CCN-Based Therapeutic Peptides Modify Pancreatic Ductal Adenocarcinoma Microenvironment and Decrease Tumor Growth in Combination with Chemotherapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Tumor Cells
2.3. Proliferation Assay
2.4. ELISA Assays
2.5. In Vivo Studies
2.6. Pharmacokinetics Studies
2.7. Histological and Immunohistochemical Analysis
2.8. Statistical Analysis
3. Results
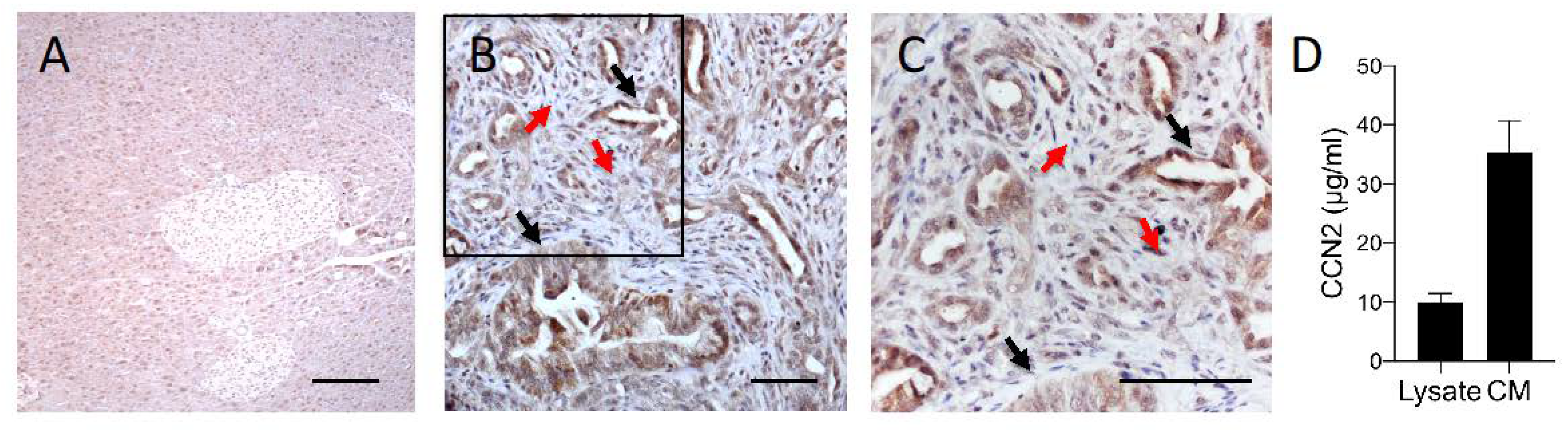
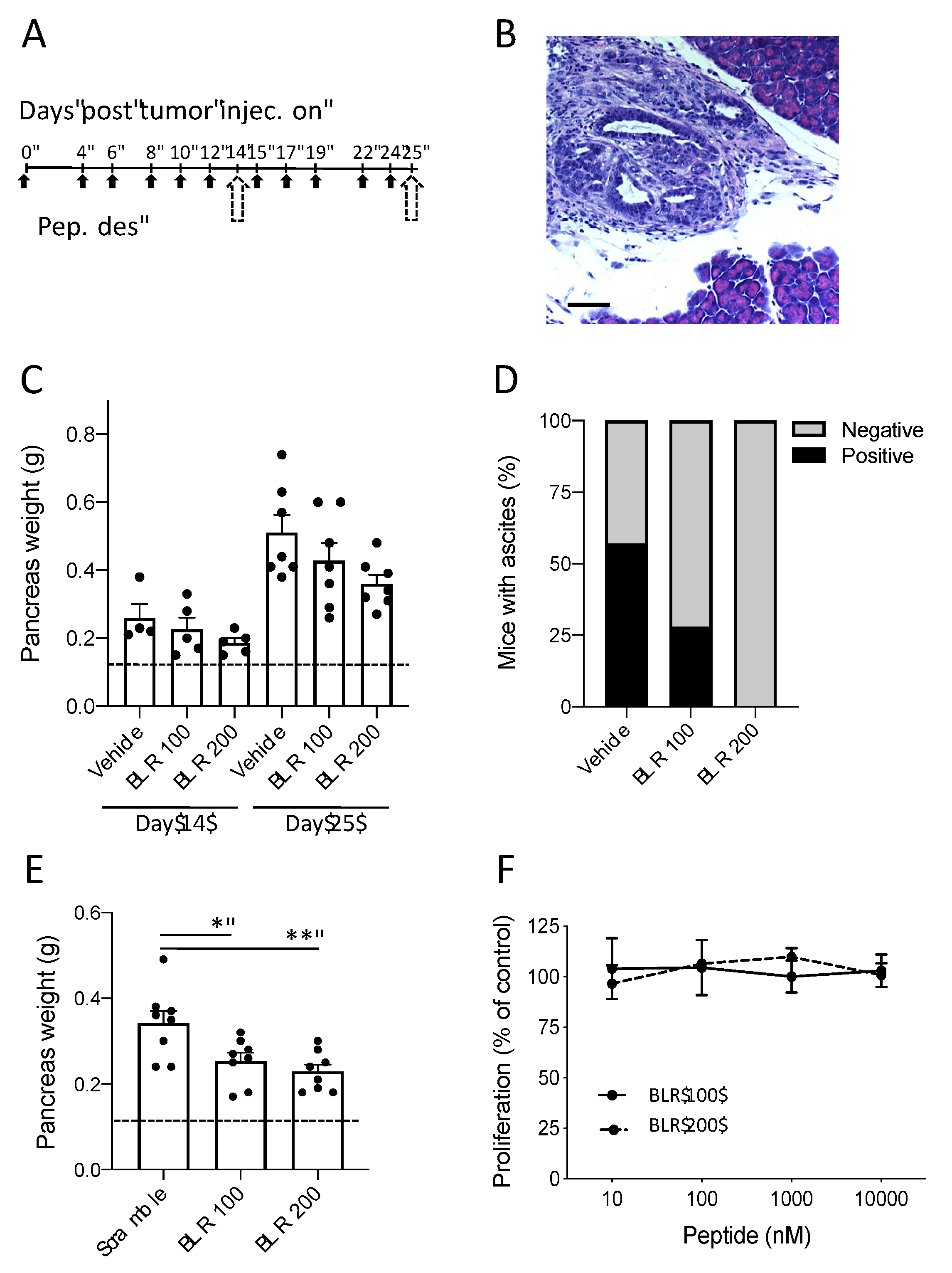
3.1. Effect of CCN-Targeting Peptides (BLR100 and BLR200) on the Orthotopic Growth of PDAC
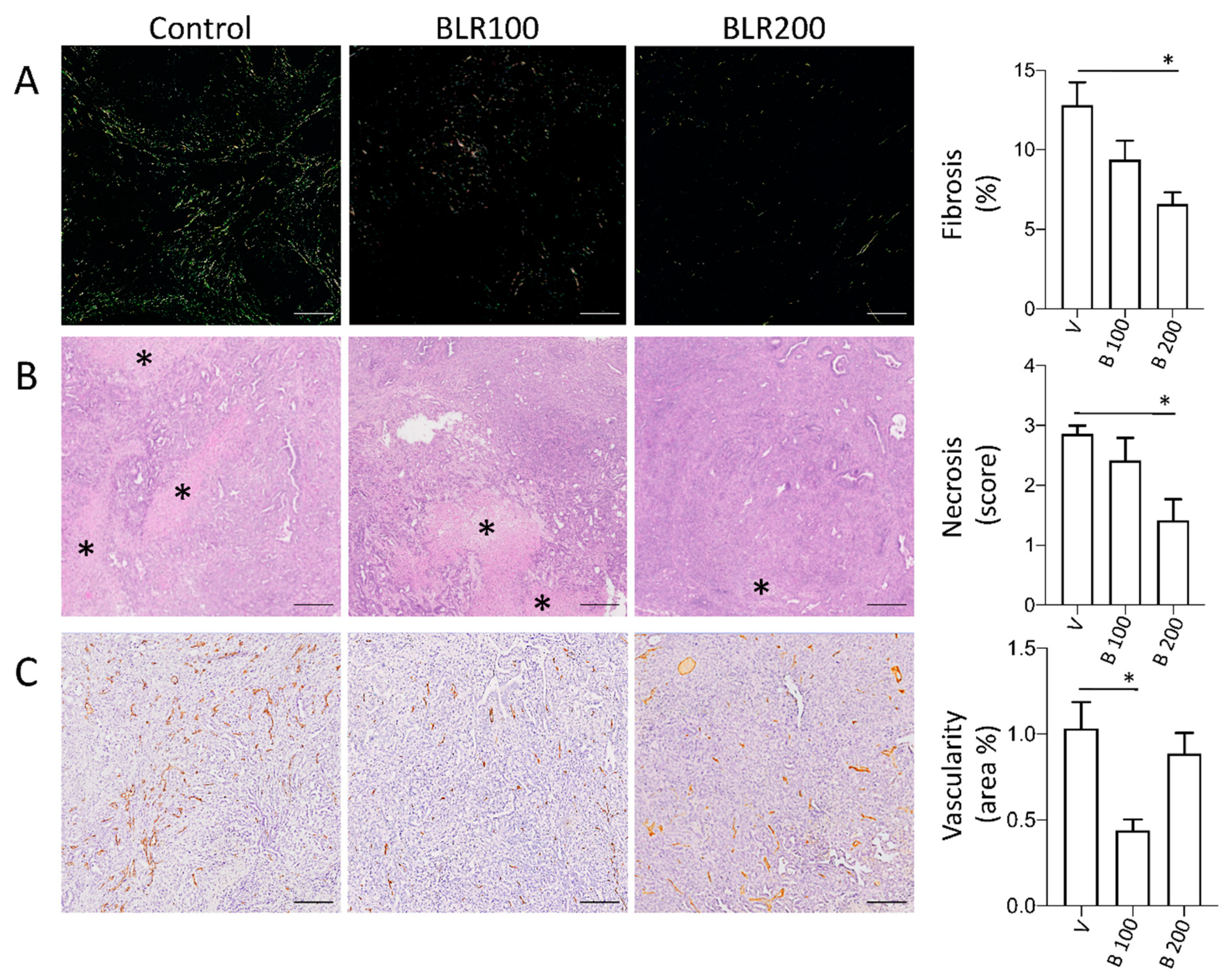
3.2. BLR100 and BLR200 Reorganize the Tumor Microenvironment
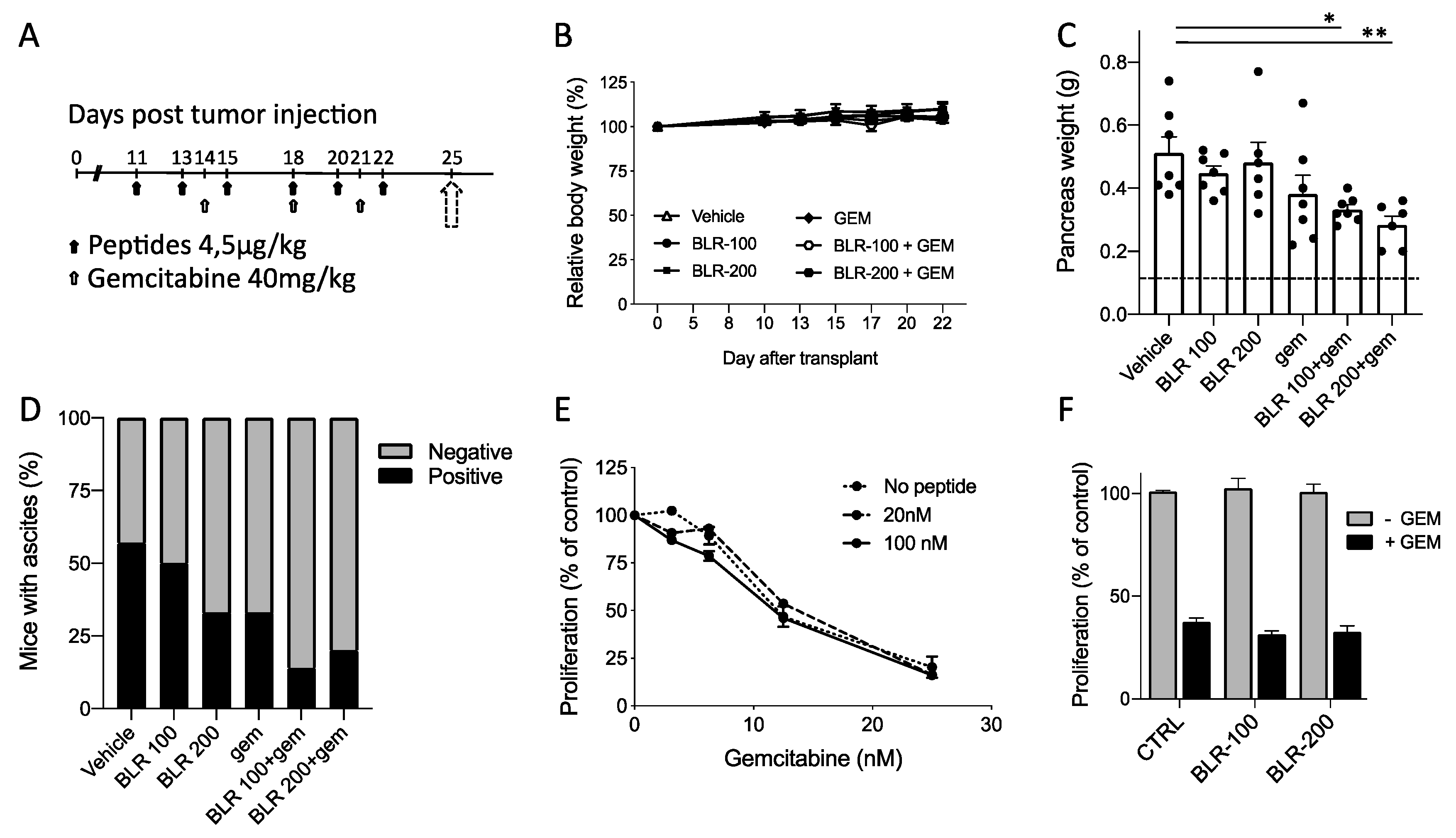
3.3. Antineoplastic activity of BLR100 and BLR200 in Combination with Chemotherapy
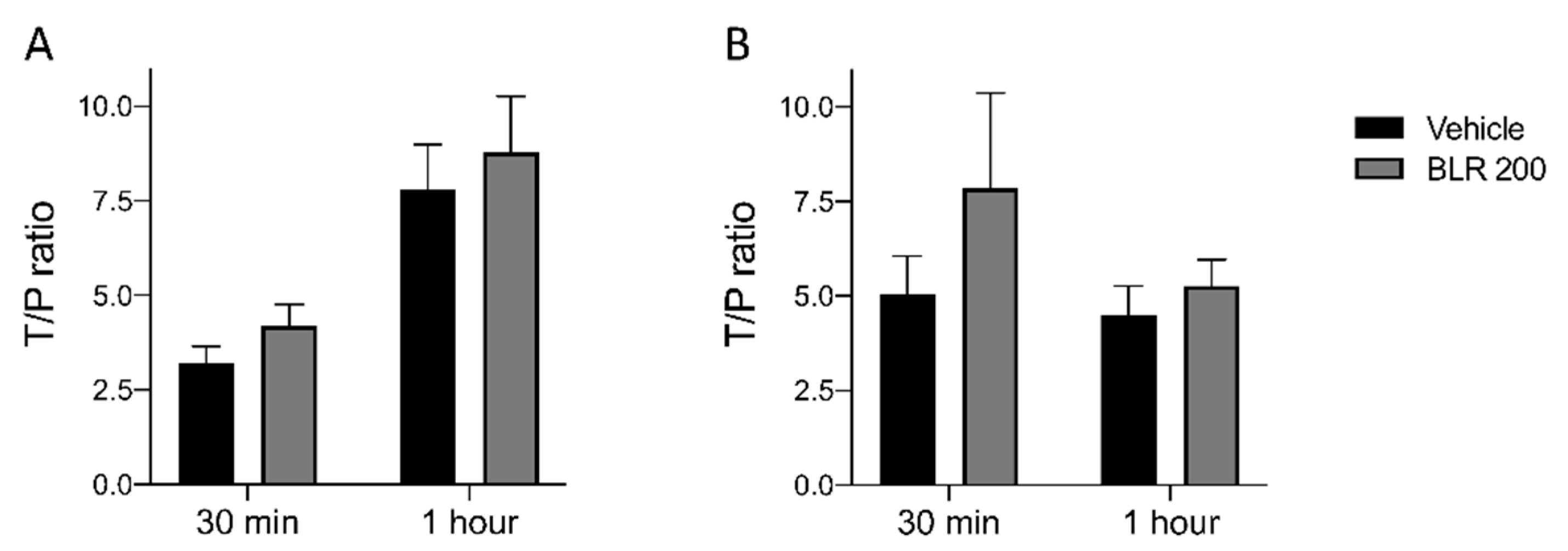
3.4. Pharmacokinetic Studies
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers (Basel) 2018, 10, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merika, E.E.; Syrigos, K.N.; Saif, M.W. Desmoplasia in pancreatic cancer. Can we fight it? Gastroenterol. Res. Pract. 2012, 2012, 781765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafaro, K.J.; Melstrom, L.G. The Paradoxical Web of Pancreatic Cancer Tumor Microenvironment. Am. J. Pathol. 2019, 189, 44–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neesse, A.; Algül, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Belotti, D.; Foglieni, C.; Resovi, A.; Giavazzi, R.; Taraboletti, G. Targeting angiogenesis with compounds from the extracellular matrix. Int. J. Biochem. Cell Biol. 2011, 43, 1674–1685. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.F. Cell surface receptors for CCN proteins. J. Cell Commun. Signal. 2016, 10, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Makino, Y.; Hikita, H.; Kodama, T.; Shigekawa, M.; Yamada, R.; Sakamori, R.; Eguchi, H.; Morii, E.; Yokoi, H.; Mukoyama, M.; et al. CTGF Mediates Tumor-Stroma Interactions between Hepatoma Cells and Hepatic Stellate Cells to Accelerate HCC Progression. Cancer Res. 2018, 78, 4902–4914. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.K.; Maity, G.; Haque, I.; Ghosh, A.; Sarkar, S.; Gupta, V.; Campbell, D.R.; Von Hoff, D.; Banerjee, S. Human pancreatic cancer progression: An anarchy among CCN-siblings. J. Cell Commun. Signal. 2016, 10, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Wenger, C.; Ellenrieder, V.; Alber, B.; Lacher, U.; Menke, A.; Hameister, H.; Wilda, M.; Iwamura, T.; Beger, H.G.; Adler, G.; et al. Expression and differential regulation of connective tissue growth factor in pancreatic cancer cells. Oncogene 1999, 18, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- di Mola, F.F.; Friess, H.; Martignoni, M.E.; Di Sebastiano, P.; Zimmermann, A.; Innocenti, P.; Graber, H.; Gold, L.I.; Korc, M.; Büchler, M.W. Connective tissue growth factor is a regulator for fibrosis in human chronic pancreatitis. Ann. Surg. 1999, 230, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Denichilo, M.; Cortes, P.; Baker, C.; Grondin, J.M.; Yee, J.; Narins, R.G. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J. Am. Soc. Nephrol. 2000, 11, 25–38. [Google Scholar] [PubMed]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Rodrigues-Diez, R.R.; Rodrigues-Diez, R.; Falke, L.L.; Mezzano, S.; Ortiz, A.; Egido, J.; Goldschmeding, R.; Ruiz-Ortega, M. Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J. Pathol. 2018, 244, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cui, H.; Wu, S. CTGF: A potential therapeutic target for Bronchopulmonary dysplasia. Eur. J. Pharm. 2019, 860, 172588. [Google Scholar] [CrossRef] [PubMed]
- Dorn, L.E.; Petrosino, J.M.; Wright, P.; Accornero, F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J. Mol. Cell. Cardiol. 2018, 121, 205–211. [Google Scholar] [CrossRef]
- Kubota, S.; Takigawa, M. Cellular and molecular actions of CCN2/CTGF and its role under physiological and pathological conditions. Clin. Sci. 2015, 128, 181–196. [Google Scholar] [CrossRef]
- Bennewith, K.L.; Huang, X.; Ham, C.M.; Graves, E.E.; Erler, J.T.; Kambham, N.; Feazell, J.; Yang, G.P.; Koong, A.; Giaccia, A.J. The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 2009, 69, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Resovi, A.; Bani, M.R.; Porcu, L.; Anastasia, A.; Minoli, L.; Allavena, P.; Cappello, P.; Novelli, F.; Scarpa, A.; Morandi, E.; et al. Soluble stroma-related biomarkers of pancreatic cancer. Embo Mol. Med. 2018, 10, e8741. [Google Scholar] [CrossRef]
- Aikawa, T.; Gunn, J.; Spong, S.M.; Klaus, S.J.; Korc, M. Connective tissue growth factor-specific antibody attenuates tumor growth, metastasis, and angiogenesis in an orthotopic mouse model of pancreatic cancer. Mol. Cancer 2006, 5, 1108–1116. [Google Scholar] [CrossRef] [Green Version]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riser, B.L.; Najmabadi, F.; Perbal, B.; Rambow, J.A.; Riser, M.L.; Sukowski, E.; Yeger, H.; Riser, S.C.; Peterson, D.R. CCN3/CCN2 regulation and the fibrosis of diabetic renal disease. J. Cell Commun. Signal. 2010, 4, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riser, B.L.; Najmabadi, F.; Perbal, B.; Peterson, D.R.; Rambow, J.A.; Riser, M.L.; Sukowski, E.; Yeger, H.; Riser, S.C. CCN3 (NOV) is a negative regulator of CCN2 (CTGF) and a novel endogenous inhibitor of the fibrotic pathway in an in vitro model of renal disease. Am. J. Pathol. 2009, 174, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riser, B.L.; Najmabadi, F.; Garchow, K.; Barnes, J.L.; Peterson, D.R.; Sukowski, E.J. Treatment with the matricellular protein CCN3 blocks and/or reverses fibrosis development in obesity with diabetic nephropathy. Am. J. Pathol. 2014, 184, 2908–2921. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Barnes, J.L.; Varani, J. Balanced regulation of the CCN family of matricellular proteins: A novel approach to the prevention and treatment of fibrosis and cancer. J. Cell Commun. Signal. 2015, 9, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.a.H.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [Green Version]
- Bapiro, T.E.; Richards, F.M.; Goldgraben, M.A.; Olive, K.P.; Madhu, B.; Frese, K.K.; Cook, N.; Jacobetz, M.A.; Smith, D.-M.; Tuveson, D.A.; et al. A novel method for quantification of gemcitabine and its metabolites 2′,2′-difluorodeoxyuridine and gemcitabine triphosphate in tumour tissue by LC-MS/MS: Comparison with (19)F NMR spectroscopy. Cancer Chemother. Pharm. 2011, 68, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Shi, S.; Meng, Q.; Liang, D.; Ji, S.; Zhang, B.; Qin, Y.; Xu, J.; Ni, Q.; Yu, X. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: Where we are and where we are going. Exp. Mol. Med. 2017, 49, e406. [Google Scholar] [CrossRef] [Green Version]
- Hall-Glenn, F.; De Young, R.A.; Huang, B.-L.; van Handel, B.; Hofmann, J.J.; Chen, T.T.; Choi, A.; Ong, J.R.; Benya, P.D.; Mikkola, H.; et al. CCN2/connective tissue growth factor is essential for pericyte adhesion and endothelial basement membrane formation during angiogenesis. PLoS ONE 2012, 7, e30562. [Google Scholar] [CrossRef] [Green Version]
- Chaqour, B. Caught between a “Rho” and a hard place: Are CCN1/CYR61 and CCN2/CTGF the arbiters of microvascular stiffness? J. Cell Commun. Signal. 2019. [Google Scholar] [CrossRef]
- Toda, N.; Mori, K.; Kasahara, M.; Koga, K.; Ishii, A.; Mori, K.P.; Osaki, K.; Mukoyama, M.; Yanagita, M.; Yokoi, H. Deletion of connective tissue growth factor ameliorates peritoneal fibrosis by inhibiting angiogenesis and inflammation. Nephrol. Dial. Transpl. 2018, 33, 943–953. [Google Scholar] [CrossRef]
- Sakai, N.; Nakamura, M.; Lipson, K.E.; Miyake, T.; Kamikawa, Y.; Sagara, A.; Shinozaki, Y.; Kitajima, S.; Toyama, T.; Hara, A.; et al. Inhibition of CTGF ameliorates peritoneal fibrosis through suppression of fibroblast and myofibroblast accumulation and angiogenesis. Sci. Rep. 2017, 7, 5392. [Google Scholar] [CrossRef] [PubMed]
- Zervos, E.E.; Osborne, D.; Boe, B.A.; Luzardo, G.; Goldin, S.B.; Rosemurgy, A.S. Prognostic significance of new onset ascites in patients with pancreatic cancer. World J. Surg. Oncol. 2006, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ren, Y.; Kang, L.; Zhang, L. Overexpression of CCN3 inhibits inflammation and progression of atherosclerosis in apolipoprotein E-deficient mice. PLoS ONE 2014, 9, e94912. [Google Scholar] [CrossRef]
- Haque, I.; Mehta, S.; Majumder, M.; Dhar, K.; De, A.; McGregor, D.; Van Veldhuizen, P.J.; Banerjee, S.K.; Banerjee, S. Cyr61/CCN1 signaling is critical for epithelial-mesenchymal transition and stemness and promotes pancreatic carcinogenesis. Mol. Cancer 2011, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Maity, G.; Ghosh, A.; Gupta, V.; Haque, I.; Sarkar, S.; Das, A.; Dhar, K.; Bhavanasi, S.; Gunewardena, S.S.; Von Hoff, D.D.; et al. CYR61/CCN1 Regulates dCK and CTGF and Causes Gemcitabine-resistant Phenotype in Pancreatic Ductal Adenocarcinoma. Mol. Cancer 2019, 18, 788–800. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Deng, Y.; Mao, Z.; Ma, X.; Fan, X.; Cui, L.; Qu, J.; Xie, D.; Zhang, J. CCN1 promotes tumorigenicity through Rac1/Akt/NF-κB signaling pathway in pancreatic cancer. Tumour. Biol. 2012, 33, 1745–1758. [Google Scholar] [CrossRef]
- Cesca, M.; Morosi, L.; Berndt, A.; Fuso Nerini, I.; Frapolli, R.; Richter, P.; Decio, A.; Dirsch, O.; Micotti, E.; Giordano, S.; et al. Bevacizumab-Induced Inhibition of Angiogenesis Promotes a More Homogeneous Intratumoral Distribution of Paclitaxel, Improving the Antitumor Response. Mol. Cancer 2016, 15, 125–135. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Resovi, A.; Borsotti, P.; Ceruti, T.; Passoni, A.; Zucchetti, M.; Berndt, A.; Riser, B.L.; Taraboletti, G.; Belotti, D. CCN-Based Therapeutic Peptides Modify Pancreatic Ductal Adenocarcinoma Microenvironment and Decrease Tumor Growth in Combination with Chemotherapy. Cells 2020, 9, 952. https://doi.org/10.3390/cells9040952
Resovi A, Borsotti P, Ceruti T, Passoni A, Zucchetti M, Berndt A, Riser BL, Taraboletti G, Belotti D. CCN-Based Therapeutic Peptides Modify Pancreatic Ductal Adenocarcinoma Microenvironment and Decrease Tumor Growth in Combination with Chemotherapy. Cells. 2020; 9(4):952. https://doi.org/10.3390/cells9040952
Chicago/Turabian StyleResovi, Andrea, Patrizia Borsotti, Tommaso Ceruti, Alice Passoni, Massimo Zucchetti, Alexander Berndt, Bruce L. Riser, Giulia Taraboletti, and Dorina Belotti. 2020. "CCN-Based Therapeutic Peptides Modify Pancreatic Ductal Adenocarcinoma Microenvironment and Decrease Tumor Growth in Combination with Chemotherapy" Cells 9, no. 4: 952. https://doi.org/10.3390/cells9040952