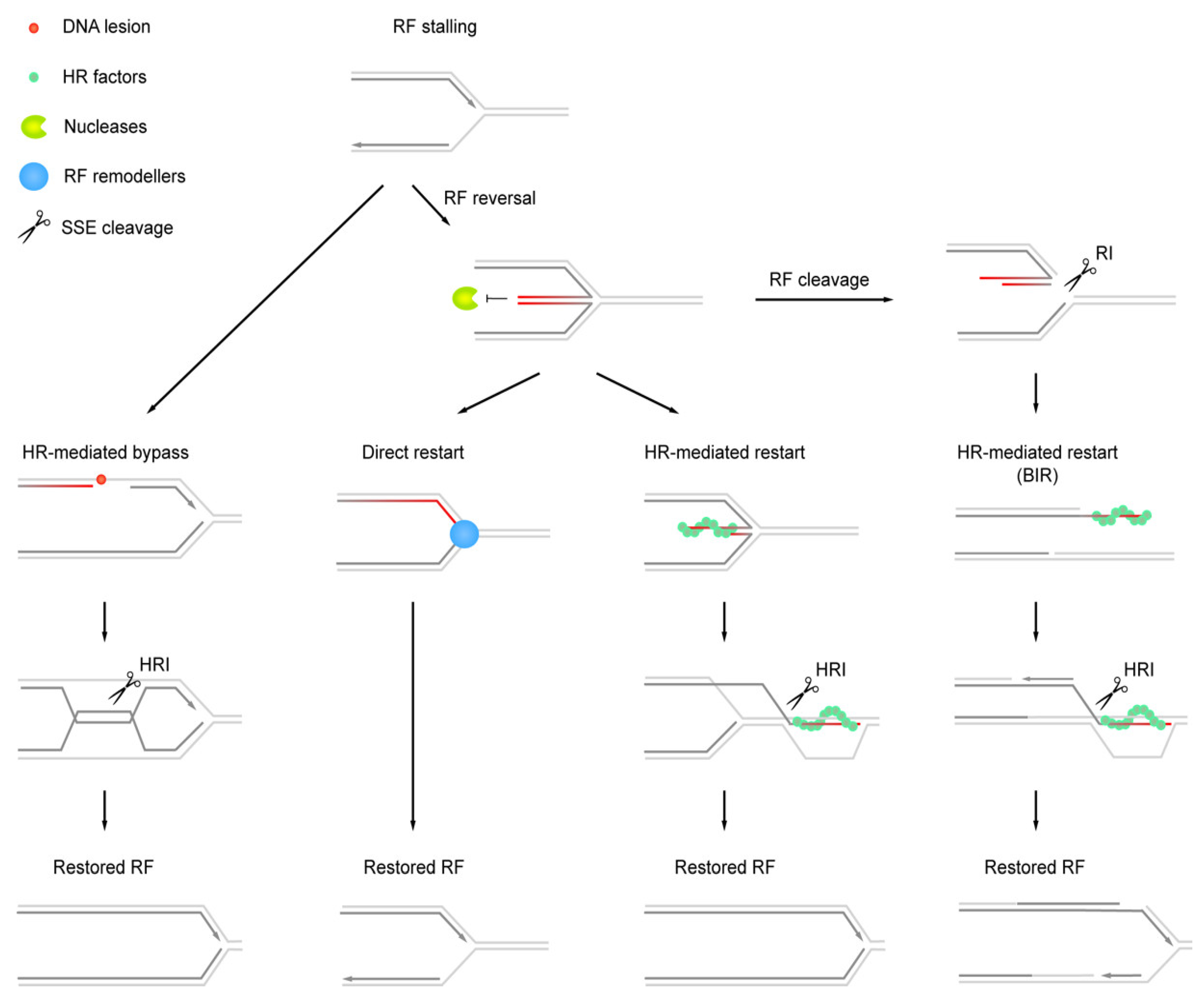
3. Preventing Underreplication in the Face of Replication Stress
4. Structure-Specific Endonucleases and Their Roles in Protecting Cells from Chromosomal Underreplication
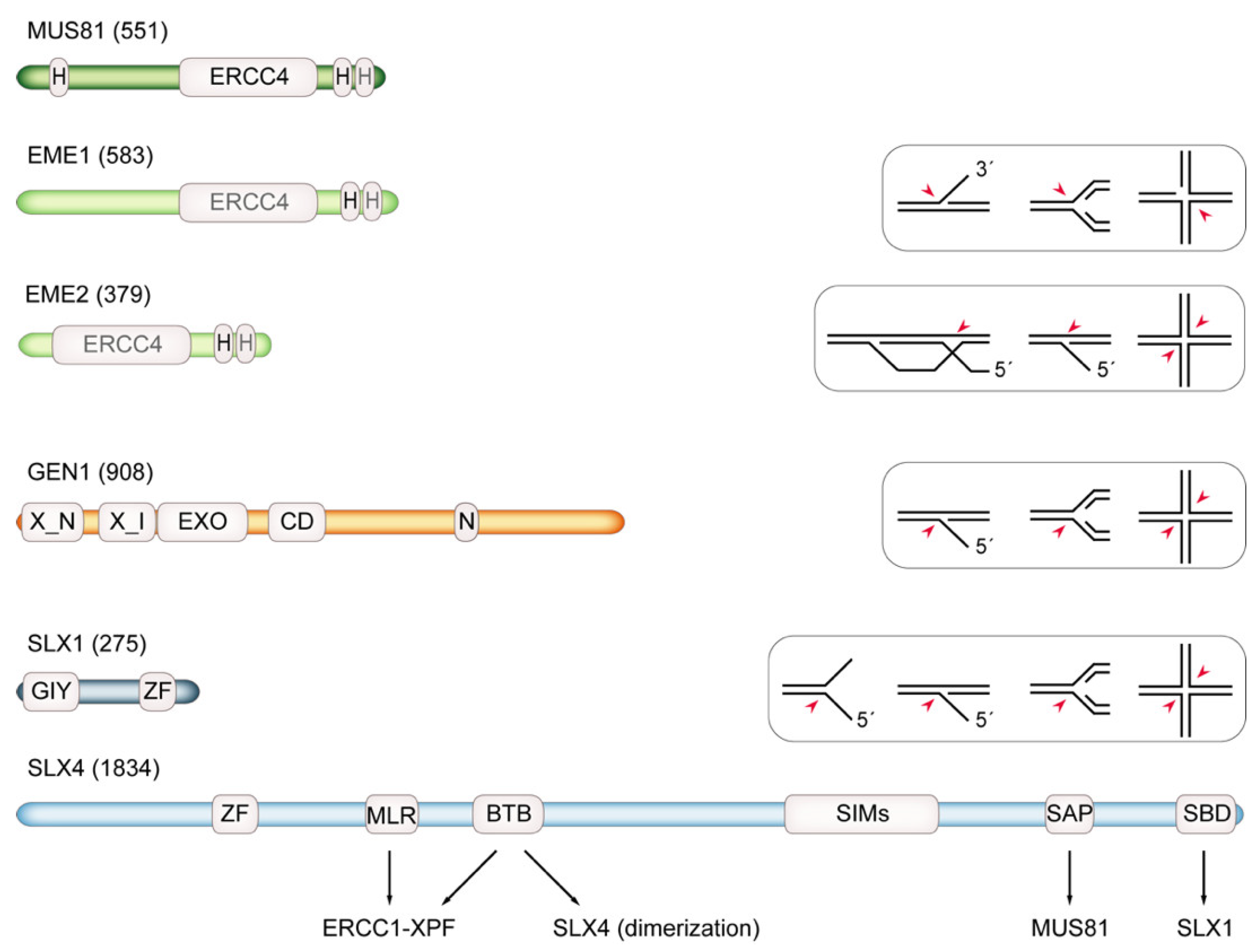
5. Structure-Specific Endonucleases: Substrate Spectrum and Cell-Cycle Regulation
5.1. MUS81
5.2. Slx1-Slx4/SLX1-SLX4
5.3. The SMX Tri-Nuclease Complex
5.4. Yen1/GEN1
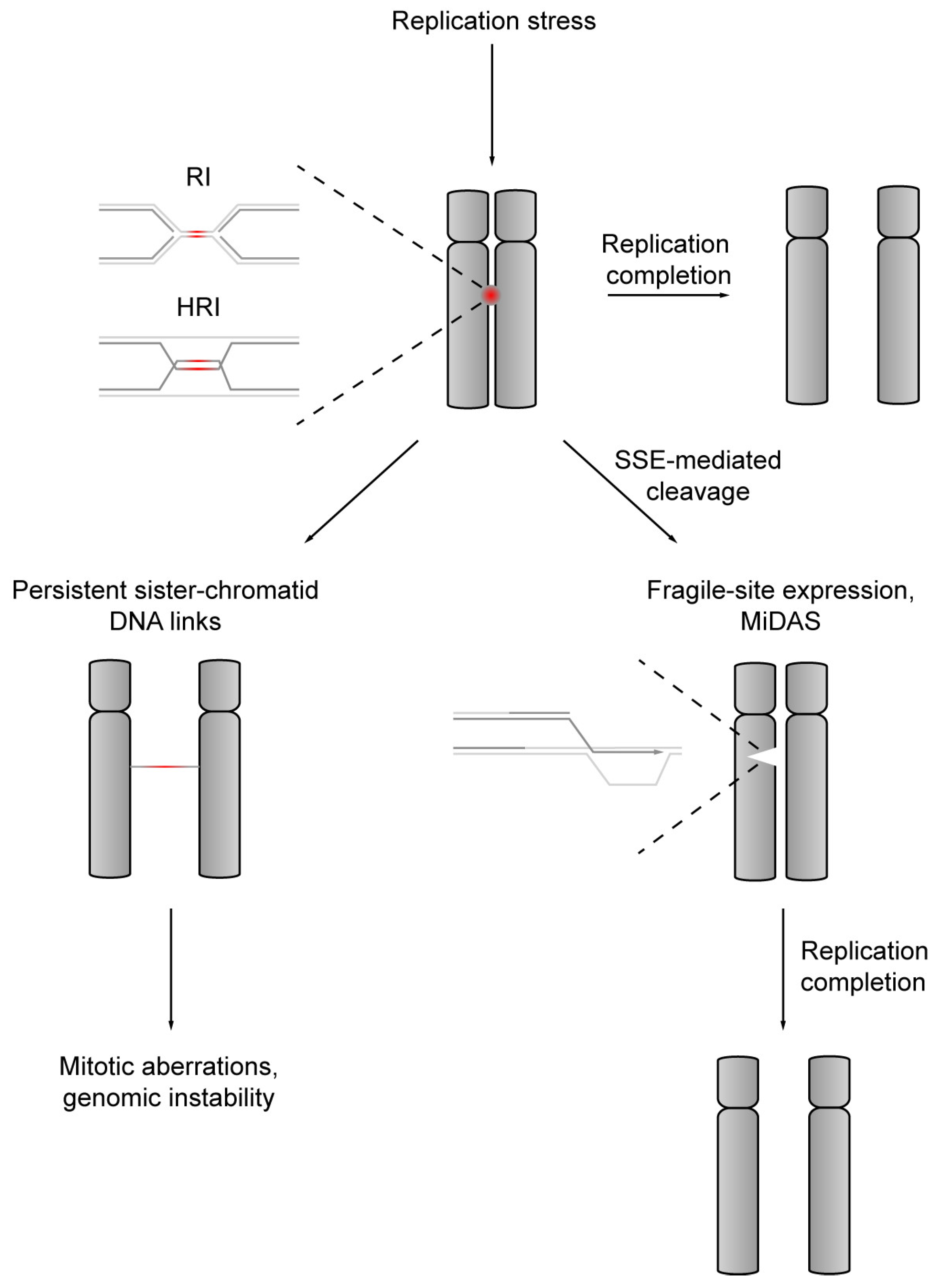
6. Holliday Junction Resolution by Structure-Specific Endonucleases Facilitates Chromosome Segregation
7. Structure-Specific Endonucleases Cleave DNA Replication Intermediates to Promote Cell Viability
8. Structure-Specific Endonuclease-Mediated Cleavage of DNA Replication Intermediates Initiates DNA Repair Synthesis in Mitosis
9. Structure-Specific Endonuclease Targets Arising at Stalled Replication Forks
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holliday, R. A mechanism for gene conversion in fungi. Genet. Res. 1964, 5, 282–304. [Google Scholar] [CrossRef]
- Liu, Y.; West, S.C. Happy Hollidays: 40th anniversary of the Holliday junction. Nat. Rev. Mol. Cell Biol. 2004, 5, 937–944. [Google Scholar] [CrossRef]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, H.D.M.; West, S.C. Holliday junction resolvases. Cold Spring Harb. Perspect. Biol. 2014, 6, a023192. [Google Scholar] [CrossRef] [PubMed]
- Rass, U. Resolving branched DNA intermediates with structure-specific nucleases during replication in eukaryotes. Chromosoma 2013, 122, 499–515. [Google Scholar] [CrossRef]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replicating large genomes: Divide and conquer. Mol. Cell 2016, 62, 756–765. [Google Scholar] [CrossRef]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef]
- Newman, T.J.; Mamun, M.A.; Nieduszynski, C.A.; Blow, J.J. Replisome stall events have shaped the distribution of replication origins in the genomes of yeasts. Nucleic Acids Res. 2013, 41, 9705–9718. [Google Scholar] [CrossRef]
- Kawabata, T.; Luebben, S.W.; Yamaguchi, S.; Ilves, I.; Matise, I.; Buske, T.; Botchan, M.R.; Shima, N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell 2011, 41, 543–553. [Google Scholar] [CrossRef]
- Bellelli, R.; Borel, V.; Logan, C.; Svendsen, J.; Cox, D.E.; Nye, E.; Metcalfe, K.; O’Connell, S.M.; Stamp, G.; Flynn, H.R.; et al. Polε instability drives replication stress, abnormal development, and tumorigenesis. Mol. Cell 2018, 70, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef]
- Woodward, A.M.; Göhler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Mendez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- Kunnev, D.; Rusiniak, M.E.; Kudla, A.; Freeland, A.; Cady, G.K.; Pruitt, S.C. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 2010, 29, 3630–3638. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Petermann, E.; Gillespie, D.A.F.; Caldecott, K.W.; Jackson, D.A. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007, 26, 2719–2731. [Google Scholar] [CrossRef]
- Syljuåsen, R.G.; Sørensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Christensen, P.U.; Bentley, N.J.; Martinho, R.G.; Nielsen, O.; Carr, A.M. Mik1 levels accumulate in S phase and may mediate an intrinsic link between S phase and mitosis. Proc. Natl. Acad. Sci. USA 2000, 97, 2579–2584. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA replication determines timing of mitosis by restricting CDK1 and PLK1 activation. Mol. Cell 2018, 71, 117–128. [Google Scholar] [CrossRef]
- Sugimoto, K.; Shimomura, T.; Hashimoto, K.; Araki, H.; Sugino, A.; Matsumoto, K. Rfc5, a small subunit of replication factor C complex, couples DNA replication and mitosis in budding yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 7048–7052. [Google Scholar] [CrossRef]
- Hofmann, J.F.; Beach, D. Cdt1 is an essential target of the Cdc10/Sct1 transcription factor: Requirement for DNA replication and inhibition of mitosis. EMBO J. 1994, 13, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Piatti, S.; Lengauer, C.; Nasmyth, K. Cdc6 is an unstable protein whose de novo synthesis in G1 is important for the onset of S phase and for preventing a “reductional” anaphase in the budding yeast Saccharomyces cerevisiae. EMBO J. 1995, 14, 3788–3799. [Google Scholar] [CrossRef]
- Bastos de Oliveira, F.M.; Kim, D.; Cussiol, J.R.; Das, J.; Jeong, M.C.; Doerfler, L.; Schmidt, K.H.; Yu, H.; Smolka, M.B. Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol. Cell 2015, 57, 1124–1132. [Google Scholar] [CrossRef]
- Eykelenboom, J.K.; Harte, E.C.; Canavan, L.; Pastor-Peidro, A.; Calvo-Asensio, I.; Llorens-Agost, M.; Lowndes, N.F. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 2013, 5, 1095–1107. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef]
- Iyer, D.R.; Rhind, N. The intra-S checkpoint responses to DNA damage. Genes 2017, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Cha, R.S.; Kleckner, N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 2002, 297, 602–606. [Google Scholar] [CrossRef]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef]
- Shastri, N.; Tsai, Y.-C.; Hile, S.; Jordan, D.; Powell, B.; Chen, J.; Maloney, D.; Dose, M.; Lo, Y.; Anastassiadis, T.; et al. Genome-wide identification of structure-forming repeats as principal sites of fork collapse upon ATR inhibition. Mol. Cell 2018, 72, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Sridharan, S.; van Wietmarschen, N.; Maman, Y.; Callén, E.; Stanlie, A.; Wu, W.; Wu, X.; Day, A.; Wong, N.; et al. Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell 2018, 174, 1127–1142. [Google Scholar] [CrossRef]
- Tercero, J.A.; Longhese, M.P.; Diffley, J.F.X. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Tercero, J.A.; Diffley, J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001, 412, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newlon, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef]
- Froget, B.; Blaisonneau, J.; Lambert, S.; Baldacci, G. Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Mol. Biol. Cell 2008, 19, 445–456. [Google Scholar] [CrossRef]
- El-Shemerly, M.; Hess, D.; Pyakurel, A.K.; Moselhy, S.; Ferrari, S. ATR-dependent pathways control hEXO1 stability in response to stalled forks. Nucleic Acids Res. 2008, 36, 511–519. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Zanini, I.M.Y.; Herrador, R.; Lopes, M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Cotta-Ramusino, C.; Fachinetti, D.; Lucca, C.; Doksani, Y.; Lopes, M.; Sogo, J.; Foiani, M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 2005, 17, 153–159. [Google Scholar] [CrossRef]
- Segurado, M.; Diffley, J.F.X. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008, 22, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Seiler, J.A.; Conti, C.; Syed, A.; Aladjem, M.I.; Pommier, Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: Single-cell and -DNA fiber analyses. Mol. Cell. Biol. 2007, 27, 5806–5818. [Google Scholar] [CrossRef]
- Santocanale, C.; Diffley, J.F. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 1998, 395, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Shirahige, K.; Hori, Y.; Shiraishi, K.; Yamashita, M.; Takahashi, K.; Obuse, C.; Tsurimoto, T.; Yoshikawa, H. Regulation of DNA-replication origins during cell-cycle progression. Nature 1998, 395, 618–621. [Google Scholar] [CrossRef]
- Dimitrova, D.S.; Gilbert, D.M. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat. Cell Biol. 2000, 2, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Yekezare, M.; Gómez-González, B.; Diffley, J.F.X. Controlling DNA replication origins in response to DNA damage—inhibit globally, activate locally. J. Cell. Sci. 2013, 126, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, K.; Walworth, N.; Booher, R.; Dembski, M.; Kirschner, M.; Beach, D. mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell 1991, 64, 1111–1122. [Google Scholar] [CrossRef]
- Boddy, M.N.; Furnari, B.; Mondesert, O.; Russell, P. Replication checkpoint enforced by kinases Cds1 and Chk1. Science 1998, 280, 909–912. [Google Scholar] [CrossRef]
- Zeng, Y.; Forbes, K.C.; Wu, Z.; Moreno, S.; Piwnica-Worms, H.; Enoch, T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature 1998, 395, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Furnari, B.; Rhind, N.; Russell, P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 1997, 277, 1495–1497. [Google Scholar] [CrossRef]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Palou, G.; Palou, R.; Zeng, F.; Vashisht, A.A.; Wohlschlegel, J.A.; Quintana, D.G. Three different pathways prevent chromosome segregation in the presence of DNA damage or replication stress in budding yeast. PLoS Genet. 2015, 11, e1005468. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, M.; Albergante, L.; Moreno, A.; Carrington, J.T.; Blow, J.J.; Newman, T.J. Inevitability and containment of replication errors for eukaryotic genome lengths spanning megabase to gigabase. Proc. Natl. Acad. Sci. USA 2016, 113, E5765–E5774. [Google Scholar] [CrossRef]
- Moreno, A.; Carrington, J.T.; Albergante, L.; Al Mamun, M.; Haagensen, E.J.; Komseli, E.-S.; Gorgoulis, V.G.; Newman, T.J.; Blow, J.J. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5757–E5764. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rosell, J.; De Piccoli, G.; Cordón-Preciado, V.; Farmer, S.; Jarmuz, A.; Machín, F.; Pasero, P.; Lisby, M.; Haber, J.E.; Aragón, L. Anaphase onset before complete DNA replication with intact checkpoint responses. Science 2007, 315, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.; Körner, R.; Hofmann, K.; Nigg, E.A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 2007, 128, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-L.; North, P.S.; Hickson, I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007, 26, 3397–3409. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef]
- Fernández-Casañas, M.; Chan, K.-L. The Unresolved Problem of DNA Bridging. Genes 2018, 9, 623. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef]
- Durkin, S.G.; Ragland, R.L.; Arlt, M.F.; Mulle, J.G.; Warren, S.T.; Glover, T.W. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc. Natl. Acad. Sci. USA 2008, 105, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Mulle, J.G.; Schaibley, V.M.; Ragland, R.L.; Durkin, S.G.; Warren, S.T.; Glover, T.W. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009, 84, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Le Beau, M.M.; Rassool, F.V.; Neilly, M.E.; Espinosa, R.; Glover, T.W.; Smith, D.I.; McKeithan, T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998, 7, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef]
- West, S.C. Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet. 1997, 31, 213–244. [Google Scholar] [CrossRef]
- Kim, S.M.; Forsburg, S.L. Regulation of structure-specific endonucleases in replication stress. Genes 2018, 9, 634. [Google Scholar] [CrossRef]
- Lee, S.-H.; Princz, L.N.; Klügel, M.F.; Habermann, B.; Pfander, B.; Biertümpfel, C. Human Holliday junction resolvase GEN1 uses a chromodomain for efficient DNA recognition and cleavage. eLife 2015, 4, 213. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, J.-H.; Takedachi, A.; Naim, V.; Scaglione, S.; Chawhan, C.; Lovera, Y.; Despras, E.; Kuraoka, I.; Kannouche, P.; Rosselli, F.; et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol. Cell 2015, 57, 123–137. [Google Scholar] [CrossRef]
- Mullen, J.R.; Kaliraman, V.; Ibrahim, S.S.; Brill, S.J. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 2001, 157, 103–118. [Google Scholar] [PubMed]
- Fabre, F.; Chan, A.; Heyer, W.-D.; Gangloff, S. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. USA 2002, 99, 16887–16892. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.; Murray-Rust, J.; Lally, J.; Rudolf, J.; Fadden, A.; Knowles, P.P.; White, M.F.; McDonald, N.Q. Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. 2005, 24, 895–905. [Google Scholar] [CrossRef]
- Nishino, T.; Komori, K.; Ishino, Y.; Morikawa, K. Structural and functional analyses of an archaeal XPF/Rad1/Mus81 nuclease: asymmetric DNA binding and cleavage mechanisms. Structure 2005, 13, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Constantinou, A.; West, S.C. Identification and characterization of the human mus81-eme1 endonuclease. J. Biol. Chem. 2003, 278, 25172–25178. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Ling, C.; Coulthard, R.; Yan, Z.; Xue, Y.; Meetei, A.R.; Laghmani, E.H.; Joenje, H.; McDonald, N.; de Winter, J.P.; et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell 2007, 25, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Boddy, M.N.; Gaillard, P.H.; McDonald, W.H.; Shanahan, P.; Yates, J.R.; Russell, P. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 2001, 107, 537–548. [Google Scholar] [CrossRef]
- Kaliraman, V.; Mullen, J.R.; Fricke, W.M.; Bastin-Shanower, S.A.; Brill, S.J. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 2001, 15, 2730–2740. [Google Scholar] [CrossRef]
- Chen, X.B.; Melchionna, R.; Denis, C.M.; Gaillard, P.H.; Blasina, A.; Van de Weyer, I.; Boddy, M.N.; Russell, P.; Vialard, J.; McGowan, C.H. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell 2001, 8, 1117–1127. [Google Scholar] [CrossRef]
- Oğrünç, M.; Sancar, A. Identification and characterization of human MUS81-MMS4 structure-specific endonuclease. J. Biol. Chem. 2003, 278, 21715–21720. [Google Scholar] [CrossRef] [PubMed]
- Fricke, W.M.; Bastin-Shanower, S.A.; Brill, S.J. Substrate specificity of the Saccharomyces cerevisiae Mus81-Mms4 endonuclease. DNA Repair 2005, 4, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Ehmsen, K.T.; Heyer, W.-D. A junction branch point adjacent to a DNA backbone nick directs substrate cleavage by Saccharomyces cerevisiae Mus81-Mms4. Nucleic Acids Res. 2009, 37, 2026–2036. [Google Scholar] [CrossRef] [PubMed]
- Amangyeld, T.; Shin, Y.-K.; Lee, M.; Kwon, B.; Seo, Y.-S. Human MUS81-EME2 can cleave a variety of DNA structures including intact Holliday junction and nicked duplex. Nucleic Acids Res. 2014, 42, 5846–5862. [Google Scholar] [CrossRef]
- Pepe, A.; West, S.C. Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res. 2014, 42, 3833–3845. [Google Scholar] [CrossRef]
- Pepe, A.; West, S.C. MUS81-EME2 promotes replication fork restart. Cell Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.; Blanco, M.G.; Maslen, S.; Skehel, J.M.; West, S.C. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 2011, 147, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Dehé, P.-M.; Coulon, S.; Scaglione, S.; Shanahan, P.; Takedachi, A.; Wohlschlegel, J.A.; Yates, J.R.; Llorente, B.; Russell, P.; Gaillard, P.-H.L. Regulation of Mus81-Eme1 Holliday junction resolvase in response to DNA damage. Nat. Struct. Mol. Biol. 2013, 20, 598–603. [Google Scholar] [CrossRef]
- Gallo-Fernández, M.; Saugar, I.; Ortiz-Bazán, M.Á.; Vázquez, M.V.; Tercero, J.A. Cell cycle-dependent regulation of the nuclease activity of Mus81–Eme1/Mms4. Nucleic Acids Res. 2012, 40, 8325–8335. [Google Scholar] [CrossRef] [PubMed]
- Princz, L.N.; Wild, P.; Bittmann, J.; Aguado, F.J.; Blanco, M.G.; Matos, J.; Pfander, B. Dbf4-dependent kinase and the Rtt107 scaffold promote Mus81-Mms4 resolvase activation during mitosis. EMBO J. 2017, 36, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.; Blanco, M.G.; West, S.C. Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep. 2013, 4, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Szakal, B.; Branzei, D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013, 32, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Sebesta, M.; Urulangodi, M.; Stefanovie, B.; Szakal, B.; Pacesa, M.; Lisby, M.; Branzei, D.; Krejci, L. Esc2 promotes Mus81 complex-activity via its SUMO-like and DNA binding domains. Nucleic Acids Res. 2017, 45, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Sisakova, A.; Altmannova, V.; Sebesta, M.; Krejci, L. Role of PCNA and RFC in promoting Mus81-complex activity. BMC Biol. 2017, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, H.D.M.; Sarbajna, S.; Matos, J.; West, S.C. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday Junction resolution in human Cells. Mol. Cell 2013, 52, 234–247. [Google Scholar] [CrossRef]
- Duda, H.; Arter, M.; Gloggnitzer, J.; Teloni, F.; Wild, P.; Blanco, M.G.; Altmeyer, M.; Matos, J. A Mechanism for controlled breakage of under-replicated chromosomes during mitosis. Dev. Cell 2016, 39, 740–755. [Google Scholar] [CrossRef]
- Fekairi, S.; Scaglione, S.; Chahwan, C.; Taylor, E.R.; Tissier, A.; Coulon, S.; Dong, M.-Q.; Ruse, C.; Yates, J.R.; Russell, P.; et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 2009, 138, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, I.M.; Hain, K.; Déclais, A.-C.; Gardiner, M.; Toh, G.W.; Sanchez-Pulido, L.; Heuckmann, J.M.; Toth, R.; Macartney, T.; Eppink, B.; et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 2009, 35, 116–127. [Google Scholar] [CrossRef]
- Svendsen, J.M.; Smogorzewska, A.; Sowa, M.E.; O’Connell, B.C.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 2009, 138, 63–77. [Google Scholar] [CrossRef]
- Wyatt, H.D.M.; Laister, R.C.; Martin, S.R.; Arrowsmith, C.H.; West, S.C. The SMX DNA repair Tri-nuclease. Mol. Cell 2017, 65, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wan, B.; Sarkar, J.; Horvath, K.; Wu, J.; Chen, Y.; Cheng, G.; Wan, K.; Chin, P.; Lei, M.; et al. Dimerization of SLX4 contributes to functioning of the SLX4-nuclease complex. Nucleic Acids Res. 2016, 44, 4871–4880. [Google Scholar] [CrossRef]
- Coulon, S.; Gaillard, P.-H.L.; Chahwan, C.; McDonald, W.H.; Yates, J.R.; Russell, P. Slx1-Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol. Biol. Cell 2004, 15, 71–80. [Google Scholar] [CrossRef]
- Fricke, W.M.; Brill, S.J. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003, 17, 1768–1778. [Google Scholar] [CrossRef]
- Kaliraman, V.; Brill, S.J. Role of SGS1 and SLX4 in maintaining rDNA structure in Saccharomyces cerevisiae. Curr. Genet. 2002, 41, 389–400. [Google Scholar] [CrossRef]
- Coulon, S.; Noguchi, E.; Noguchi, C.; Du, L.-L.; Nakamura, T.M.; Russell, P. Rad22Rad52-dependent repair of ribosomal DNA repeats cleaved by Slx1-Slx4 endonuclease. Mol. Biol. Cell 2006, 17, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Sarbajna, S.; Davies, D.; West, S.C. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014, 28, 1124–1136. [Google Scholar] [CrossRef]
- Garner, E.; Kim, Y.; Lach, F.P.; Kottemann, M.C.; Smogorzewska, A. Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 are essential for the resolution of replication-induced Holliday junctions. Cell Rep. 2013, 5, 207–215. [Google Scholar] [CrossRef]
- Guervilly, J.-H.; Gaillard, P.H. SLX4: Multitasking to maintain genome stability. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 475–514. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lach, F.P.; Desetty, R.; Hanenberg, H.; Auerbach, A.D.; Smogorzewska, A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011, 43, 142–146. [Google Scholar] [CrossRef]
- Stoepker, C.; Hain, K.; Schuster, B.; Hilhorst-Hofstee, Y.; Rooimans, M.A.; Steltenpool, J.; Oostra, A.B.; Eirich, K.; Korthof, E.T.; Nieuwint, A.W.M.; et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011, 43, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Gaur, V.; Wyatt, H.D.M.; Komorowska, W.; Szczepanowski, R.H.; de Sanctis, D.; Gorecka, K.M.; West, S.C.; Nowotny, M. Structural and mechanistic analysis of the Slx1-Slx4 endonuclease. Cell Rep. 2015, 10, 1467–1476. [Google Scholar] [CrossRef]
- Castor, D.; Nair, N.; Déclais, A.-C.; Lachaud, C.; Toth, R.; Macartney, T.J.; Lilley, D.M.J.; Arthur, J.S.C.; Rouse, J. Cooperative control of holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol. Cell 2013, 52, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Gwon, G.H.; Jo, A.; Baek, K.; Jin, K.S.; Fu, Y.; Lee, J.-B.; Kim, Y.; Cho, Y. Crystal structures of the structure-selective nuclease Mus81-Eme1 bound to flap DNA substrates. EMBO J. 2014, 33, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Palma, A.; Pugliese, G.M.; Murfuni, I.; Marabitti, V.; Malacaria, E.; Rinalducci, S.; Minoprio, A.; Sanchez, M.; Mazzei, F.; Zolla, L.; et al. Phosphorylation by CK2 regulates MUS81/EME1 in mitosis and after replication stress. Nucleic Acids Res. 2018, 46, 5109–5124. [Google Scholar] [CrossRef]
- Saugar, I.; Jiménez-Martín, A.; Tercero, J.A. Subnuclear relocalization of structure-specific endonucleases in response to DNA damage. Cell Rep. 2017, 20, 1553–1562. [Google Scholar] [CrossRef]
- Schwartz, E.K.; Wright, W.D.; Ehmsen, K.T.; Evans, J.E.; Stahlberg, H.; Heyer, W.-D. Mus81-Mms4 functions as a single heterodimer to cleave nicked intermediates in recombinational DNA repair. Mol. Cell. Biol. 2012, 32, 3065–3080. [Google Scholar] [CrossRef] [PubMed]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Almeida, B.S.; Smolka, M.B. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell 2010, 39, 300–306. [Google Scholar] [CrossRef]
- Gritenaite, D.; Princz, L.N.; Szakal, B.; Bantele, S.C.S.; Wendeler, L.; Schilbach, S.; Habermann, B.H.; Matos, J.; Lisby, M.; Branzei, D.; et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014, 28, 1604–1619. [Google Scholar] [CrossRef] [PubMed]
- Princz, L.N.; Gritenaite, D.; Pfander, B. The Slx4-Dpb11 scaffold complex: coordinating the response to replication fork stalling in S-phase and the subsequent mitosis. Cell Cycle 2014, 14, 488–494. [Google Scholar] [CrossRef]
- Ip, S.C.Y.; Rass, U.; Blanco, M.G.; Flynn, H.R.; Skehel, J.M.; West, S.C. Identification of Holliday junction resolvases from humans and yeast. Nature 2008, 456, 357–361. [Google Scholar] [CrossRef]
- West, S.C. The search for a human Holliday junction resolvase. Biochem. Soc. Trans. 2009, 37, 519–526. [Google Scholar] [CrossRef]
- Lieber, M.R. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays 1997, 19, 233–240. [Google Scholar] [CrossRef]
- Bailly, A.P.; Freeman, A.; Hall, J.; Déclais, A.-C.; Alpi, A.; Lilley, D.M.J.; Ahmed, S.; Gartner, A. The Caenorhabditis elegans homolog of Gen1/Yen1 resolvases links DNA damage signaling to DNA double-strand break repair. PLoS Genet. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Yang, Y.; Ishino, S.; Yamagami, T.; Kumamaru, T.; Satoh, H.; Ishino, Y. The OsGEN-L protein from Oryza sativa possesses Holliday junction resolvase activity as well as 5’-flap endonuclease activity. J. Biochem. 2012, 151, 317–327. [Google Scholar] [CrossRef]
- Bauknecht, M.; Kobbe, D. AtGEN1 and AtSEND1, two paralogs in Arabidopsis, possess Holliday junction resolvase activity. Plant Physiol. 2014, 166, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Kuo, H.K.; Savukoski, D.; Brodsky, M.H.; Sekelsky, J. Three structure-selective endonucleases are essential in the absence of BLM helicase in Drosophila. PLoS Genet. 2011, 7, e1002315. [Google Scholar] [CrossRef]
- Lorenz, A.; West, S.C.; Whitby, M.C. The human Holliday junction resolvase GEN1 rescues the meiotic phenotype of a Schizosaccharomyces pombe mus81 mutant. Nucleic Acids Res. 2010, 38, 1866–1873. [Google Scholar] [CrossRef]
- Freeman, A.D.J.; Liu, Y.; Déclais, A.-C.; Gartner, A.; Lilley, D.M.J. GEN1 from a thermophilic fungus is functionally closely similar to non-eukaryotic junction-resolving enzymes. J. Mol. Biol. 2014, 426, 3946–3959. [Google Scholar] [CrossRef] [PubMed]
- Shah Punatar, R.; Martin, M.J.; Wyatt, H.D.M.; Chan, Y.W.; West, S.C. Resolution of single and double Holliday junction recombination intermediates by GEN1. Proc. Natl. Acad. Sci. USA 2017, 114, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Rass, U.; Compton, S.A.; Matos, J.; Singleton, M.R.; Ip, S.C.Y.; Blanco, M.G.; Griffith, J.D.; West, S.C. Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 2010, 24, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.W.; West, S. GEN1 promotes Holliday junction resolution by a coordinated nick and counter-nick mechanism. Nucleic Acids Res. 2015, 43, 10882–10892. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Freeman, A.D.J.; Déclais, A.-C.; Wilson, T.J.; Gartner, A.; Lilley, D.M.J. Crystal structure of a eukaryotic GEN1 resolving enzyme bound to DNA. Cell Rep. 2015, 13, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Sobhy, M.A.; Bralic, A.; Raducanu, V.-S.; Takahashi, M.; Tehseen, M.; Rashid, F.; Zaher, M.S.; Hamdan, S.M. Resolution of the Holliday junction recombination intermediate by human GEN1 at the single-molecule level. Nucleic Acids Res. 2018, 5, 282. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.G.; Matos, J.; West, S.C. Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol. Cell 2014, 54, 94–106. [Google Scholar] [CrossRef]
- García-Luis, J.; Clemente-Blanco, A.; Aragón, L.; Machín, F. Cdc14 targets the Holliday junction resolvase Yen1 to the nucleus in early anaphase. Cell Cycle 2014, 13, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Hasebe, M.; Tomita, M.; Yanagawa, H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA 2009, 106, 10171–10176. [Google Scholar] [CrossRef] [PubMed]
- Talhaoui, I.; Bernal, M.; Mullen, J.R.; Dorison, H.; Palancade, B.; Brill, S.J.; Mazón, G. Slx5-Slx8 ubiquitin ligase targets active pools of the Yen1 nuclease to limit crossover formation. Nat. Commun. 2018, 9, 5016. [Google Scholar] [CrossRef]
- Chan, Y.W.; West, S.C. Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nat. Commun. 2014, 5, 4844. [Google Scholar] [CrossRef] [PubMed]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair 2016, 44, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Agmon, N.; Yovel, M.; Harari, Y.; Liefshitz, B.; Kupiec, M. The role of Holliday junction resolvases in the repair of spontaneous and induced DNA damage. Nucleic Acids Res. 2011, 39, 7009–7019. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.G.; Matos, J.; Rass, U.; Ip, S.C.Y.; West, S.C. Functional overlap between the structure-specific nucleases Yen1 and Mus81-Mms4 for DNA-damage repair in S. cerevisiae. DNA Repair 2010, 9, 394–402. [Google Scholar] [CrossRef]
- Ho, C.K.; Mazón, G.; Lam, A.F.; Symington, L.S. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol. Cell 2010, 40, 988–1000. [Google Scholar] [CrossRef]
- Tay, Y.D.; Wu, L. Overlapping roles for Yen1 and Mus81 in cellular Holliday junction processing. J. Biol. Chem. 2010, 285, 11427–11432. [Google Scholar] [CrossRef] [PubMed]
- Boddy, M.N.; Lopez-Girona, A.; Shanahan, P.; Interthal, H.; Heyer, W.D.; Russell, P. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol. Cell. Biol. 2000, 20, 8758–8766. [Google Scholar] [CrossRef]
- García-Luis, J.; Machín, F. Mus81-Mms4 and Yen1 resolve a novel anaphase bridge formed by noncanonical Holliday junctions. Nat. Commun. 2014, 5, 5652. [Google Scholar] [CrossRef]
- Ying, S.; Minocherhomji, S.; Chan, K.-L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Chan, Y.W.; Fugger, K.; West, S.C. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol. 2018, 20, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Eissler, C.L.; Mazón, G.; Powers, B.L.; Savinov, S.N.; Symington, L.S.; Hall, M.C. The Cdk/cDc14 module controls activation of the Yen1 Holliday junction resolvase to promote genome stability. Mol. Cell 2014, 54, 80–93. [Google Scholar] [CrossRef]
- Bizard, A.H.; Hickson, I.D. The dissolution of double Holliday junctions. Cold Spring Harb. Perspect. Biol. 2014, 6, a016477. [Google Scholar] [CrossRef] [PubMed]
- Técher, H.; Koundrioukoff, S.; Carignon, S.; Wilhelm, T.; Millot, G.A.; Lopez, B.S.; Brison, O.; Debatisse, M. Signaling from Mus81-Eme2-dependent DNA damage elicited by Chk1 deficiency modulates replication fork speed and origin usage. Cell Rep. 2016, 14, 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; Blasius, M.; Guerini, I.; Jackson, S.P. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS ONE 2011, 6, e23517. [Google Scholar] [CrossRef] [PubMed]
- Szmyd, R.; Niska-Blakie, J.; Diril, M.K.; Renck Nunes, P.; Tzelepis, K.; Lacroix, A.; van Hul, N.; Deng, L.-W.; Matos, J.; Dreesen, O.; et al. Premature activation of Cdk1 leads to mitotic events in S phase and embryonic lethality. Oncogene 2018, 448, 811. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Nähse, V.; Larsen, M.S.Y.; Groth, P.; Clancy, T.; Lees, M.; Jørgensen, M.; Helleday, T.; Syljuåsen, R.G.; Sørensen, C.S. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J. Cell Biol. 2010, 188, 629–638. [Google Scholar] [CrossRef]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef]
- Domínguez-Kelly, R.; Martín, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.J.; Medema, R.H.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Modesti, M.; Maas, A.; Wyman, C.; Essers, J.; Kanaar, R. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 2006, 25, 4921–4932. [Google Scholar] [CrossRef]
- Kramara, J.; Osia, B.; Malkova, A. Break-induced replication: The where, the why, and the how. Trends Genet. 2018, 34, 518–531. [Google Scholar] [CrossRef]
- Xu, Y.; Ning, S.; Wei, Z.; Xu, R.; Xu, X.; Xing, M.; Guo, R.; Xu, D. 53BP1 and BRCA1 control pathway choice for stalled replication restart. eLlife 2017, 6, 35897. [Google Scholar] [CrossRef]
- Shimura, T.; Torres, M.J.; Martin, M.M.; Rao, V.A.; Pommier, Y.; Katsura, M.; Miyagawa, K.; Aladjem, M.I. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J. Mol. Biol. 2008, 375, 1152–1164. [Google Scholar] [CrossRef] [PubMed]
- Regairaz, M.; Zhang, Y.-W.; Fu, H.; Agama, K.K.; Tata, N.; Agrawal, S.; Aladjem, M.I.; Pommier, Y. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol. 2011, 195, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Murfuni, I.; Nicolai, S.; Baldari, S.; Crescenzi, M.; Bignami, M.; Franchitto, A.; Pichierri, P. The WRN and MUS81 proteins limit cell death and genome instability following oncogene activation. Oncogene 2013, 32, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Ochiai, Y.; Noma, N.; Oikawa, T.; Sano, Y.; Fukumoto, M. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle 2013, 12, 773–782. [Google Scholar] [CrossRef]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.-S.; Tarsounas, M. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef]
- Fugger, K.; Chu, W.K.; Haahr, P.; Kousholt, A.N.; Beck, H.; Payne, M.J.; Hanada, K.; Hickson, I.D.; Sørensen, C.S. FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat. Commun. 2013, 4, 1423. [Google Scholar] [CrossRef]
- Kurashima, K.; Sekimoto, T.; Oda, T.; Kawabata, T.; Hanaoka, F.; Yamashita, T. Polη, a Y-family translesion synthesis polymerase, promotes cellular tolerance of Myc-induced replication stress. J. Cell. Sci. 2018, 131, jcs212183. [Google Scholar] [CrossRef]
- Mayle, R.; Campbell, I.M.; Beck, C.R.; Yu, Y.; Wilson, M.; Shaw, C.A.; Bjergbaek, L.; Lupski, J.R.; Ira, G. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 2015, 349, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Chan, Y.W.; West, S.C. A new class of ultrafine anaphase bridges generated by homologous recombination. Cell Cycle 2018, 17, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Addis Jones, O.; Chan, K.-L. 53BP1 can limit sister-chromatid rupture and rearrangements driven by a distinct ultrafine DNA bridging-breakage process. Nat. Commun. 2018, 9, 677. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Di Marco, S.; Hasanova, Z.; Kanagaraj, R.; Chappidi, N.; Altmannova, V.; Menon, S.; Sedlackova, H.; Langhoff, J.; Surendranath, K.; Hühn, D.; Bhowmick, R.; Marini, V.; Ferrari, S.; Hickson, I.D.; et al. RECQ5 helicase cooperates with MUS81 endonuclease in processing stalled replication forks at common fragile sites during mitosis. Mol. Cell 2017, 66, 658–671. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef]
- Downing, B.; Morgan, R.; VanHulle, K.; Deem, A.; Malkova, A. Large inverted repeats in the vicinity of a single double-strand break strongly affect repair in yeast diploids lacking Rad51. Mutat. Res. 2008, 645, 9–18. [Google Scholar] [CrossRef]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef]
- Ottaviani, D.; LeCain, M.; Sheer, D. The role of microhomology in genomic structural variation. Trends Genet. 2014, 30, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Özer, Ö.; Bhowmick, R.; Liu, Y.; Hickson, I.D. Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 2018, 9, 15836–15846. [Google Scholar] [CrossRef]
- Budd, M.E.; Tong, A.H.Y.; Polaczek, P.; Peng, X.; Boone, C.; Campbell, J.L. A network of multi-tasking proteins at the DNA replication fork preserves genome stability. PLoS Genet. 2005, 1, e61. [Google Scholar] [CrossRef] [PubMed]
- Falquet, B.; Rass, U. A new role for Holliday junction resolvase Yen1 in processing DNA replication intermediates exposes Dna2 as an accessory replicative helicase. Microb. Cell 2017, 4, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Ölmezer, G.; Levikova, M.; Klein, D.; Falquet, B.; Fontana, G.A.; Cejka, P.; Rass, U. Replication intermediates that escape Dna2 activity are processed by Holliday junction resolvase Yen1. Nat. Commun. 2016, 7, 13157. [Google Scholar] [CrossRef]
- Duxin, J.P.; Dao, B.; Martinsson, P.; Rajala, N.; Guittat, L.; Campbell, J.L.; Spelbrink, J.N.; Stewart, S.A. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell. Biol. 2009, 29, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Moore, H.R.; Sidorova, J.; Karanja, K.; Honaker, Y.; Dao, B.; Piwnica-Worms, H.; Campbell, J.L.; Monnat, R.J.; Stewart, S.A. Okazaki fragment processing-independent role for human Dna2 enzyme during DNA replication. J. Biol. Chem. 2012, 287, 21980–21991. [Google Scholar] [CrossRef]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef]
- Li, Z.; Liu, B.; Jin, W.; Wu, X.; Zhou, M.; Liu, V.Z.; Goel, A.; Shen, Z.; Zheng, L.; Shen, B. hDNA2 nuclease/helicase promotes centromeric DNA replication and genome stability. EMBO J. 2018, e96729. [Google Scholar] [CrossRef]
- Michel, A.H.; Hatakeyama, R.; Kimmig, P.; Arter, M.; Peter, M.; Matos, J.; De Virgilio, C.; Kornmann, B. Functional mapping of yeast genomes by saturated transposition. eLife 2017, 6, E3179. [Google Scholar] [CrossRef]
- Alderton, G.K.; Joenje, H.; Varon, R.; Børglum, A.D.; Jeggo, P.A.; O’Driscoll, M. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum. Mol. Genet. 2004, 13, 3127–3138. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Faqeih, E.; Ansari, S.; Abdel-Salam, G.; Al-Hassnan, Z.N.; Al-Shidi, T.; Alomar, R.; Sogaty, S.; Alkuraya, F.S. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014, 24, 291–299. [Google Scholar] [CrossRef]
- Peng, G.; Dai, H.; Zhang, W.; Hsieh, H.-J.; Pan, M.-R.; Park, Y.-Y.; Tsai, R.Y.-L.; Bedrosian, I.; Lee, J.-S.; Ira, G.; et al. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res. 2012, 72, 2802–2813. [Google Scholar] [CrossRef] [PubMed]
- Strauss, C.; Kornowski, M.; Benvenisty, A.; Shahar, A.; Masury, H.; Ben-Porath, I.; Ravid, T.; Arbel-Eden, A.; Goldberg, M. The DNA2 nuclease/helicase is an estrogen-dependent gene mutated in breast and ovarian cancers. Oncotarget 2014, 5, 9396–9409. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication fork reversal: Players and guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef]
- Mutreja, K.; Krietsch, J.; Hess, J.; Ursich, S.; Berti, M.; Roessler, F.K.; Zellweger, R.; Patra, M.; Gasser, G.; Lopes, M. ATR-mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at DNA interstrand cross-links. Cell Rep. 2018, 24, 2629–2642. [Google Scholar] [CrossRef]
- Menin, L.; Ursich, S.; Trovesi, C.; Zellweger, R.; Lopes, M.; Longhese, M.P.; Clerici, M. Tel1/ATM prevents degradation of replication forks that reverse after topoisomerase poisoning. EMBO Rep. 2018, 19, e45535. [Google Scholar] [CrossRef]
- Bhat, K.P.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX modulates RAD51 activity to control replication fork protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Bhat, K.P.; Le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol. Cell 2017, 67, 374–386. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Higgs, M.R.; Reynolds, J.J.; Winczura, A.; Blackford, A.N.; Borel, V.; Miller, E.S.; Zlatanou, A.; Nieminuszczy, J.; Ryan, E.L.; Davies, N.J.; et al. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell 2015, 59, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Przetocka, S.; Porro, A.; Bolck, H.A.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.-F.; D’Andrea, A.D.; Ceccaldi, R.; et al. CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 2018, 72, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Manosas, M.; Perumal, S.K.; Croquette, V.; Benkovic, S.J. Direct observation of stalled fork restart via fork regression in the T4 replication system. Science 2012, 338, 1217–1220. [Google Scholar] [CrossRef]
- Manosas, M.; Perumal, S.K.; Bianco, P.R.; Bianco, P.; Ritort, F.; Benkovic, S.J.; Croquette, V. RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat. Commun. 2013, 4, 2368. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falquet, B.; Rass, U. Structure-Specific Endonucleases and the Resolution of Chromosome Underreplication. Genes 2019, 10, 232. https://doi.org/10.3390/genes10030232
Falquet B, Rass U. Structure-Specific Endonucleases and the Resolution of Chromosome Underreplication. Genes. 2019; 10(3):232. https://doi.org/10.3390/genes10030232
Chicago/Turabian StyleFalquet, Benoît, and Ulrich Rass. 2019. "Structure-Specific Endonucleases and the Resolution of Chromosome Underreplication" Genes 10, no. 3: 232. https://doi.org/10.3390/genes10030232