Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants

 ,
, 
Abstract
:1. Overview on Cytokines
2. The Concept of Onco-Inflammation
3. MPNs as a Model of Onco-Inflammatory Disorders
Role of the Megakaryocytic Clone in Cytokine Production in MPNs
4. Cytokine Profile in MPN Patients
4.1. Circulating and Bone Marrow Cytokine Levels
4.2. Cytokine Gene Expression Profiling
5. Correlations of Cytokine Profile with Symptoms and Thrombosis
6. Correlations of Cytokine Profile with Driver Mutations
7. Host Genetic Variants Predisposing to Chronic Inflammatory State in MPNs
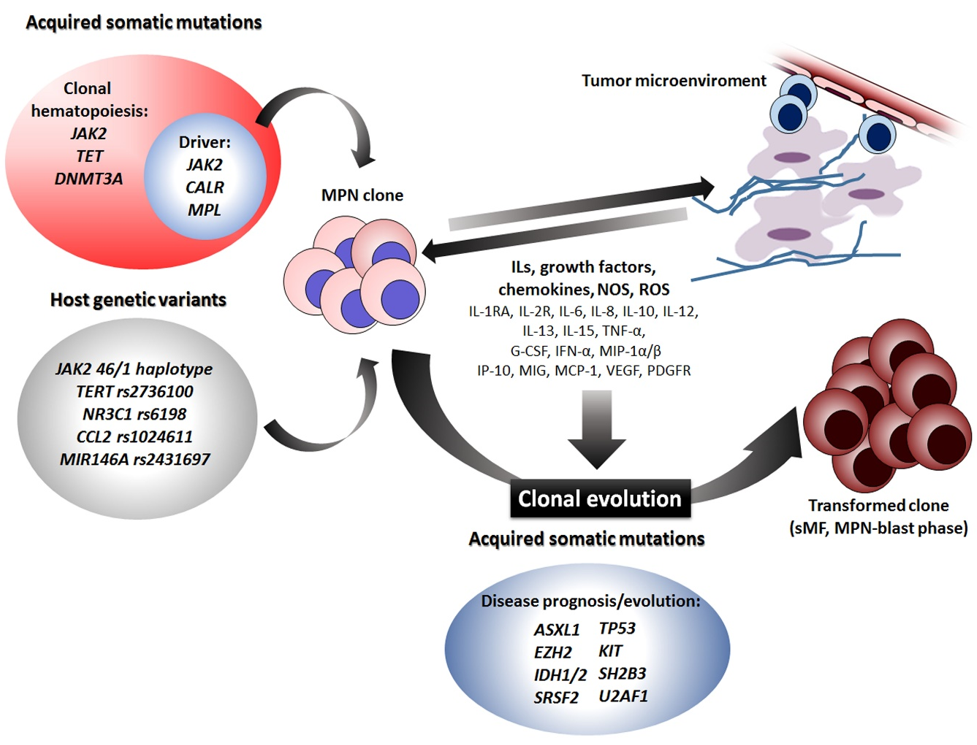
8. Clonal Hematopoiesis, Cytokines, and Host Genetic Background
9. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ramani, T.; Auletta, C.S.; Weinstock, D.; Mounho-Zamora, B.; Ryan, P.C.; Salcedo, T.W.; Bannish, G. Cytokines: The Good, the Bad, and the Deadly. Int. J. Toxicol. 2015, 34, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Historical insights into cytokines. Eur. J. Immunol. 2007, 37, S34–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Landskron, G.; De La Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Bergers, G. Tumors vs. Chronic Wounds: An Immune Cell’s Perspective. Front. Immunol. 2019, 10, 2178. [Google Scholar] [CrossRef] [Green Version]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, inflammation and cancer. Semin. Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef]
- Barosi, G. An Immune Dysregulation in MPN. Curr. Hematol. Malign Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [Green Version]
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Yogarajah, M.; Tefferi, A. Leukemic Transformation in Myeloproliferative Neoplasms. Mayo Clin. Proc. 2017, 92, 1118–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- Sapre, M.; Tremblay, D.; Wilck, E.; James, A.; Leiter, A.; Coltoff, A.; Koshy, A.G.; Kremyanskaya, M.; Hoffman, R.; Mascarenhas, J.O.; et al. Metabolic Effects of JAK1/2 Inhibition in Patients with Myeloproliferative Neoplasms. Sci. Rep. 2019, 9, 16609. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.; Teo, S.-S.; Tiedt, R.; Passweg, J.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation ofJAK2in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Mascarenhas, J.; Mughal, T.I.; Verstovsek, S. Biology and Clinical Management of Myeloproliferative Neoplasms and Development of the JAK Inhibitor Ruxolitinib. Curr. Med. Chem. 2012, 19, 4399–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibundgut, E.O.; Horn, M.P.; Brunold, C.; Pfanner-Meyer, B.; Marti, D.; Hirsiger, H.; Tobler, A.; Zwicky, C. Hematopoietic and endothelial progenitor cell trafficking in patients with myeloproliferative diseases. Haematologica 2006, 91, 1465–1472. [Google Scholar]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Levine, R.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.; Wolanskyj, A.P.; Hogan, W.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Them, N.C.C.; Berg, T.; Vladimer, G.I.; Bagienski, K.; Milanesi, C.; Casetti, I.C.; Sant’Antonio, E.; Ferretti, V.V.; Schischlik, F.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Pecquet, C.; Chachoua, I.; Roy, A.; Balligand, T.; Vertenoeil, G.; Leroy, E.; Albu, R.-I.; Defour, J.-P.; Nivarthi, H.; Hug, E.; et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood 2019, 133, 2669–2681. [Google Scholar] [CrossRef]
- Lanikova, L.; Babosova, O.; Prchal, J.T. Experimental Modeling of Myeloproliferative Neoplasms. Genes 2019, 10, 813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Vannucchi, A.M. Genetic Risk Assessment in Myeloproliferative Neoplasms. Mayo Clin. Proc. 2017, 92, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Desterke, C.; Martinaud, C.; Ruzehaji, N.; Le Bousse-Kerdilès, M.-C. Inflammation as a Keystone of Bone Marrow Stroma Alterations in Primary Myelofibrosis. Mediat. Inflamm. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lussana, F.; Carobbio, A.; Salmoiraghi, S.; Guglielmelli, P.; Vannucchi, A.M.; Bottazzi, B.; Leone, R.; Mantovani, A.; Barbui, T.; Rambaldi, A. Driver mutations (JAK2V617F, MPLW515L/K or CALR), pentraxin-3 and C-reactive protein in essential thrombocythemia and polycythemia vera. J. Hematol. Oncol. 2017, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Di Marcantonio, D.; Martinez, E.; Sidoli, S.; Vadaketh, J.; Nieborowska-Skorska, M.; Gupta, A.; Meadows, J.M.; Ferraro, F.; Masselli, E.; Challen, G.A.; et al. Protein Kinase C Epsilon Is a Key Regulator of Mitochondrial Redox Homeostasis in Acute Myeloid Leukemia. Clin. Cancer Res. 2017, 24, 608–618. [Google Scholar] [CrossRef] [Green Version]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Wajant, H. The Fas Signaling Pathway: More Than a Paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Mirandola, P.; Sponzilli, I.; Gobbi, G.; Marmiroli, S.; Rinaldi, L.; Binazzi, R.; Piccari, G.G.; Ramazzotti, G.; Gaboardi, G.C.; Cocco, L.; et al. Anticancer agents sensitize osteosarcoma cells to TNF-related apoptosis-inducing ligand downmodulating IAP family proteins. Int. J. Oncol. 2006, 28, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Mirandola, P.; Gobbi, G.; Ponti, C.; Sponzilli, I.; Cocco, L.; Vitale, M. PKCepsilon controls protection against TRAIL in erythroid progenitors. Blood 2006, 107, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Gobbi, G.; Mirandola, P.; Sponzilli, I.; Micheloni, C.; Malinverno, C.; Cocco, L.; Vitale, M. Timing and expression level of protein kinase C epsilon regulate the megakaryocytic differentiation of human CD34 cells. Stem Cells 2007, 25, 2322–2329. [Google Scholar] [CrossRef] [PubMed]
- Ciurea, S.O.; Merchant, D.; Mahmud, N.; Ishii, T.; Zhao, Y.; Hu, W.; Bruno, E.; Barosi, G.; Xu, M.; Hoffman, R. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood 2007, 110, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Masselli, E.; Carubbi, C.; Gobbi, G.; Mirandola, P.; Galli, D.; Martini, S.; Bonomini, S.; Crugnola, M.; Craviotto, L.; Aversa, F.; et al. Protein kinase Cepsilon inhibition restores megakaryocytic differentiation of hematopoietic progenitors from primary myelofibrosis patients. Leukemia 2015, 29, 2192–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tognon, R.; Gasparotto, E.P.L.; Leroy, J.M.G.; De Oliveira, G.L.V.; Neves, R.P.; Carrara, R.D.C.V.; Kashima, S.; Covas, D.T.; Santana, M.; Souto, E.X.; et al. Differential expression of apoptosis-related genes from death receptor pathway in chronic myeloproliferative diseases. J. Clin. Pathol. 2010, 64, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Thiele, J.; Vannucchi, A.M.; Tefferi, A. Myeloproliferative neoplasms: Morphology and clinical practice. Am. J. Hematol. 2016, 91, 430–433. [Google Scholar] [CrossRef] [Green Version]
- Carubbi, C.; Masselli, E.; Martini, S.; Galli, D.; Aversa, F.; Mirandola, P.; Italiano, J.E., Jr.; Gobbi, G.; Vitale, M. Human thrombopoiesis depends on Protein kinase Cdelta/protein kinase Cepsilon functional couple. Haematologica 2016, 101, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Balduini, A.; Badalucco, S.; Pugliano, M.T.; Baev, D.; De Silvestri, A.; Cattaneo, M.; Rosti, V.; Barosi, G. In Vitro Megakaryocyte Differentiation and Proplatelet Formation in Ph-Negative Classical Myeloproliferative Neoplasms: Distinct Patterns in the Different Clinical Phenotypes. PLoS ONE 2011, 6, e21015. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, M.; Falanga, A. Thrombosis in Myeloproliferative Neoplasms. Semin. Thromb. Hemost. 2014, 40, 348–358. [Google Scholar] [CrossRef]
- Parashar, Y.; Kushwaha, R.; Kumar, A.; Agarwal, K.; Singh, U.S.; Jain, M.; Verma, S.P.; Tripathi, A.K. Haemostatic Profile in Patients of Myeloproliferative Neoplasms-A Tertiary Care Centre Experience. J. Clin. Diagn. Res. 2016, 10, EC01–EC04. [Google Scholar] [CrossRef]
- Carubbi, C.; Masselli, E.; Nouvenne, A.; Russo, M.; Galli, D.; Mirandola, P.; Gobbi, G.; Vitale, M. Laboratory diagnostics of inherited platelet disorders. Clin. Chem. Lab. Med. 2014, 52, 1091–1106. [Google Scholar] [CrossRef] [Green Version]
- Marneth, A.E.; Mullally, A. Busy signal: Platelet-derived growth factor activation in myelofibrosis. Haematologica 2020, 105, 1988–1990. [Google Scholar] [CrossRef] [PubMed]
- Woods, B.; Chen, W.; Chiu, S.; Marinaccio, C.; Fu, C.; Gu, L.; Bulic, M.; Yang, Q.; Zouak, A.; Jia, S.; et al. Activation of JAK/STAT Signaling in Megakaryocytes Sustains Myeloproliferation In Vivo. Clin. Cancer Res. 2019, 25, 5901–5912. [Google Scholar] [CrossRef] [PubMed]
- Malara, A.; Abbonante, V.; Zingariello, M.; Migliaccio, A.R.; Balduini, A. Megakaryocyte Contribution to Bone Marrow Fibrosis: Many Arrows in the Quiver. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingariello, M.; Martelli, F.; Verachi, P.; Bardelli, C.; Gobbo, F.; Mazzarini, M.; Migliaccio, A.R. Novel targets to cure primary myelofibrosis from studies on Gata1 low mice. IUBMB Life 2019, 72, 131–141. [Google Scholar] [CrossRef]
- Zingariello, M.; Martelli, F.; Ciaffoni, F.; Masiello, F.; Ghinassi, B.; D’Amore, E.; Massa, M.; Barosi, G.; Sancillo, L.; Li, X.; et al. Characterization of the TGF-beta1 signaling abnormalities in the Gata1low mouse model of myelofibrosis. Blood 2013, 121, 3345–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingariello, M.; Ruggeri, A.; Martelli, F.; Marra, M.; Sancillo, L.; Ceglia, I.; Rana, R.A.; Migliaccio, A.R. A novel interaction between megakaryocytes and activated fibrocytes increases TGF-beta bioavailability in the Gata1(low) mouse model of myelofibrosis. Am. J. Blood Res. 2015, 5, 34–61. [Google Scholar]
- Spangrude, G.J.; Lewandowski, D.; Martelli, F.; Marra, M.; Zingariello, M.; Sancillo, L.; Rana, R.A.; Migliaccio, A.R. P-Selectin Sustains Extramedullary Hematopoiesis in theGata1lowModel of Myelofibrosis. STEM CELLS 2015, 34, 67–82. [Google Scholar] [CrossRef]
- Ceglia, I.; Dueck, A.C.; Masiello, F.; Martelli, F.; He, W.; Federici, G.; Petricoin, E.F.; Zeuner, A.; Iancu-Rubin, C.; Weinberg, R.; et al. Preclinical rationale for TGF-β inhibition as a therapeutic target for the treatment of myelofibrosis. Exp. Hematol. 2016, 44, 1138–1155. [Google Scholar] [CrossRef] [Green Version]
- Kramer, F.; Dernedde, J.; Mezheyeuski, A.; Tauber, R.; Micke, P.; Kappert, K. Platelet-derived growth factor receptor beta activation and regulation in murine myelofibrosis. Haematologica 2020, 105, 2083–2094. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Hsu, H.-C.; Tsai, W.-H.; Jiang, M.-L.; Ho, C.-H.; Hsu, M.-L.; Ho, C.-K.; Wang, S.-Y. Circulating levels of thrombopoietic and inflammatory cytokines in patients with clonal and reactive thrombocytosis. J. Lab. Clin. Med. 1999, 134, 392–397. [Google Scholar] [CrossRef]
- Bourantas, K.L.; Hatzimichael, E.C.; Makis, A.C.; Chaidos, A.; Kapsali, E.D.; Tsiara, S.; Mavridis, A. Serum beta-2-microglobulin, TNF-alpha and interleukins in myeloproliferative disorders. Eur. J. Haematol. 1999, 63, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Panteli, K.E.; Hatzimichael, E.; Bouranta, P.K.; Katsaraki, A.; Seferiadis, K.; Stebbing, J.; Bourantas, K.L. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br. J. Haematol. 2005, 130, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene 2010, 30, 990–1001. [Google Scholar] [CrossRef] [Green Version]
- Hermouet, S.; Godard, A.; Pineau, D.; Corre, I.; Raher, S.; Lippert, E.; Jacques, Y. Abnormal production of interleukin (IL)-11 and IL-8 in polycythaemia vera. Cytokine 2002, 20, 178–183. [Google Scholar] [CrossRef]
- Gangemi, S.; Allegra, A.; Pace, E.; Alonci, A.; Ferraro, M.; Petrungaro, A.; Saitta, S.; Gerace, D.; Russo, S.; Penna, G.; et al. Evaluation of interleukin-23 plasma levels in patients with polycythemia vera and essential thrombocythemia. Cell. Immunol. 2012, 278, 91–94. [Google Scholar] [CrossRef]
- Ho, C.-L.; Lasho, T.L.; Butterfield, J.H.; Tefferi, A. Global cytokine analysis in myeloproliferative disorders. Leuk. Res. 2007, 31, 1389–1392. [Google Scholar] [CrossRef]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef]
- Pardanani, A.; Begna, K.; Finke, C.; Lasho, T.; Tefferi, A. Circulating levels of MCP-1, sIL-2R, IL-15, and IL-8 predict anemia response to pomalidomide therapy in myelofibrosis. Am. J. Hematol. 2011, 86, 343–345. [Google Scholar] [CrossRef]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; Tefferi, A. Plasma cytokines in polycythemia vera: Phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am. J. Hematol. 2012, 87, 1003–1005. [Google Scholar] [CrossRef]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Cacemiro, M.C.; Cominal, J.G.; Tognon, R.; Nunes, N.D.S.; Simões, B.P.; De Figueiredo-Pontes, L.L.; Catto, L.F.B.; Traina, F.; Souto, E.X.; Zambuzi, F.A.; et al. Philadelphia-negative myeloproliferative neoplasms as disorders marked by cytokine modulation. Hematol. Transfus. Cell Ther. 2018, 40, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Mambet, C.; Necula, L.G.; Mihai, S.; Matei, L.; Bleotu, C.; Chivu-Economescu, M.; Stanca, O.; Tatic, A.; Berbec, N.; Tanase, C.; et al. Increased Dkk-1 plasma levels may discriminate disease subtypes in myeloproliferative neoplasms. J. Cell. Mol. Med. 2018, 22, 4005–4011. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Campanelli, R.; Catarsi, P.; De Amici, M.; Abba, C.; Viarengo, G.; Villani, L.; Gale, R.P.; Rosti, V.; Massa, M. Plasma sIL-2Ralpha levels are associated with disease progression in myelofibrosis with JAK2(V617F) but not CALR mutation. Leuk. Res. 2020, 90, 106319. [Google Scholar] [CrossRef] [PubMed]
- Øbro, N.F.; Grinfeld, J.; Belmonte, M.; Irvine, M.; Shepherd, M.S.; Rao, T.N.; Karow, A.; Riedel, L.M.; Harris, O.B.; Baxter, E.J.; et al. Longitudinal Cytokine Profiling Identifies GRO-α and EGF as Potential Biomarkers of Disease Progression in Essential Thrombocythemia. HemaSphere 2020, 4, e371. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Cytokine Profiling as a Novel Complementary Tool to Predict Prognosis in MPNs? HemaSphere 2020, 4, e407. [Google Scholar] [CrossRef]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Whole-blood transcriptional profiling of interferon-inducible genes identifies highly upregulated IFI27 in primary myelofibrosis. Eur. J. Haematol. 2011, 87, 54–60. [Google Scholar] [CrossRef]
- Kabanova, S. Gene expression analysis of human red blood cells. Int. J. Med. Sci. 2009, 6, 156. [Google Scholar] [CrossRef] [Green Version]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leuk. Res. 2012, 36, 1387–1392. [Google Scholar] [CrossRef]
- Wong, W.J.; Baltay, M.; Getz, A.; Fuhrman, K.; Aster, J.C.; Hasserjian, R.P.; Pozdnyakova, O.A. Gene expression profiling distinguishes prefibrotic from overtly fibrotic myeloproliferative neoplasms and identifies disease subsets with distinct inflammatory signatures. PLoS ONE 2019, 14, e0216810. [Google Scholar] [CrossRef] [Green Version]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Miller, C.B.; Thyne, M.; Mangan, J.; Goldberger, S.; Fazal, S.; Ma, X.; Wilson, W.; Paranagama, D.; Dubinski, D.; et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: The MPN Landmark survey. BMC Cancer 2016, 16, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherber, R.M.; Geyer, H.L.; Mesa, R.A. Quality of Life in MPN Comes of Age as a Therapeutic Target. Curr. Hematol. Malign Rep. 2014, 9, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Guglielmelli, P.; Salmoiraghi, S.; Rosti, V.; Rambaldi, A.; Vannucchi, A.M.; Barosi, G. Elevated C-reactive protein is associated with shortened leukemia-free survival in patients with myelofibrosis. Leukemia 2013, 27, 2084–2086. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2010, 96, 315–318. [Google Scholar] [CrossRef]
- Masselli, E.; Carubbi, C.; Pozzi, G.; Martini, S.; Aversa, F.; Galli, D.; Gobbi, G.; Mirandola, P.; Vitale, M. Platelet expression of PKCepsilon oncoprotein in myelofibrosis is associated with disease severity and thrombotic risk. Ann. Transl. Med. 2017, 5, 273. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Masarova, L.; Bose, P.; Verstovsek, S. The Rationale for Immunotherapy in Myeloproliferative Neoplasms. Curr. Hematol. Malign Rep. 2019, 14, 310–327. [Google Scholar] [CrossRef]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef] [Green Version]
- Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. The JAK2 GGCC (46/1) Haplotype in Myeloproliferative Neoplasms: Causal or Random? Int. J. Mol. Sci. 2018, 19, 1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahajevszky, S.; Andrikovics, H.; Batai, A.; Adam, E.; Bors, A.; Csomor, J.; Gopcsa, L.; Koszarska, M.; Kozma, A.; Lovas, N.; et al. The prognostic impact of germline 46/1 haplotype of Janus kinase 2 in cytogenetically normal acute myeloid leukemia. Haematologica 2011, 96, 1613–1618. [Google Scholar]
- Ferguson, L.; Han, D.Y.; Fraser, A.G.; Huebner, C.; Lam, W.J.; Morgan, A.R.; Duan, H.; Karunasinghe, N. Genetic factors in chronic inflammation: Single nucleotide polymorphisms in the STAT-JAK pathway, susceptibility to DNA damage and Crohn’s disease in a New Zealand population. Mutat. Res. Mol. Mech. Mutagen. 2010, 690, 108–115. [Google Scholar] [CrossRef]
- Hermouet, S.; Vilaine, M. The JAK2 46/1 haplotype: A marker of inappropriate myelomonocytic response to cytokine stimulation, leading to increased risk of inflammation, myeloid neoplasm, and impaired defense against infection? Haematologica 2011, 96, 1575–1579. [Google Scholar] [CrossRef]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Fu, P.; Pang, Y.; Liu, C.; Shao, Z.; Zhu, J.; Li, J.; Wang, T.; Zhang, X.; Liu, J. TERT rs2736100T/G polymorphism upregulates interleukin 6 expression in non-small cell lung cancer especially in adenocarcinoma. Tumor Boil. 2014, 35, 4667–4672. [Google Scholar] [CrossRef]
- Kino, T.; Su, Y.A.; Chrousos, G.P. Human glucocorticoid receptor isoform beta: Recent understanding of its potential implications in physiology and pathophysiology. Cell Mol. Life Sci. 2009, 66, 3435–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRijk, R.H.; Schaaf, M.J.; Turner, G.; Datson, N.A.; Vreugdenhil, E.; Cidlowski, J.; De Kloet, E.R.; Emery, P.; Sternberg, E.M.; Detera-Wadleigh, S.D. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J. Rheumatol. 2001, 28, 2383–2388. [Google Scholar]
- Varricchio, L.; Masselli, E.; Alfani, E.; Battistini, A.; Migliaccio, G.; Vannucchi, A.M.; Zhang, W.; Rondelli, D.; Godbold, J.; Ghinassi, B.; et al. The dominant negative beta isoform of the glucocorticoid receptor is uniquely expressed in erythroid cells expanded from polycythemia vera patients. Blood 2011, 118, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Poletto, V.; Rosti, V.; Villani, L.; Catarsi, P.; Carolei, A.; Campanelli, R.; Massa, M.; Martinetti, M.; Viarengo, G.; Malovini, A.; et al. A3669G polymorphism of glucocorticoid receptor is a susceptibility allele for primary myelofibrosis and contributes to phenotypic diversity and blast transformation. Blood 2012, 120, 3112–3117. [Google Scholar] [CrossRef] [Green Version]
- Masselli, E.; Carubbi, C.; Cambò, B.; Pozzi, G.; Gobbi, G.; Mirandola, P.; Follini, E.; Pagliaro, L.; Di Marcantonio, D.; Bonatti, F.; et al. The -2518 A/G polymorphism of the monocyte chemoattractant protein-1 as a candidate genetic predisposition factor for secondary myelofibrosis and biomarker of disease severity. Leukemia 2018, 32, 2266–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melgarejo, E.; Medina, M. Ángel; Sánchez-Jiménez, F.; Urdiales, J.L. Monocyte chemoattractant protein-1: A key mediator in inflammatory processes. Int. J. Biochem. Cell Boil. 2009, 41, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Lu, L.; Saxena, R. A Novel Polymorphism in the MCP-1 Gene Regulatory Region That Influences MCP-1 Expression. Biochem. Biophys. Res. Commun. 1999, 259, 344–348. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.H.; Yang, Q.; Kathiresan, S.; Cupples, L.A.; Massaro, J.; Keaney, J.F.J.; Larson, M.G.; Vasan, R.S.; Hirschhorn, J.N.; O’Donnell, C.J.; et al. CCL2 Polymorphisms Are Associated With Serum Monocyte Chemoattractant Protein-1 Levels and Myocardial Infarction in the Framingham Heart Study. Circulation 2005, 112, 1113–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Marín, F.; Arroyo, A.B.; Bellosillo, B.; Cuenca, E.J.; Zamora, L.; Hernández-Rivas, J.M.; Hernández-Boluda, J.C.; Fernandez-Rodriguez, C.; Luño, E.; Hernandez, C.G.; et al. miR-146a rs2431697 identifies myeloproliferative neoplasm patients with higher secondary myelofibrosis progression risk. Leukemia 2020, 1–12. [Google Scholar] [CrossRef]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef]
- Sano, S.; Wang, Y.; Yura, Y.; Sano, M.; Oshima, K.; Yang, Y.; Katanasaka, Y.; Min, K.-D.; Matsuura, S.; Ravid, K.; et al. JAK2 V617F -Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl. Sci. 2019, 4, 684–697. [Google Scholar] [CrossRef]
- Fabre, M.A.; McKerrell, T.; Zwiebel, M.; Vijayabaskar, M.S.; Park, N.R.; Wells, P.M.; Rad, R.; Deloukas, P.; Small, K.S.; Steves, C.J.; et al. Concordance for clonal hematopoiesis is limited in elderly twins. Blood 2020, 135, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.W.; Pedersen, D.A.; Larsen, L.A.; Husby, S.; Clemmensen, S.B.; Hjelmborg, J.B.; Favero, F.; Weischenfeldt, J.; Christensen, K.; Grønbæk, K. Clonal hematopoiesis in elderly twins: Concordance, discordance, and mortality. Blood 2020, 135, 261–268. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| ET | PV | PMF | References | ||
|---|---|---|---|---|---|
| Pro-Inflammatory Cytokines | L-1α | = | = | = | [62,63] |
| IL-1β | ↑/= | ↑ | ↑/= | [62,63,68,72,80] | |
| IL-2 | ↑ | ↑ | ↑/= | [62,63,68] | |
| IL-2R | ↑ | ↑ | ↑ | [62,63,68,73,74] | |
| IL-5 | ↑/= | ↑ | = | [68,70,72] | |
| IL-6 | ↑/= | ↑ | ↑ | [61,62,63,68,70,72] | |
| sIL-6 | ↑ | nd | nd | [61] | |
| IL-7 | nd | ↑ | = | [68,70] | |
| IL-12 | ↑ | ↑ | ↑ | [68,70,72] | |
| IL-13 | nd | ↑ | ↑ | [68,70] | |
| IL-15 | nd | nd | ↑ | [68] | |
| IL-17 | = | = | ↑/= | [68,72] | |
| IL-23 | = | ↑ | nd | [66] | |
| TNF-α | ↑/= | ↑ | ↑ | [68,72,75,80] | |
| INF-α | ↑ | ↑ | ↑ | [68,72] | |
| INF-γ | = | ↑ | ↓/↑ | [68,70,72] | |
| Anti-Inflammatory Cytokines | IL-1RA | nd | ↑ | ↑ | [68,70] |
| IL-4 | ↑ | ↑ | ↑/= | [68,72,79] | |
| IL-6 | ↑/= | ↑ | ↑ | [61,68,70,72] | |
| IL-10 | ↑/= | ↑/= | ↑ | [62,66,68,72,79] | |
| IL-11 | nd | ↑ | nd | [64,65] | |
| IL-13 | nd | ↑ | ↑ | [68,70,72] | |
| Chemokines | MCP-1 | ↑/= | ↑/= | ↑/= | [64,65,68,70,72,80] |
| MIP-1α | ↑ | ↑ | ↑ | [67,68,70,72] | |
| MIP-1β | ↑ | ↑ | ↑/= | [68,70,72] | |
| IL-8 | ↑ | ↑ | ↑ | [61,64,65,68,70,75] | |
| RANTES | ↑ | =/↓ | ↑/= | [68,70,72] | |
| IP-9 | ↑ | ↑ | ↑ | [73] | |
| IP-10 | = | ↑ | ↑ | [68,70,72,75,80] | |
| MIG | nd | ↑ | ↑ | [68,70,79] | |
| GRO-α | ↑ | = | = | [75] | |
| CCL11 | ↑ | ↑ | = | [68,75] | |
| Growth Factors | GM-CSF | ↑ | ↑ | ↑/= | [70,72] |
| G-CSF | nd | nd | ↑ | [68,79] | |
| HGF | nd | ↑ | ↑ | [64,65,68,70,79] | |
| PDGF | ↑ | ↑ | ↑ | [73,80] | |
| VEGF | = | ↑/= | ↑ | [68,70,79] | |
| EGF | ↑ | ↓/↑ | ↑ | [70,73,75,79] | |
| FGF | nd | nd | = | [68] | |
| TPO | = | = | ↑ | [61,63] | |
| SCF | ↑ | nd | nd | [61] | |
| TGF-β | = | = | ↑ | [80] | |
| Pro-Fibrotic Cytokines | MCP-1 | ↑/= | ↑/= | ↑/= | [64,65,68,70,72,80] |
| IL-8 | ↑ | ↑ | ↑ | [61,64,65,68,70,75] | |
| PDGF | ↑ | ↑ | ↑ | [73,80] | |
| EGF | ↑ | ↓/↑ | ↑ | [70,73,75,79] | |
| FGF | nd | nd | = | [68] | |
| TGFβ | = | = | ↑ | [80] |
| PMF | References | ||
|---|---|---|---|
| Pro-Inflammatory Cytokines | IL-1α | = | [63] |
| IL-1β | ↑/= | [70,80] | |
| IL-2 | ↑ | [63] | |
| IL-2R | ↑ | [63,70] | |
| IL-5 | nd | ||
| IL-6 | ↑ | [63] | |
| sIL-6 | nd | ||
| IL-7 | ↓ | [70] | |
| IL-12 | ↑ | [70,72] | |
| IL-13 | nd | ||
| IL-15 | nd | ||
| IL-17 | ↑ | [72] | |
| IL-23 | nd | ||
| TNF-α | ↑ | [72,80] | |
| INF-α | ↑ | [70,72] | |
| INF-γ | ↓/↑ | [70,72] | |
| Anti-inflammatory Cytokines | IL-1RA | ↑ | [70] |
| IL-4 | ↑ | [72] | |
| IL-6 | nd | ||
| IL-10 | ↑ | [70] | |
| IL-11 | nd | ||
| IL-13 | nd | ||
| Chemokines | MCP-1 | ↑/= | [70,72,80] |
| MIP-1α | ↑/↓ | [70] | |
| MIP-1β | ↑ | [72] | |
| IL-8 | nd | ||
| RANTES | ↑ | [70] | |
| IP-10 | ↑/↓ | [70,72] | |
| MIG | ↓ | [70] | |
| GRO-α | ↓ | [75] | |
| CCL11 | ↓ | [70,75] | |
| Growth Factors | GM-CSF | ↑/↓ | [70,72] |
| G-CSF | nd | ||
| HGF | nd | ||
| PDGF | nd | ||
| VEGF | ↓ | [70,79] | |
| EGF | ↑/↓ | [70,75] | |
| FGF | ↑ | [70] | |
| TPO | ↑ | [63] | |
| SCF | nd | ||
| TGF-β | ↑ | [80] | |
| Pro-Fibrotic Cytokines | MCP-1 | ↑/= | [72,80] |
| IL-8 | nd | ||
| PDGF | nd | ||
| EGF | ↑/↓ | [70,75] | |
| FGF | ↑ | [70] | |
| TGFβ | ↑ | [80] |
| ET | References | ||
|---|---|---|---|
| Pro-Inflammatory Cytokines | L-1α | = | [62,63] |
| IL-1β | = | [71,72] | |
| IL-2 | = | [63] | |
| IL-2R | = | [63] | |
| IL-5 | = | [72] | |
| IL-6 | = | [71,72] | |
| IL-7 | nd | ||
| IL-12 | nd | ||
| IL-13 | nd | ||
| IL-15 | nd | ||
| IL-17 | = | [72] | |
| IL-23 | ↓ | [66] | |
| TNF-α | = | [71,72] | |
| INF-α | = | [72] | |
| INF-γ | ↑/= | [71,72] | |
| Anti-Inflammatory Cytokines | IL-1RA | nd | |
| IL-4 | ↑/= | [71,72] | |
| IL-6 | nd | ||
| IL-10 | = | [66,71,72] | |
| IL-11 | nd | ||
| IL-13 | nd | ||
| Chemokines | MCP-1 | ↑ | [71] |
| MIP-1α | = | [72] | |
| MIP-1β | nd | ||
| IL-8 | ↑ | [71] | |
| RANTES | ↑ | [72] | |
| IP-10 | = | [72] | |
| MIG | nd | ||
| GRO-α | ↑ | [75] | |
| CCL11 | = | [75] | |
| Growth Factors | GM-CSF | ↑/= | [71,72] |
| G-CSF | nd | ||
| HGF | nd | ||
| PDGF | ↑ | [71] | |
| VEGF | ↑ | [71] | |
| EGF | = | [75] | |
| FGF | nd | ||
| TPO | = | [63] | |
| SCF | nd | ||
| TGF-β | = | [67] | |
| Pro-Fibrotic Cytokines | MCP-1 | ↑ | [71] |
| IL-8 | ↑ | [71] | |
| PDGF | ↑ | [71] | |
| EGF | = | [75] | |
| FGF | nd | ||
| TGFβ | = | [67] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masselli, E.; Pozzi, G.; Gobbi, G.; Merighi, S.; Gessi, S.; Vitale, M.; Carubbi, C. Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants. Cells 2020, 9, 2136. https://doi.org/10.3390/cells9092136
Masselli E, Pozzi G, Gobbi G, Merighi S, Gessi S, Vitale M, Carubbi C. Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants. Cells. 2020; 9(9):2136. https://doi.org/10.3390/cells9092136
Chicago/Turabian StyleMasselli, Elena, Giulia Pozzi, Giuliana Gobbi, Stefania Merighi, Stefania Gessi, Marco Vitale, and Cecilia Carubbi. 2020. "Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants" Cells 9, no. 9: 2136. https://doi.org/10.3390/cells9092136