miR-9-Mediated Inhibition of EFEMP1 Contributes to the Acquisition of Pro-Tumoral Properties in Normal Fibroblasts

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. In-Silico Analysis to Define Caf and NF EFEMP1 Expression Portraits
2.2. Cell Culture and Primary Fibroblasts Isolation
2.3. MiRNA Mimics and siRNA Transient Transfection
2.4. Cloning and Mutagenesis
2.5. Luciferase Reporter Assay
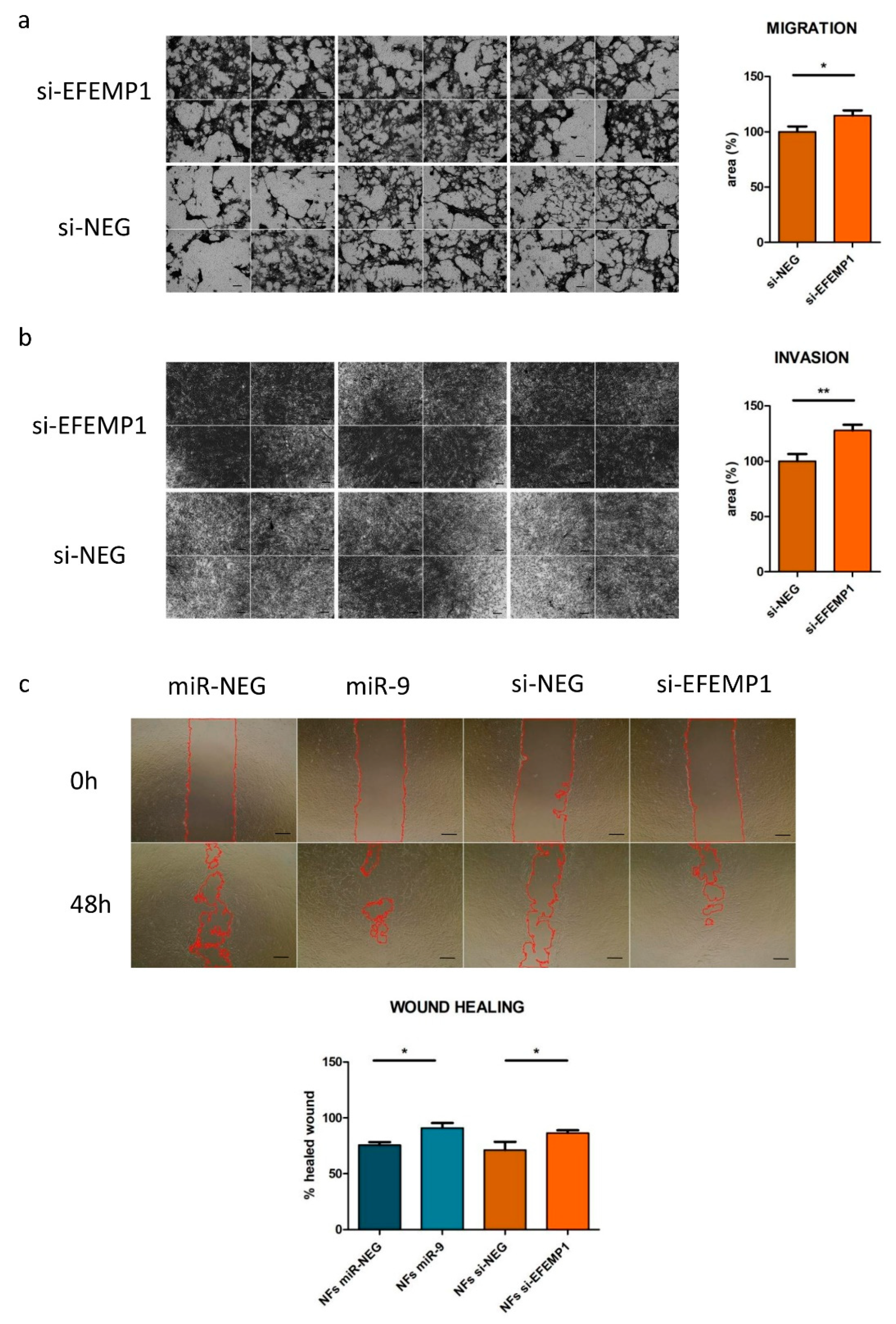
2.6. Motility Assays
2.7. Protein Extraction and Western Blot
2.8. Immunohistochemistry
2.9. Tumor Cell Conditioning and Resistance Test
2.10. Mining Data to Evaluate Correlation of MiR-9 Expression and Cisplatin Response
3. Results
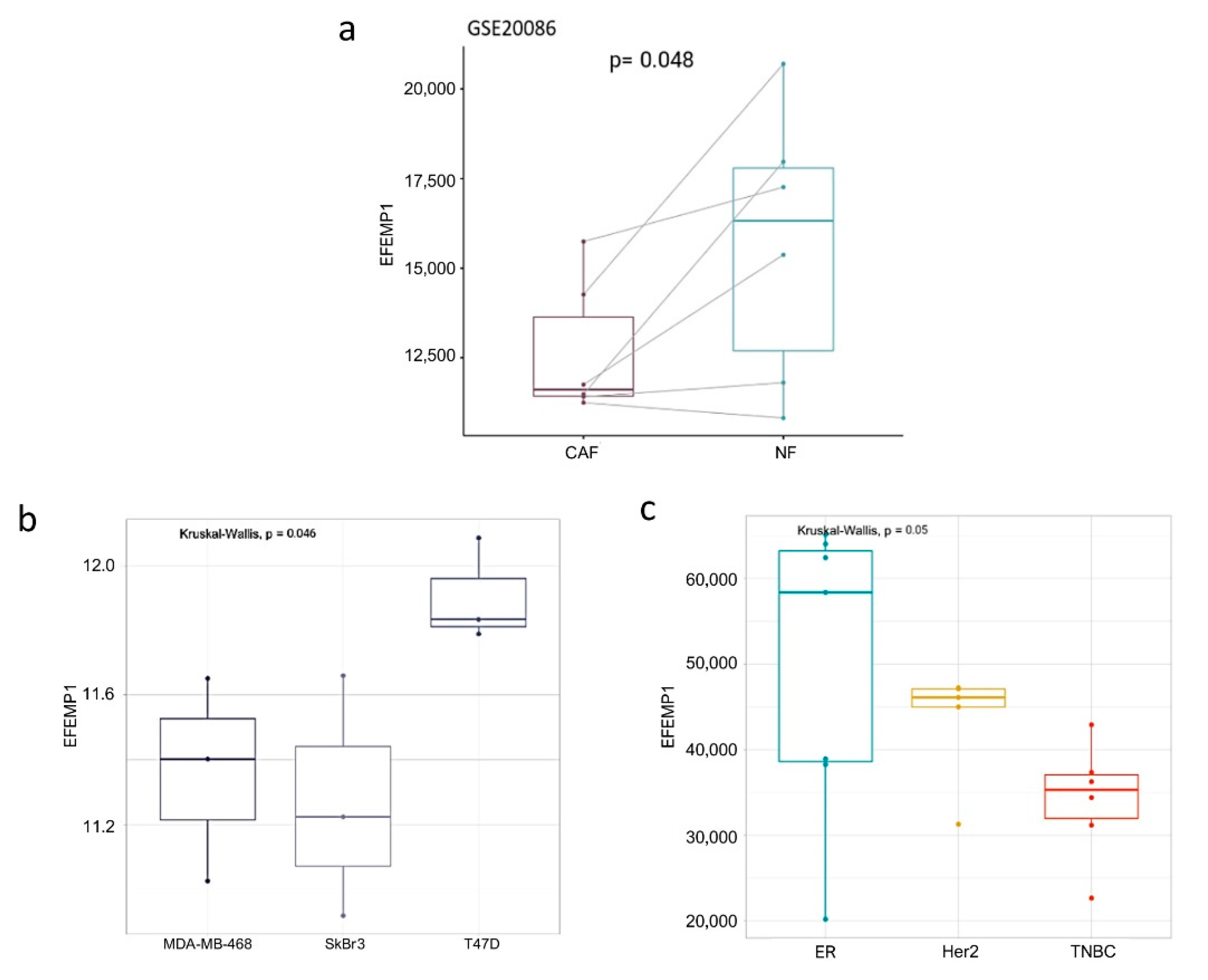
3.1. In-Silico Evaluation of EFEMP1 Levels in CAFs
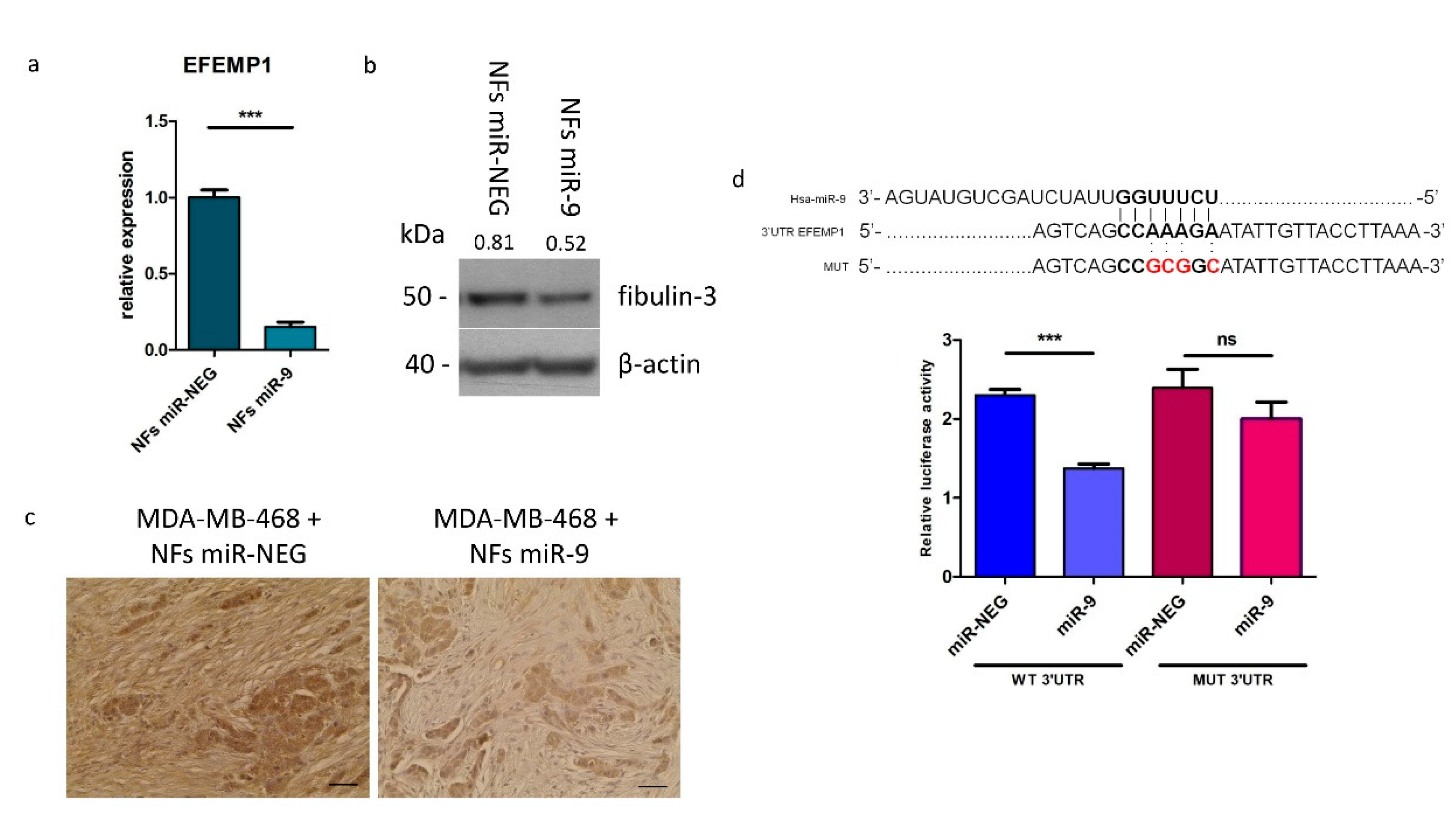
3.2. MiR-9 Directly Targets EFEMP1 and Affects Protein Levels In Vitro and In Vivo
3.3. EFEMP1 Silencing Recapitulates miR-9-Induced CAF-Like Features in Normal Fibroblasts
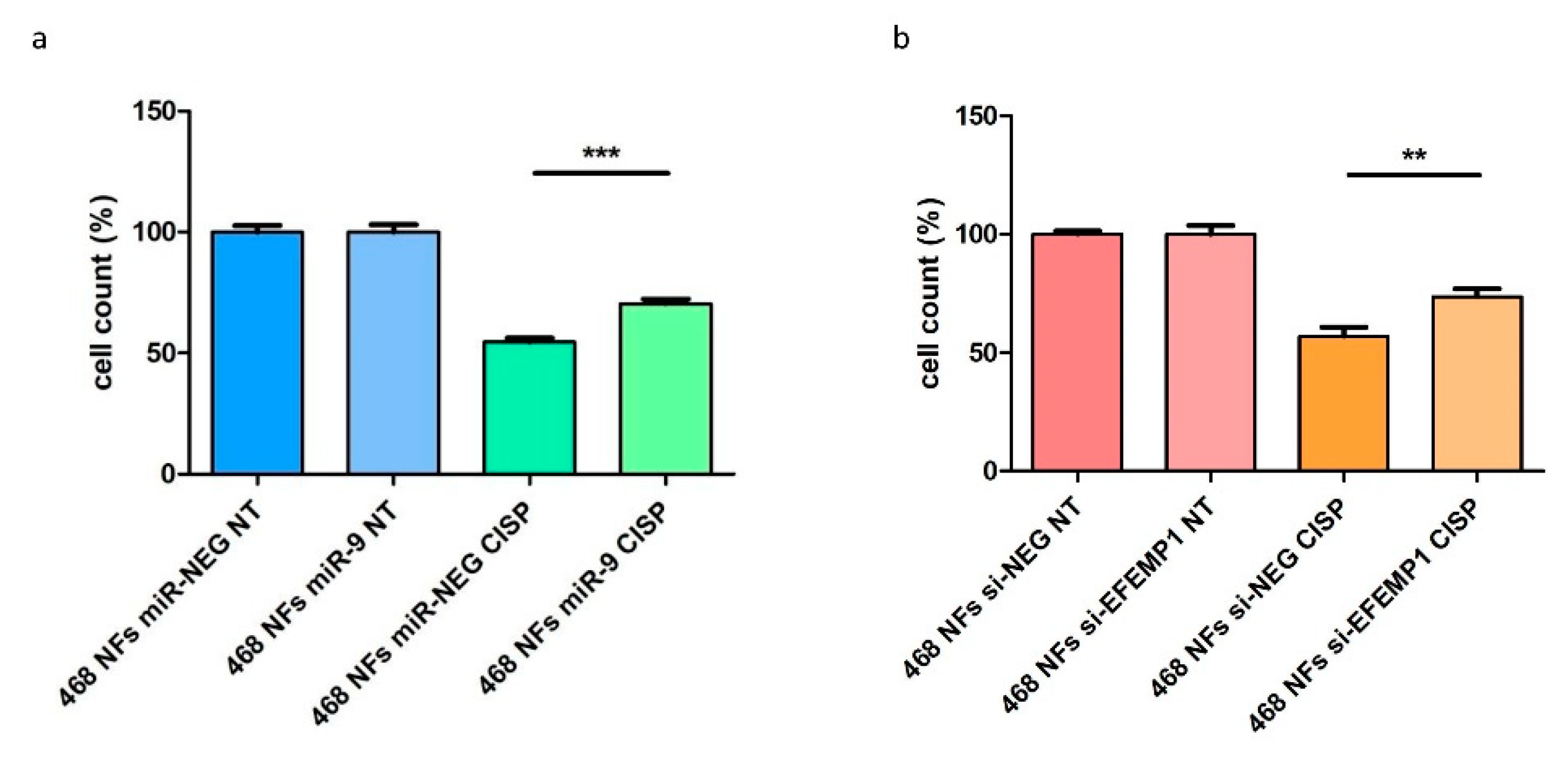
3.4. CAF-Like Properties Induced by miR-9/si-EFEMP1-Transfection Reduce MDA-MB-468 Cell Sensitivity to Cisplatin
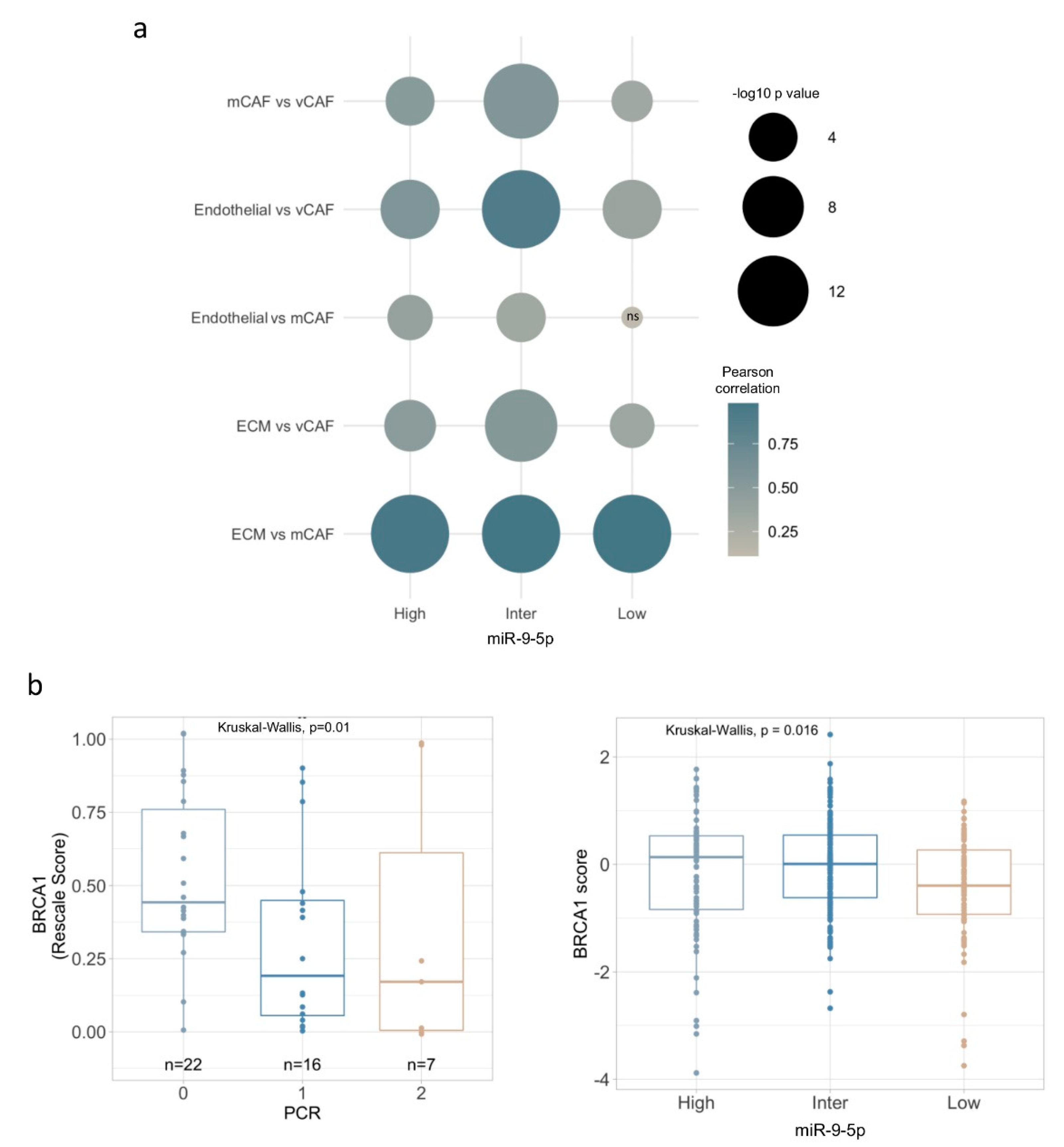
3.5. Characterization of miR-9/CAF Axis on TNBC Biology and Chemotherapy Response by Mining mRNA and miRNA Expression Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huet, E.; Jaroz, C.; Nguyen, H.Q.; Belkacemi, Y.; de la Taille, A.; Stavrinides, V.; Whitaker, H. Stroma in normal and cancer wound healing. FEBS J. 2019, 286, 2909–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.; Liu, B.; Defilippis, R.A.; Chang, H.; Rabban, J.T.; Karnezis, A.N.; Tjoe, J.A.; Marx, J.; Parvin, B.; Tlsty, T.D. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 2013, 15, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE 2009, 4, e7965. [Google Scholar] [CrossRef] [Green Version]
- Hembruff, S.L.; Jokar, I.; Yang, L.; Cheng, N. Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia 2010, 12, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Yu, T.; Di, G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res. 2017, 29, 237–252. [Google Scholar] [CrossRef]
- Kogure, A.; Kosaka, N.; Ochiya, T. Cross-talk between cancer cells and their neighbors via miRNA in extracellular vesicles: An emerging player in cancer metastasis. J. Biomed. Sci. 2019, 26, 7. [Google Scholar] [CrossRef]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’Ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M.G.; et al. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis 2016, 7, e2312. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zeng, Z.; Wang, J.; Wu, Y.; Chen, W.; Zheng, L.; Xi, T.; Wang, A.; Lu, Y. MicroRNA-9 and breast cancer. Biomed. Pharmacother. 2020, 122, 109687. [Google Scholar] [CrossRef] [PubMed]
- De Vega, S.; Iwamoto, T.; Yamada, Y. Fibulins: Multiple roles in matrix structures and tissue functions. Cell. Mol. Life Sci. 2009, 66, 1890–1902. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Marmorstein, L.Y. Focus on molecules: Fibulin-3 (EFEMP1). Exp. Eye Res. 2010, 90, 374–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; Liu, J.; Chen, J.; Gatza, M.L.; Blobe, G.C. Fibulin-3 is a novel TGF-β pathway inhibitor in the breast cancer microenvironment. Oncogene 2015, 34, 5635–5647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun 2018, 9, 5150. [Google Scholar] [CrossRef] [Green Version]
- Winslow, S.; Lindquist, K.E.; Edsjö, A.; Larsson, C. The expression pattern of matrix-producing tumor stroma is of prognostic importance in breast cancer. BMC Cancer 2016, 16, 841. [Google Scholar] [CrossRef] [Green Version]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Wirapati, P.; Becette, V.; André, S.; Piccart, M.; Campone, M.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Tobin, N.P.; Wennmalm, K.; Lindström, L.S.; Foukakis, T.; He, L.; Genové, G.; Östman, A.; Landberg, G.; Betsholtz, C.; Bergh, J. An Endothelial Gene Signature Score Predicts Poor Outcome in Patients with Endocrine-Treated, Low Genomic Grade Breast Tumors. Clin. Cancer Res. 2016, 22, 2417–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkbak, N.J.; Li, Y.; Pathania, S.; Greene-Colozzi, A.; Dreze, M.; Bowman-Colin, C.; Sztupinszki, Z.; Krzystanek, M.; Diossy, M.; Tung, N.; et al. Overexpression of BLM promotes DNA damage and increased sensitivity to platinum salts in triple-negative breast and serous ovarian cancers. Ann. Oncol. 2018, 29, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.-O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Turner, N.C.; Reis-Filho, J.S.; Russell, A.M.; Springall, R.J.; Ryder, K.; Steele, D.; Savage, K.; Gillett, C.E.; Schmitt, F.C.; Ashworth, A.; et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 2007, 26, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/Bcl-2 signalling. Cell Death Dis. 2019, 10, 375. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Ren, Y.; Geng, H.; Zhang, Q.; Cao, L.; Meng, Z.; Wu, X.; Xu, M.; Xu, K. Cancer-associated fibroblasts contribute to cisplatin resistance by modulating ANXA3 in lung cancer cells. Cancer Sci. 2019, 110, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Kadel, D.; Zhang, Y.; Sun, H.-R.; Zhao, Y.; Dong, Q.-Z.; Qin, L.-X. Current perspectives of cancer-associated fibroblast in therapeutic resistance: Potential mechanism and future strategy. Cell Biol. Toxicol. 2019, 35, 407–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.P.; Harper, A.; Malcolm, J.; McAndrews, M.S.; Mockus, S.M.; Patterson, S.E.; Reynolds, T.; Baker, E.J.; Bult, C.J.; Chesler, E.J.; et al. Cisplatin-resistant triple-negative breast cancer subtypes: Multiple mechanisms of resistance. BMC Cancer 2019, 19, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, P.; Almeida, F. Role of Exosomal miRNAs and the Tumor Microenvironment in Drug Resistance. Cells 2020, 9, 1450. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [Green Version]
- D’Ippolito, E.; Plantamura, I.; Bongiovanni, L.; Casalini, P.; Baroni, S.; Piovan, C.; Orlandi, R.; Gualeni, A.V.; Gloghini, A.; Rossini, A.; et al. miR-9 and miR-200 Regulate PDGFRβ-Mediated Endothelial Differentiation of Tumor Cells in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 5562–5572. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D.; et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012, 31, 3513–3523. [Google Scholar] [CrossRef]
- Cataldo, A.; Romero-Cordoba, S.; Plantamura, I.; Cosentino, G.; Hidalgo-Miranda, A.; Tagliabue, E.; Iorio, M.V. MiR-302b as a Combinatorial Therapeutic Approach to Improve Cisplatin Chemotherapy Efficacy in Human Triple-Negative Breast Cancer. Cancers 2020, 12, 2261. [Google Scholar] [CrossRef]
- Hwang, C.-F.; Chien, C.-Y.; Huang, S.-C.; Yin, Y.-F.; Huang, C.-C.; Fang, F.-M.; Tsai, H.-T.; Su, L.-J.; Chen, C.-H. Fibulin-3 is associated with tumour progression and a poor prognosis in nasopharyngeal carcinomas and inhibits cell migration and invasion via suppressed AKT activity. J. Pathol. 2010, 222, 367–379. [Google Scholar] [CrossRef]
- Almeida, M.; Costa, V.L.; Costa, N.R.; Ramalho-Carvalho, J.; Baptista, T.; Ribeiro, F.R.; Paulo, P.; Teixeira, M.R.; Oliveira, J.; Lothe, R.A.; et al. Epigenetic regulation of EFEMP1 in prostate cancer: Biological relevance and clinical potential. J. Cell. Mol. Med. 2014, 18, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Zhang, M.; Liu, L.; Lu, S.; Zhang, C.Z.; Yun, J. Decrease of fibulin-3 in hepatocellular carcinoma indicates poor prognosis. PLoS ONE 2013, 8, e70511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Pioli, P.D.; Siegel, E.; Zhang, Q.; Nelson, J.; Chaturbedi, A.; Mathews, M.S.; Ro, D.I.; Alkafeef, S.; Hsu, N.; et al. EFEMP1 suppresses malignant glioma growth and exerts its action within the tumor extracellular compartment. Mol. Cancer 2011, 10, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.-L.; Hou, Y.-P.; Yu, S.-P.; Chen, S.-G.; Huang, J.-T.; Luo, T.; Kong, L.-P.; Xu, J.; Wang, H.-Q. EFEMP1 expression promotes angiogenesis and accelerates the growth of cervical cancer in vivo. Gynecol. Oncol. 2011, 121, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Camaj, P.; Seeliger, H.; Ischenko, I.; Krebs, S.; Blum, H.; De Toni, E.N.; Faktorova, D.; Jauch, K.-W.; Bruns, C.J. EFEMP1 binds the EGF receptor and activates MAPK and Akt pathways in pancreatic carcinoma cells. Biol. Chem. 2009, 390, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Fang, S.; Wang, M.; Wang, Q.; Fang, R.; Chen, J. EFEMP1 promotes ovarian cancer cell growth, invasion and metastasis via activated the AKT pathway. Oncotarget 2016, 7, 47938–47953. [Google Scholar] [CrossRef] [Green Version]
- Sadr-Nabavi, A.; Ramser, J.; Volkmann, J.; Naehrig, J.; Wiesmann, F.; Betz, B.; Hellebrand, H.; Engert, S.; Seitz, S.; Kreutzfeld, R.; et al. Decreased expression of angiogenesis antagonist EFEMP1 in sporadic breast cancer is caused by aberrant promoter methylation and points to an impact of EFEMP1 as molecular biomarker. Int. J. Cancer 2009, 124, 1727–1735. [Google Scholar] [CrossRef]
- Noonan, M.M.; Dragan, M.; Mehta, M.M.; Hess, D.A.; Brackstone, M.; Tuck, A.B.; Viswakarma, N.; Rana, A.; Babwah, A.V.; Wondisford, F.E.; et al. The matrix protein Fibulin-3 promotes KISS1R induced triple negative breast cancer cell invasion. Oncotarget 2018, 9, 30034–30052. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3′UTR EFEMP1 Forward | 5′-AATTGCTAGCTTGACAATAATAGTGGGGCCA-3′ |
| 3′UTR EFEMP1 Reverse | 5′-AATTTCTAGATGCCCACTTTATACCATGG-3′ |
| pmirGLO Forward | 5′-CGCGAGATTCTCATTAAGGCC-3′ |
| pmirGLO Reverse | 5′-CAACTCAGCTTCCTTTCGG-3′ |
| MiR-9 binding site | 5′-CCAAAGA-3′ |
| 3′UTR EFEMP1 MUT Forward | 5′-ATAAAATAGTGCTTTAAGGTAACAATATCGTGTCGCTGACTTAAA TGCCTGTGGTTGACTCT-3′ |
| 3′UTR EFEMP1 MUT Reverse | 5′-AGAGTCAACCACAGGCATTTAAGTCAGCGACACGATATTGTTAC CTTAAAGCACTATTTTAT-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosentino, G.; Romero-Cordoba, S.; Plantamura, I.; Cataldo, A.; Iorio, M.V. miR-9-Mediated Inhibition of EFEMP1 Contributes to the Acquisition of Pro-Tumoral Properties in Normal Fibroblasts. Cells 2020, 9, 2143. https://doi.org/10.3390/cells9092143
Cosentino G, Romero-Cordoba S, Plantamura I, Cataldo A, Iorio MV. miR-9-Mediated Inhibition of EFEMP1 Contributes to the Acquisition of Pro-Tumoral Properties in Normal Fibroblasts. Cells. 2020; 9(9):2143. https://doi.org/10.3390/cells9092143
Chicago/Turabian StyleCosentino, Giulia, Sandra Romero-Cordoba, Ilaria Plantamura, Alessandra Cataldo, and Marilena V. Iorio. 2020. "miR-9-Mediated Inhibition of EFEMP1 Contributes to the Acquisition of Pro-Tumoral Properties in Normal Fibroblasts" Cells 9, no. 9: 2143. https://doi.org/10.3390/cells9092143