The Rationale of Neprilysin Inhibition in Prevention of Myocardial Ischemia-Reperfusion Injury during ST-Elevation Myocardial Infarction

 , , , and
, , , and 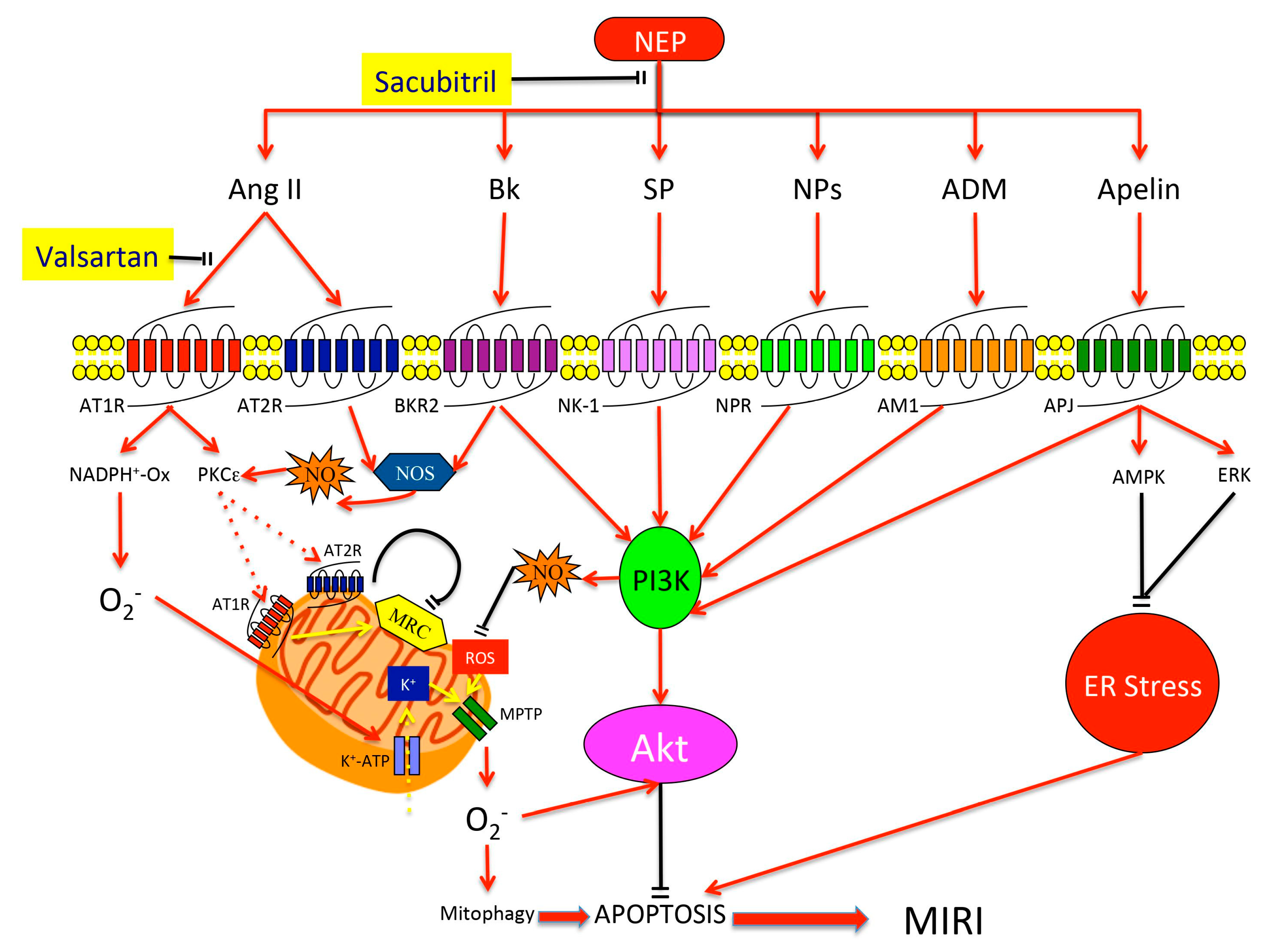{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathophysiology of Myocardial and Coronary Microvascular Reperfusion Injury
2.1. Apoptosis
2.2. Autophagy and Mitophagy
2.3. Defects in Calcium Handling
2.4. Inflammation
2.5. Cross-Talk between MIRI and Coronary Microvascular IRI
2.6. Cardioprotective Molecular Mechanisms: RISK and SAFE Pathways
3. Neprilysin
4. NEP Substrates and Their Cardioprotective Effects
4.1. Natriuretic Peptides (NPs)
4.2. Angiotensin (Ang)
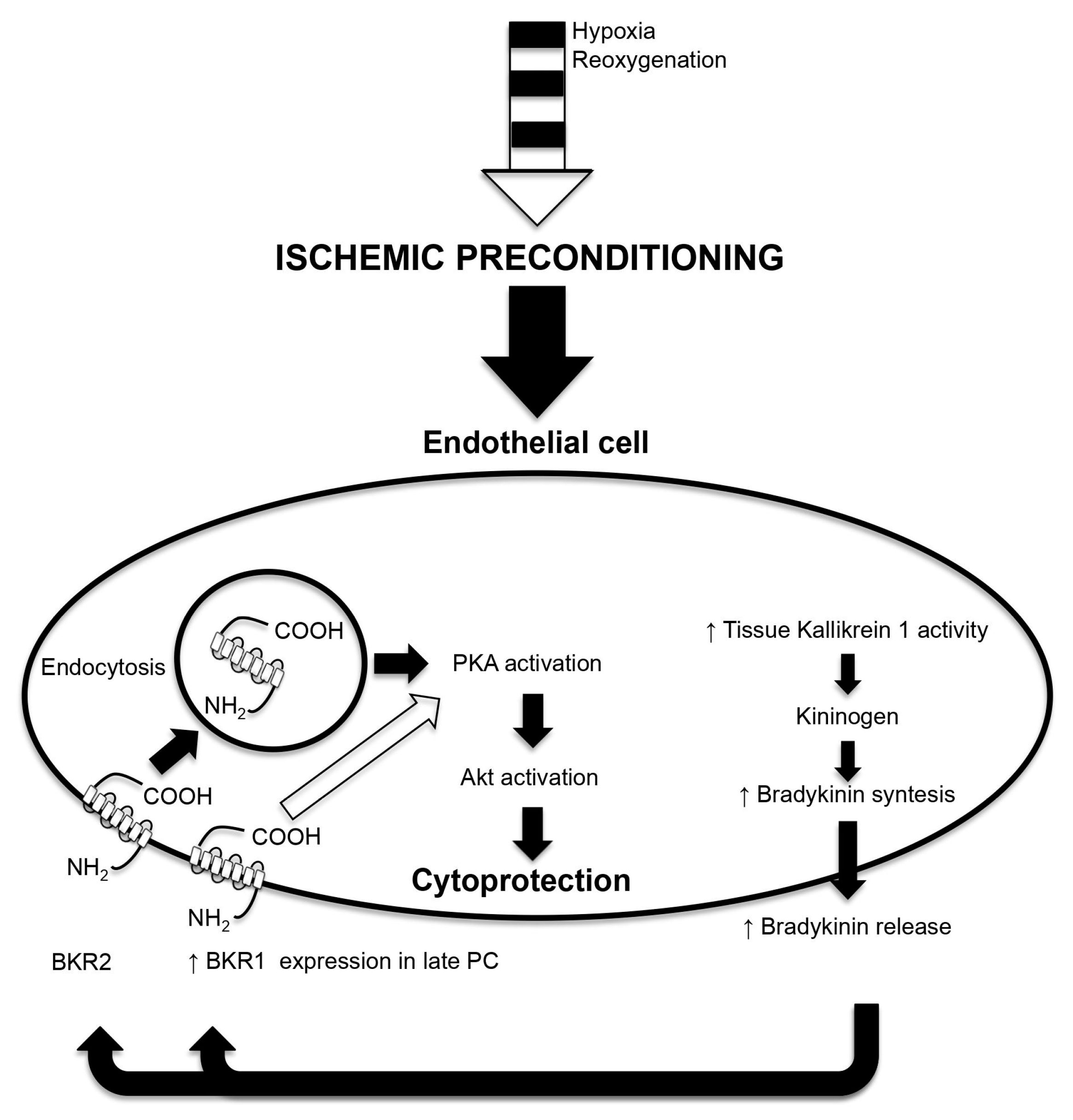
4.3. Bradykinin (Bk)
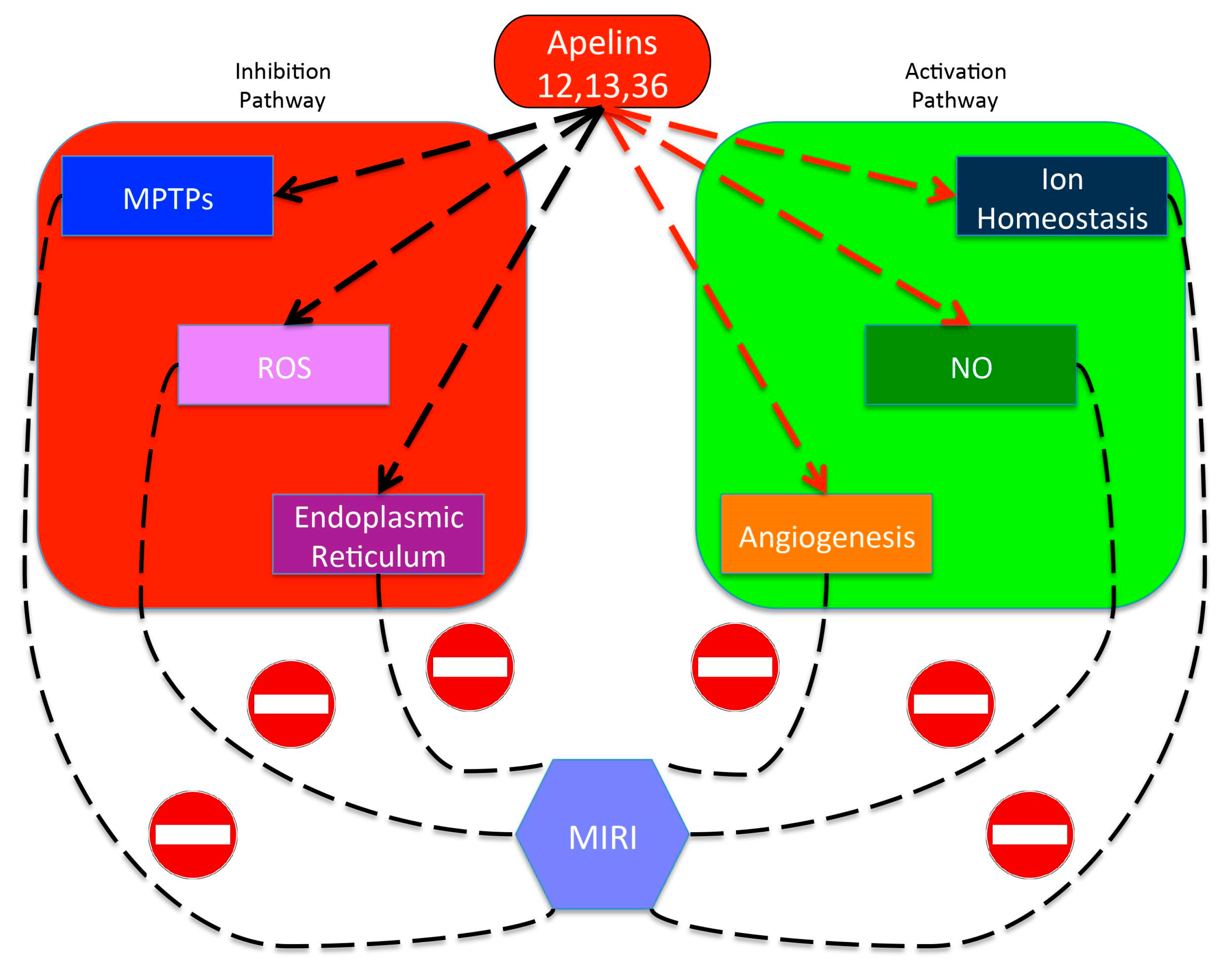
4.4. Apelin
4.5. Substance P (SP)
4.6. Adrenomedullin (ADM)
5. Clinical Perspectives for NEP Inhibition in MIRI Prevention
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Buono, F.; Spinelli, L.; Giallauria, F.; di Panzillo, E.A.; di Marino, S.; Ferrara, F.; Vigorito, C.; Trimarco, B.; Morisco, C. Usefulness of satisfactory control of low-density lipoprotein cholesterol to predict left ventricular remodeling after a first ST-elevation myocardial infarction successfully reperfused. Am. J. Cardiol. 2011, 107, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Giallauria, F.; Cirillo, P.; Lucci, R.; Pacileo, M.; de Lorenzo, A.; D’Agostino, M.; Moschella, S.; Psaroudaki, M.; del Forno, D.; Orio, F.; et al. Left ventricular remodelling in patients with moderate systolic dysfunction after myocardial infarction: Favourable effects of exercise training and predictive role of N-terminal pro-brain natriuretic peptide. Eur. J. Cardiovasc. Prev. Rehabil. 2008, 15, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Savoye, C.; Equine, O.; Tricot, O.; Nugue, O.; Segrestin, B.; Sautiere, K.; Elkohen, M.; Pretorian, E.M.; Taghipour, K.; Philias, A.; et al. Left ventricular remodeling after anterior wall acute myocardial infarction in modern clinical practice (from the REmodelage VEntriculaire [REVE] study group). Am. J. Cardiol. 2006, 98, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Gersh, B.J. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: A continual challenge. Eur. Heart J. 2017, 38, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Van der Bijl, P.; Abou, R.; Goedemans, L.; Gersh, B.J.; Holmes, D.R., Jr.; Marsan, N.A.; Delgado, V.; Bax, J.J. Left Ventricular Post-Infarct Remodeling: Implications for Systolic Function Improvement and Outcomes in the Modern Era. JACC Heart Fail. 2020, 8, 131–140. [Google Scholar] [CrossRef]
- Heusch, G.; Libby, P.; Gersh, B.; Yellon, D.; Böhm, M.; Lopaschuk, G.; Opie, L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014, 383, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Ibanez, B.; Heusch, G.; Ovize, M.; van de Werf, F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef] [Green Version]
- Schomig, A.; Mehilli, J.; Antoniucci, D.; Ndrepepa, G.; Markwardt, C.; di Pede, F.; Nekolla, S.G.; Schlotterbeck, K.; Schuhlen, H.; Pache, J.; et al. Mechanical reperfusion in patients with acute myocardial infarction presenting more than 12 hours from symptom onset: A randomized controlled trial. JAMA 2005, 293, 2865–2872. [Google Scholar] [CrossRef] [Green Version]
- Bolli, R. Myocardial ‘stunning’ in man. Circulation 1992, 86, 1671–1691. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G. The Coronary Circulation as a Target of Cardioprotection. Circ. Res. 2016, 118, 1643–1658. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Coronary microvascular obstruction: The new frontier in cardioprotection. Basic Res. Cardiol. 2019, 114, 45. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Lukovic, D.; Zlabinger, K.; Zimba, A.; Lorant, D.; Goliasch, G.; Winkler, J.; Pils, D.; Auer, K.; jan Ankersmit, H.; et al. Sequential activation of different pathway networks in ischemia-affected and non-affected myocardium, inducing intrinsic remote conditioning to prevent left ventricular remodeling. Sci. Rep. 2017, 7, 43958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kutala, V.K.; Khan, M.; Angelos, M.G.; Kuppusamy, P. Role of oxygen in postischemic myocardial injury. Antioxid. Redox Signal. 2007, 9, 1193–1206. [Google Scholar] [CrossRef]
- Ladilov, Y.; Efe, O.; Schafer, C.; Rother, B.; Kasseckert, S.; Abdallah, Y.; Meuter, K.; Schluter, K.D.; Piper, H.M. Reoxygenation-induced rigor-type contracture. J. Mol. Cell. Cardiol. 2003, 35, 1481–1490. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Abellan, A.; Miro-Casas, E.; Garcia-Dorado, D. Opening of mitochondrial permeability transition pore induces hypercontracture in Ca2+ overloaded cardiac myocytes. Basic Res. Cardiol. 2007, 102, 542–552. [Google Scholar] [CrossRef]
- Heusch, G.; Boengler, K.; Schulz, R. Inhibition of mitochondrial permeability transition pore opening: The Holy Grail of cardioprotection. Basic Res. Cardiol. 2010, 105, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Shanmuganathan, S.; Hausenloy, D.J.; Duchen, M.R.; Yellon, D.M. Mitochondrial permeability transition pore as a target for cardioprotection in the human heart. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H237–H242. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharm. 2000, 130, 197–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Gallego, C.G.; Vahl, T.P.; Goliasch, G.; Picatoste, B.; Arias, T.; Ishikawa, K.; Njerve, I.U.; Sanz, J.; Narula, J.; Sengupta, P.P.; et al. Sphingosine-1-Phosphate Receptor Agonist Fingolimod Increases Myocardial Salvage and Decreases Adverse Postinfarction Left Ventricular Remodeling in a Porcine Model of Ischemia/Reperfusion. Circulation 2016, 133, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Dong, Y.; Undyala, V.V.; Whittaker, P. Autophagy as a therapeutic target for ischaemia /reperfusion injury? Concepts, controversies, and challenges. Cardiovasc. Res. 2012, 94, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Sala-Mercado, J.A.; Wider, J.; Undyala, V.V.; Jahania, S.; Yoo, W.; Mentzer, R.M., Jr.; Gottlieb, R.A.; Przyklenk, K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation 2010, 122, S179–S184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Nah, J.; Oka, S.I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, J.; Yu, S.; Luo, Z.; Hua, F.; Yuan, L.; Zhou, Z.; Liu, Q.; Du, X.; Chen, S.; et al. Protective Effect of Sevoflurane Postconditioning against Cardiac Ischemia/Reperfusion Injury via Ameliorating Mitochondrial Impairment, Oxidative Stress and Rescuing Autophagic Clearance. PLoS ONE 2015, 10, e0134666. [Google Scholar] [CrossRef]
- Ke, J.; Yao, B.; Li, T.; Cui, S.; Ding, H. A2 Adenosine Receptor-mediated Cardioprotection Against Reperfusion Injury in Rat Hearts Is Associated With Autophagy Downregulation. J. Cardiovasc. Pharmacol. 2015, 66, 25–34. [Google Scholar] [CrossRef]
- Ma, S.; Wang, Y.; Chen, Y.; Cao, F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim. Biophys. Acta 2015, 1852, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Manning, A.S.; Hearse, D.J. Reperfusion-induced arrhythmias: Mechanisms and prevention. J. Mol. Cell. Cardiol. 1984, 16, 497–518. [Google Scholar] [CrossRef]
- Bolli, R.; Marban, E. Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 1999, 79, 609–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entman, M.L.; Michael, L.; Rossen, R.D.; Dreyer, W.J.; Anderson, D.C.; Taylor, A.A.; Smith, C.W. Inflammation in the course of early myocardial ischemia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1991, 5, 2529–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, A.; De Rochemont, W.D.M.; Korb, G. Blood supply of the myocardium after temporary coronary occlusion. Circ. Res. 1966, 19, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukreja, R.C.; Janin, Y. Reperfusion Injury: Basic Concepts and Protection Strategies. J. Thromb. Thrombolysis 1997, 4, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Morishige, N.; Masuda, M.; Lin, W.; Wieland, W.; Thone, F.; Mubagwa, K.; Borgers, M.; Flameng, W. R56865, a Na(+)- and Ca(2+)-overload inhibitor, reduces myocardial ischemia-reperfusion injury in blood-perfused rabbit hearts. J. Mol. Cell. Cardiol. 1993, 25, 1445–1459. [Google Scholar] [CrossRef]
- Askenasy, N. Is cytotoxic cellular edema real? The effect of calcium ion on water homeostasis in the rat heart. Cardiovasc. Toxicol. 2001, 1, 21–34. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Iaccarino, G. Inflammation and Cardiovascular Diseases: The Most Recent Findings. Int. J. Mol. Sci. 2019, 20, 3879. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Li, J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clinical Medicine Insights. Cardiology 2016, 10, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. Febs Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehmcke, S.; Mörgelin, M.; Herwald, H. Activation of the human contact system on neutrophil extracellular traps. J. Innate Immun. 2009, 1, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhou, X.; Ji, W.-J.; Lu, R.-Y.; Zhang, Y.; Zhang, Y.-D.; Ma, Y.-Q.; Zhao, J.-H.; Li, Y.-M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol Heart Circ. Physiol 2015, 308, H500–H509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigante, B.; Bellis, A.; Visconti, R.; Marino, M.; Morisco, C.; Trimarco, V.; Galasso, G.; Piscione, F.; de Luca, N.; Prince, J.A.; et al. Retrospective analysis of coagulation factor II receptor (F2R) sequence variation and coronary heart disease in hypertensive patients. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1213–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigante, B.; Vikstrom, M.; Meuzelaar, L.S.; Chernogubova, E.; Silveira, A.; Hooft, F.V.; Hamsten, A.; de Faire, U. Variants in the coagulation factor 2 receptor (F2R) gene influence the risk of myocardial infarction in men through an interaction with interleukin 6 serum levels. Thromb. Haemost. 2009, 101, 943–953. [Google Scholar]
- Heusch, G.; Kleinbongard, P.; Skyschally, A.; Levkau, B.; Schulz, R.; Erbel, R. The coronary circulation in cardioprotection: More than just one confounder. Cardiovasc. Res. 2012, 94, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Dauber, I.M.; VanBenthuysen, K.M.; McMurtry, I.F.; Wheeler, G.S.; Lesnefsky, E.J.; Horwitz, L.D.; Weil, J.V. Functional coronary microvascular injury evident as increased permeability due to brief ischemia and reperfusion. Circ. Res. 1990, 66, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Dorado, D.; Andres-Villarreal, M.; Ruiz-Meana, M.; Inserte, J.; Barba, I. Myocardial edema: A translational view. J. Mol. Cell. Cardiol. 2012, 52, 931–939. [Google Scholar] [CrossRef]
- Heusch, G.; Kleinbongard, P.; Bose, D.; Levkau, B.; Haude, M.; Schulz, R.; Erbel, R. Coronary microembolization: From bedside to bench and back to bedside. Circulation 2009, 120, 1822–1836. [Google Scholar] [CrossRef]
- Bolli, R.; Triana, J.F.; Jeroudi, M.O. Prolonged impairment of coronary vasodilation after reversible ischemia. Evidence for microvascular “stunning”. Circ. Res. 1990, 67, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Ehring, T.; Krajcar, M.; Baumgart, D.; Kompa, S.; Hummelgen, M.; Heusch, G. Cholinergic and alpha-adrenergic coronary vasomotion [corrected] with increasing ischemia-reperfusion injury. Am. J. Physiol. 1995, 268, H886–H894. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.J.; O’Rourke, S.T.; Pelc, L.R.; Warltier, D.C. Myocardial and endothelial dysfunction after multiple, brief coronary occlusions: Role of oxygen radicals. Am. J. Physiol. 1992, 263, H1703–H1709. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Baars, T.; Mohlenkamp, S.; Kahlert, P.; Erbel, R.; Heusch, G. Aspirate from human stented native coronary arteries vs. saphenous vein grafts: More endothelin but less particulate debris. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1222–H1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinbongard, P.; Bose, D.; Baars, T.; Mohlenkamp, S.; Konorza, T.; Schoner, S.; Elter-Schulz, M.; Eggebrecht, H.; Degen, H.; Haude, M.; et al. Vasoconstrictor potential of coronary aspirate from patients undergoing stenting of saphenous vein aortocoronary bypass grafts and its pharmacological attenuation. Circ. Res. 2011, 108, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Leineweber, K.; Bose, D.; Vogelsang, M.; Haude, M.; Erbel, R.; Heusch, G. Intense vasoconstriction in response to aspirate from stented saphenous vein aortocoronary bypass grafts. J. Am. Coll. Cardiol. 2006, 47, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Barrabes, J.A.; Garcia-Dorado, D.; Mirabet, M.; Inserte, J.; Agullo, L.; Soriano, B.; Massaguer, A.; Padilla, F.; Lidon, R.M.; Soler-Soler, J. Antagonism of selectin function attenuates microvascular platelet deposition and platelet-mediated myocardial injury after transient ischemia. J. Am. Coll. Cardiol. 2005, 45, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Driesen, R.B.; Zalewski, J.; Driessche, N.V.; Vermeulen, K.; Bogaert, J.; Sipido, K.R.; van de Werf, F.; Claus, P. Histological correlate of a cardiac magnetic resonance imaged microvascular obstruction in a porcine model of ischemia-reperfusion. Cardiovasc. Pathol. 2012, 21, 129–131. [Google Scholar] [CrossRef]
- Sheridan, F.M.; Dauber, I.M.; McMurtry, I.F.; Lesnefsky, E.J.; Horwitz, L.D. Role of leukocytes in coronary vascular endothelial injury due to ischemia and reperfusion. Circ. Res. 1991, 69, 1566–1574. [Google Scholar] [CrossRef] [Green Version]
- Higginson, L.A.; White, F.; Heggtveit, H.A.; Sanders, T.M.; Bloor, C.M.; Covell, W.J. Determinants of myocardial hemorrhage after coronary reperfusion in the anesthetized dog. Circulation 1982, 65, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Robbers, L.F.; Eerenberg, E.S.; Teunissen, P.F.; Jansen, M.F.; Hollander, M.R.; Horrevoets, A.J.; Knaapen, P.; Nijveldt, R.; Heymans, M.W.; Levi, M.; et al. Magnetic resonance imaging-defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and haemorrhage. Eur. Heart J. 2013, 34, 2346–2353. [Google Scholar] [CrossRef] [Green Version]
- Kloner, R.A.; Ganote, C.E.; Jennings, R.B. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J. Clin. Investig. 1974, 54, 1496–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niccoli, G.; Burzotta, F.; Galiuto, L.; Crea, F. Myocardial no-reflow in humans. J. Am. Coll. Cardiol. 2009, 54, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Gersh, B.J.; Mehran, R.; Brodie, B.R.; Brener, S.J.; Dizon, J.M.; Lansky, A.J.; Witzenbichler, B.; Kornowski, R.; Guagliumi, G.; et al. Effect of Ischemia Duration and Door-to-Balloon Time on Myocardial Perfusion in ST-Segment Elevation Myocardial Infarction: An Analysis From HORIZONS-AMI Trial (Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction). JACC Cardiovasc. Interv. 2015, 8, 1966–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betgem, R.P.; de Waard, G.A.; Nijveldt, R.; Beek, A.M.; Escaned, J.; van Royen, N. Intramyocardial haemorrhage after acute myocardial infarction. Nat. Rev. Cardiol. 2015, 12, 156–167. [Google Scholar] [CrossRef]
- Hamirani, Y.S.; Wong, A.; Kramer, C.M.; Salerno, M. Effect of microvascular obstruction and intramyocardial hemorrhage by CMR on LV remodeling and outcomes after myocardial infarction: A systematic review and meta-analysis. JACC Cardiovasc. Imaging 2014, 7, 940–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndrepepa, G.; Tiroch, K.; Fusaro, M.; Keta, D.; Seyfarth, M.; Byrne, R.A.; Pache, J.; Alger, P.; Mehilli, J.; Schomig, A.; et al. 5-year prognostic value of no-reflow phenomenon after percutaneous coronary intervention in patients with acute myocardial infarction. J. Am. Coll. Cardiol. 2010, 55, 2383–2389. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, G.; Montone, R.A.; Ibanez, B.; Thiele, H.; Crea, F.; Heusch, G.; Bulluck, H.; Hausenloy, D.J.; Berry, C.; Stiermaier, T.; et al. Optimized Treatment of ST-Elevation Myocardial Infarction. Circ. Res. 2019, 125, 245–258. [Google Scholar] [CrossRef]
- Heusch, G.; Kleinbongard, P.; Skyschally, A. Myocardial infarction and coronary microvascular obstruction: An intimate, but complicated relationship. Basic Res. Cardiol. 2013, 108, 380. [Google Scholar] [CrossRef] [Green Version]
- Reffelmann, T.; Hale, S.L.; Li, G.; Kloner, R.A. Relationship between no reflow and infarct size as influenced by the duration of ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H766–H772. [Google Scholar] [CrossRef]
- Hori, M.; Gotoh, K.; Kitakaze, M.; Iwai, K.; Iwakura, K.; Sato, H.; Koretsune, Y.; Inoue, M.; Kitabatake, A.; Kamada, T. Role of oxygen-derived free radicals in myocardial edema and ischemia in coronary microvascular embolization. Circulation 1991, 84, 828–840. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Musiolik, J.; Gedik, N.; Skyschally, A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ. Res. 2011, 109, 1302–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocanu, M.M.; Bell, R.M.; Yellon, D.M. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J. Mol. Cell. Cardiol. 2002, 34, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Efentakis, P.; Andreadou, I.; Bibli, S.I.; Vasileiou, S.; Dagres, N.; Zoga, A.; Lougiakis, N.; Kremastinos, D.T.; Iliodromitis, E.K. Ranolazine triggers pharmacological preconditioning and postconditioning in anesthetized rabbits through activation of RISK pathway. Eur. J. Pharmacol. 2016, 789, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Kim, S.; Huh, J. Zinc plays a critical role in the cardioprotective effect of postconditioning by enhancing the activation of the RISK pathway in rat hearts. J. Mol. Cell. Cardiol. 2014, 66, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Webb, I.G.; Davidson, S.M.; Ahmed, A.I.; Clark, J.E.; Jacquet, S.; Shah, A.M.; Miura, T.; Yellon, D.M.; Avkiran, M.; et al. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ. Res. 2008, 103, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Skyschally, A.; Gent, S.; Amanakis, G.; Schulte, C.; Kleinbongard, P.; Heusch, G. Across-Species Transfer of Protection by Remote Ischemic Preconditioning With Species-Specific Myocardial Signal Transduction by Reperfusion Injury Salvage Kinase and Survival Activating Factor Enhancement Pathways. Circ. Res. 2015, 117, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Hadebe, N.; Cour, M.; Lecour, S. The SAFE pathway for cardioprotection: Is this a promising target? Arch. Kreislaufforsch. 2018, 113, 9. [Google Scholar] [CrossRef]
- Suleman, N.; Somers, S.; Smith, R.; Opie, L.H.; Lecour, S.C. Dual activation of STAT-3 and Akt is required during the trigger phase of ischaemic preconditioning. Basic Res. Cardiol. 2008, 79, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Lacerda, L.; Somers, S.; Opie, L.H.; Lecour, S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc. Res. 2009, 84, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Qi, Q.R.; Yang, Z.M. Regulation and function of signal transducer and activator of transcription 3. World J. Biol. Chem. 2014, 5, 231–239. [Google Scholar] [CrossRef]
- Huang, C.H.; Tsai, M.S.; Chiang, C.Y.; Su, Y.J.; Wang, T.D.; Chang, W.T.; Chen, H.W.; Chen, W. Activation of mitochondrial STAT-3 and reduced mitochondria damage during hypothermia treatment for post-cardiac arrest myocardial dysfunction. Basic Res. Cardiol. 2015, 110, 59. [Google Scholar] [CrossRef] [PubMed]
- Wincewicz, A.; Sulkowski, S. Stat proteins as intracellular regulators of resistance to myocardial injury in the context of cardiac remodeling and targeting for therapy. Adv. Clin. Exp. Med. 2017, 26, 703–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Hu, W.; Di, S.; Ma, Z.; Fan, C.; Wang, D.; Jiang, S.; Li, Y.; Zhou, Q.; Li, T.; et al. Tackling myocardial ischemic injury: The signal transducer and activator of transcription 3 (STAT3) at a good site. Expert Opin. Ther. Targets 2017, 21, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Kottenberg, E.; Musiolik, J.; Thielmann, M.; Jakob, H.; Peters, J.; Heusch, G. Interference of propofol with signal transducer and activator of transcription 5 activation and cardioprotection by remote ischemic preconditioning during coronary artery bypass grafting. J. Thorac. Cardiovasc. Surg. 2014, 147, 376–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Elia, E.; Iacovoni, A.; Vaduganathan, M.; Lorini, F.L.; Perlini, S.; Senni, M. Neprilysin inhibition in heart failure: Mechanisms and substrates beyond modulating natriuretic peptides. Eur. J. Heart Fail. 2017, 19, 710–717. [Google Scholar] [CrossRef] [PubMed]
- McKinnie, S.M.; Fischer, C.; Tran, K.M.; Wang, W.; Mosquera, F.; Oudit, G.Y.; Vederas, J.C. The Metalloprotease Neprilysin Degrades and Inactivates Apelin Peptides. Chembiochem A Eur. J. Chem. Biol. 2016, 17, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Vodovar, N.; Seronde, M.F.; Laribi, S.; Gayat, E.; Lassus, J.; Januzzi, J.L.; Boukef, R., Jr.; Nouira, S.; Manivet, P.; Samuel, J.L.; et al. Elevated Plasma B-Type Natriuretic Peptide Concentrations Directly Inhibit Circulating Neprilysin Activity in Heart Failure. JACC Heart Fail. 2015, 3, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Kuoppala, A.; Lindstedt, K.A.; Saarinen, J.; Kovanen, P.T.; Kokkonen, J.O. Inactivation of bradykinin by angiotensin-converting enzyme and by carboxypeptidase N in human plasma. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1069–H1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, E.; Yasue, H.; Yoshimura, M.; Ogawa, H.; Jougasaki, M.; Matsumura, T.; Mukoyama, M.; Nakao, K. Increased plasma levels of brain natriuretic peptide in patients with acute myocardial infarction. Circulation 1993, 88, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Hamamoto, H.; Honda, Y.; Hirose, M.; Furukawa, S.; Kimura, E. Changes in components of kinin system and hemodynamics in acute myocardial infarction. Am. Heart J. 1978, 95, 619–626. [Google Scholar] [CrossRef]
- Lumsden, N.G.; Khambata, R.S.; Hobbs, A.J. C-type natriuretic peptide (CNP): Cardiovascular roles and potential as a therapeutic target. Curr. Pharm. Des. 2010, 16, 4080–4088. [Google Scholar] [CrossRef] [PubMed]
- Addisu, A.; Gower, W.R., Jr.; Landon, C.S.; Dietz, J.R. B-type natriuretic peptide decreases gastric emptying and absorption. Exp. Biol. Med. 2008, 233, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Boschmann, M.; Moro, C.; Adams, F.; Heusser, K.; Franke, G.; Berlan, M.; Luft, F.C.; Lafontan, M.; Jordan, J. Lipid mobilization with physiological atrial natriuretic peptide concentrations in humans. J. Clin. Endocrinol. Metab. 2005, 90, 3622–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, J.C., Jr.; Granger, J.P.; Opgenorth, T.J. Effects of synthetic atrial natriuretic factor on renal function and renin release. Am. J. Physiol. 1984, 247, F863–F866. [Google Scholar] [CrossRef] [Green Version]
- Casserly, B.P.; Sears, E.H.; Gartman, E.J. The role of natriuretic peptides in inflammation and immunity. Recent Pat. Inflamm. Allergy Drug Discov. 2010, 4, 90–104. [Google Scholar] [CrossRef]
- Shah, S.J.; Michaels, A.D. Acute effects of intravenous nesiritide on cardiac contractility in heart failure. J. Card. Fail. 2010, 16, 720–727. [Google Scholar] [CrossRef]
- Holmes, S.J.; Espiner, E.A.; Richards, A.M.; Yandle, T.G.; Frampton, C. Renal, endocrine, and hemodynamic effects of human brain natriuretic peptide in normal man. J. Clin. Endocrinol. Metab. 1993, 76, 91–96. [Google Scholar] [CrossRef]
- Richards, A.M.; McDonald, D.; Fitzpatrick, M.A.; Nicholls, M.G.; Espiner, E.A.; Ikram, H.; Jans, S.; Grant, S.; Yandle, T. Atrial natriuretic hormone has biological effects in man at physiological plasma concentrations. J. Clin. Endocrinol. Metab. 1988, 67, 1134–1139. [Google Scholar] [CrossRef]
- Kiemer, A.K.; Weber, N.C.; Furst, R.; Bildner, N.; Kulhanek-Heinze, S.; Vollmar, A.M. Inhibition of p38 MAPK activation via induction of MKP-1: Atrial natriuretic peptide reduces TNF-alpha-induced actin polymerization and endothelial permeability. Circ. Res. 2002, 90, 874–881. [Google Scholar] [CrossRef]
- El, M.M.; Houard, X.; Rais, S.; Pasquier, C.; Oudghiri, M.; Jacob, M.P.; Meilhac, O.; Michel, J.B. Pharmacological potentiation of natriuretic peptide limits polymorphonuclear neutrophil-vascular cell interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1824–1831. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Oberwinkler, H.; Werner, F.; Gassner, B.; Nakagawa, H.; Feil, R.; Hofmann, F.; Schlossmann, J.; Dietrich, A.; Gudermann, T.; et al. Atrial natriuretic peptide-mediated inhibition of microcirculatory endothelial Ca2+ and permeability response to histamine involves cGMP-dependent protein kinase I and TRPC6 channels. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2121–2129. [Google Scholar] [CrossRef]
- Moyes, A.J.; Chu, S.M.; Aubdool, A.A.; Dukinfield, M.S.; Margulies, K.B.; Bedi, K.C.; Hodivala-Dilke, K.; Baliga, R.S.; Hobbs, A.J. C-type natriuretic peptide co-ordinates cardiac structure and function. Eur. Heart J. 2020, 41, 1006–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burley, D.S.; Baxter, G.F. B-type natriuretic peptide at early reperfusion limits infarct size in the rat isolated heart. Basic Res. Cardiol. 2007, 102, 529–541. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.P.; Yellon, D.M.; Martin, C.; Schulz, R.; Heusch, G.; Onody, A.; Ferdinandy, P.; Baxter, G.F. B-type natriuretic peptide limits infarct size in rat isolated hearts via KATP channel opening. Am. J. Physiol.. Heart Circ. Physiol. 2003, 284, H1592–H1600. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Garcia-Dorado, D.; Agullo, L.; Paniagua, A.; Soler-Soler, J. Urodilatin limits acute reperfusion injury in the isolated rat heart. Cardiovasc. Res. 2000, 45, 351–359. [Google Scholar] [CrossRef]
- Yang, X.M.; Philipp, S.; Downey, J.M.; Cohen, M.V. Atrial natriuretic peptide administered just prior to reperfusion limits infarction in rabbit hearts. Basic Res. Cardiol. 2006, 101, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Hempel, A.; Friedrich, M.; Schluter, K.D.; Forssmann, W.G.; Kuhn, M.; Piper, H.M. ANP protects against reoxygenation-induced hypercontracture in adult cardiomyocytes. Am. J. Physiol. 1997, 273, H244–H249. [Google Scholar] [CrossRef]
- Abdallah, Y.; Gkatzoflia, A.; Pieper, H.; Zoga, E.; Walther, S.; Kasseckert, S.; Schafer, M.; Schluter, K.D.; Piper, H.M.; Schafer, C. Mechanism of cGMP-mediated protection in a cellular model of myocardial reperfusion injury. Cardiovasc. Res. 2005, 66, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitakaze, M.; Asakura, M.; Kim, J.; Shintani, Y.; Asanuma, H.; Hamasaki, T.; Seguchi, O.; Myoishi, M.; Minamino, T.; Ohara, T.; et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): Two randomised trials. Lancet 2007, 370, 1483–1493. [Google Scholar] [CrossRef]
- Arakawa, K.; Himeno, H.; Kirigaya, J.; Otomo, F.; Matsushita, K.; Nakahashi, H.; Shimizu, S.; Nitta, M.; Takamizawa, T.; Yano, H.; et al. B-type natriuretic peptide as a predictor of ischemia/reperfusion injury immediately after myocardial reperfusion in patients with ST-segment elevation acute myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care 2016, 5, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Ajmani, P.; Yadav, H.N.; Singh, M.; Sharma, P.L. Possible involvement of caveolin in attenuation of cardioprotective effect of ischemic preconditioning in diabetic rat heart. BMC Cardiovasc. Disord. 2011, 11, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez, R.E.; Castro, M.; Javadov, S.; Escobales, N. Angiotensin II and ischemic preconditioning synergize to improve mitochondrial function while showing additive effects on ventricular postischemic recovery. J. Cardiovasc. Pharmacol. 2014, 64, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, M. Role of angiotensin in cardioprotective effect of ischemic preconditioning. J. Cardiovasc. Pharmacol. 1999, 33, 772–778. [Google Scholar] [CrossRef]
- Escobales, N.; Nunez, R.E.; Javadov, S. Mitochondrial angiotensin receptors and cardioprotective pathways. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1426–H1438. [Google Scholar] [CrossRef]
- Loot, A.E.; Roks, A.J.; Henning, R.H.; Tio, R.A.; Suurmeijer, J.A.; Boomsma, F.; van Gilst, W.H. Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation 2002, 105, 1548–1550. [Google Scholar] [CrossRef] [Green Version]
- Trask, A.J.; Ferrario, C.M. Angiotensin-(1-7): Pharmacology and new perspectives in cardiovascular treatments. Cardiovasc. Drug Rev. 2007, 25, 162–174. [Google Scholar] [CrossRef]
- Macedo, L.M.; Souza, A.P.; de Maria, M.L.; Borges, C.L.; Soares, C.M.; Pedrino, G.R.; Colugnati, D.B.; Santos, R.A.; Mendes, E.P.; Ferreira, A.J.; et al. Cardioprotective effects of diminazene aceturate in pressure-overloaded rat hearts. Life Sci. 2016, 155, 63–69. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, J.; Cole-Jeffrey, C.T.; Shenoy, V.; Espejo, A.; Hanna, M.; Song, C.; Pepine, C.J.; Katovich, M.J.; Raizada, M.K. Diminazene aceturate enhances angiotensin-converting enzyme 2 activity and attenuates ischemia-induced cardiac pathophysiology. Hypertension 2013, 62, 746–752. [Google Scholar] [CrossRef]
- Bessa, A.S.M.; Jesus, E.F.; Nunes, A.D.C.; Pontes, C.N.R.; Lacerda, I.S.; Costa, J.M.; Souza, E.J.; Lino-Junior, R.S.; Biancardi, M.F.; Santos, F.C.A.D.; et al. Stimulation of the ACE2/Ang-(1-7)/Mas axis in hypertensive pregnant rats attenuates cardiovascular dysfunction in adult male offspring. Hypertens. Res. 2019, 42, 1883–1893. [Google Scholar] [CrossRef]
- Singh, N.; Joshi, S.; Guo, L.; Baker, M.B.; Li, Y.; Castellano, R.K.; Raizada, M.K.; Jarajapu, Y.P. ACE2/Ang-(1-7)/Mas axis stimulates vascular repair-relevant functions of CD34+ cells. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1697–H1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneton, P.; Bloch-Faure, M.; Hagege, A.A.; Ruetten, H.; Huang, W.; Bergaya, S.; Ceiler, D.; Gehring, D.; Martins, I.; Salmon, G.; et al. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2634–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Zhang, J.J.; Chao, L.; Chao, J. Kallikrein gene delivery attenuates myocardial infarction and apoptosis after myocardial ischemia and reperfusion. Hypertension 2000, 35, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, J.; Yin, H.; Gao, L.; Hagiwara, M.; Shen, B.; Yang, Z.R.; Chao, L. Tissue kallikrein elicits cardioprotection by direct kinin b2 receptor activation independent of kinin formation. Hypertension 2008, 52, 715–720. [Google Scholar] [CrossRef]
- Heusch, G.; Boengler, K.; Schulz, R. Cardioprotection: Nitric oxide, protein kinases, and mitochondria. Circulation 2008, 118, 1915–1919. [Google Scholar] [CrossRef] [Green Version]
- Jalowy, A.; Schulz, R.; Dorge, H.; Behrends, M.; Heusch, G. Infarct size reduction by AT1-receptor blockade through a signal cascade of AT2-receptor activation, bradykinin and prostaglandins in pigs. J. Am. Coll. Cardiol. 1998, 32, 1787–1796. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Mancardi, D.; Tullio, F.; Pagliaro, P. Postconditioning and intermittent bradykinin induced cardioprotection require cyclooxygenase activation and prostacyclin release during reperfusion. Basic Res. Cardiol. 2008, 103, 368–377. [Google Scholar] [CrossRef]
- Ehring, T.; Baumgart, D.; Krajcar, M.; Hümmelgen, M.; Kompa, S.; Heusch, G. Attenuation of myocardial stunning by the ACE inhibitor ramiprilat through a signal cascade of bradykinin and prostaglandins but not nitric oxide. Circulation 1994, 90, 1368–1385. [Google Scholar] [CrossRef] [Green Version]
- Bellis, A.; Sorriento, D.; Fiordelisi, A.; Izzo, R.; Sadoshima, J.; Mauro, C.; Cerasuolo, F.; Mancusi, C.; Barbato, E.; Pilato, E.; et al. Autocrine Bradykinin Release Promotes Ischemic Preconditioning-Induced Cytoprotection in Bovine Aortic Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 2965. [Google Scholar] [CrossRef]
- Bellis, A.; Castaldo, D.; Trimarco, V.; Monti, M.G.; Chivasso, P.; Sadoshima, J.; Trimarco, B.; Morisco, C. Cross-talk between PKA and Akt protects endothelial cells from apoptosis in the late ischemic preconditioning. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1207–1212. [Google Scholar] [CrossRef]
- Campbell, D.J. Neprilysin Inhibitors and Bradykinin. Front. Med. 2018, 5, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.L.; Chen, S.R.; Scicli, G.M.; Carretero, O.A. Cardiac interstitial bradykinin release during ischemia is enhanced by ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H116–H121. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Anastasopoulos, F.; Duncan, A.M.; James, G.M.; Kladis, A.; Briscoe, T.A. Effects of neutral endopeptidase inhibition and combined angiotensin converting enzyme and neutral endopeptidase inhibition on angiotensin and bradykinin peptides in rats. J. Pharmacol. Exp. Ther. 1998, 287, 567–577. [Google Scholar] [PubMed]
- Kokkonen, J.O.; Kuoppala, A.; Saarinen, J.; Lindstedt, K.A.; Kovanen, P.T. Kallidin- and bradykinin-degrading pathways in human heart: Degradation of kallidin by aminopeptidase M-like activity and bradykinin by neutral endopeptidase. Circulation 1999, 99, 1984–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, Y.; Habata, Y.; Fukusumi, S.; Hosoya, M.; Fujii, R.; Hinuma, S.; Nishizawa, N.; Kitada, C.; Onda, H.; Nishimura, O.; et al. Molecular properties of apelin: Tissue distribution and receptor binding. Biochim. Biophys. Acta 2001, 1538, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wu, D.; Li, L.; Chen, L. Apelin/APJ System: A Novel Therapeutic Target for Myocardial Ischemia/Reperfusion Injury. Dna Cell Biol. 2016, 35, 766–775. [Google Scholar] [CrossRef]
- Yang, S.; Li, H.; Tang, L.; Ge, G.; Ma, J.; Qiao, Z.; Liu, H.; Fang, W. Apelin-13 protects the heart against ischemia-reperfusion injury through the RISK-GSK-3beta-mPTP pathway. Arch. Med. Sci. 2015, 11, 1065–1073. [Google Scholar] [CrossRef]
- Simpkin, J.C.; Yellon, D.M.; Davidson, S.M.; Lim, S.Y.; Wynne, A.M.; Smith, C.C. Apelin-13 and apelin-36 exhibit direct cardioprotective activity against ischemia-reperfusion injury. Basic Res. Cardiol. 2007, 102, 518–528. [Google Scholar] [CrossRef]
- Rahman, S.; Li, J.; Bopassa, J.C.; Umar, S.; Iorga, A.; Partownavid, P.; Eghbali, M. Phosphorylation of GSK-3beta mediates intralipid-induced cardioprotection against ischemia/reperfusion injury. Anesthesiology 2011, 115, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Schriewer, J.M.; Peek, C.B.; Bass, J.; Schumacker, P.T. ROS-mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia-reperfusion. J. Am. Heart Assoc. 2013, 2, e000159. [Google Scholar] [CrossRef] [Green Version]
- Pisarenko, O.I.; Pelogeykina, Y.A.; Zh, D.B.; Serebryakova, L.I.; Sidorova, M.V.; Az’muko, A.A.; Khatri, D.N.; Studneva, I.M.; Pal’keeva, M.E.; Tskitishvili, O.V.; et al. Limitation of myocardial infarction by a structural analog of the peptide apelin-12. Dokl. Biol. Sci. 2012, 443, 65–67. [Google Scholar] [CrossRef]
- Mottaghi, S.; Larijani, B.; Sharifi, A.M. Apelin 13: A novel approach to enhance efficacy of hypoxic preconditioned mesenchymal stem cells for cell therapy of diabetes. Med. Hypotheses 2012, 79, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Pisarenko, O.I.; Lankin, V.Z.; Konovalova, G.G.; Serebryakova, L.I.; Shulzhenko, V.S.; Timoshin, A.A.; Tskitishvili, O.V.; Pelogeykina, Y.A.; Studneva, I.M. Apelin-12 and its structural analog enhance antioxidant defense in experimental myocardial ischemia and reperfusion. Mol. Cell. Biochem. 2014, 391, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.J.; Zhang, L.K.; Wang, H.X.; Lu, L.Q.; Ma, L.Q.; Tang, C.S. Apelin protects heart against ischemia/reperfusion injury in rat. Peptides 2009, 30, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Pisarenko, O.I.; Serebryakova, L.I.; Studneva, I.M.; Pelogeykina, Y.A.; Tskitishvili, O.V.; Bespalova, Z.D.; Sidorova, M.V.; Az’muko, A.A.; Khatri, D.N.; Pal’keeva, M.E.; et al. Effects of structural analogues of apelin-12 in acute myocardial infarction in rats. J. Pharmacol. Pharmacother. 2013, 4, 198–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisarenko, O.I.; Serebriakova, L.I.; Iu, A.P.; Studneva, I.M.; Kkhatri, D.N.; Tskitishvili, O.V.; Zh, D.B.; Az’muko, A.A.; Sidorova, M.V.; Pal’keeva, M.E.; et al. Involvement of NO-dependent mechanisms of apelin action in myocardial protection against ischemia/reperfusion damage. Kardiologiia 2012, 52, 52–58. [Google Scholar]
- Pisarenko, O.I.; Iu, A.P.; Shul’zhenko, V.S.; Studneva, I.M.; Zh, D.B.; Az’muko, A.A.; Sidorova, M.V.; Pal’keeva, M.E. The influence of inhibiting no formation on metabolic recovery of ischemic rat heart by apelin-12. Biomeditsinskaia Khimiia 2012, 58, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Pisarenko, O.I.; Shulzhenko, V.S.; Iu, A.P.; Studneva, I.M.; Kkhatri, D.N.; Zh, D.B.; Az’muko, A.A.; Sidorova, M.V.; Pal’keeva, M.E. Effects of exogenous apelin-12 on functional and metabolic recovery of isolated rat heart after ischemia. Kardiologiia 2010, 50, 44–49. [Google Scholar]
- Kunduzova, O.; Alet, N.; Delesque-Touchard, N.; Millet, L.; Castan-Laurell, I.; Muller, C.; Dray, C.; Schaeffer, P.; Herault, J.P.; Savi, P.; et al. Apelin/APJ signaling system: A potential link between adipose tissue and endothelial angiogenic processes. FASEB J. 2008, 22, 4146–4153. [Google Scholar] [CrossRef]
- Li, L.; Li, L.; Xie, F.; Zhang, Z.; Guo, Y.; Tang, G.; Lv, D.; Lu, Q.; Chen, L.; Li, J. Jagged-1/Notch3 signaling transduction pathway is involved in apelin-13-induced vascular smooth muscle cells proliferation. Acta Biochim. Biophys. Sin. 2013, 45, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Su, T.; Li, F.; Li, L.; Qin, X.; Pan, W.; Feng, F.; Chen, F.; Liao, D.; Chen, L. PI3K/Akt signaling transduction pathway is involved in rat vascular smooth muscle cell proliferation induced by apelin-13. Acta Biochim. Biophys. Sin. 2010, 42, 396–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizi, Y.; Faghihi, M.; Imani, A.; Roghani, M.; Zekri, A.; Mobasheri, M.B.; Rastgar, T.; Moghimian, M. Post-infarct treatment with [Pyr(1)]apelin-13 improves myocardial function by increasing neovascularization and overexpression of angiogenic growth factors in rats. Eur. J. Pharmacol. 2015, 761, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zeng, H.; Chen, J.X. Apelin-13 increases myocardial progenitor cells and improves repair postmyocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H605–H618. [Google Scholar] [CrossRef] [PubMed]
- Kitiphongspattana, K.; Khan, T.A.; Ishii-Schrade, K.; Roe, M.W.; Philipson, L.H.; Gaskins, H.R. Protective role for nitric oxide during the endoplasmic reticulum stress response in pancreatic beta-cells. American journal of physiology. Endocrinol. Metab. 2007, 292, E1543–E1554. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Zhu, W.; Li, Y.; Xin, P.; Li, J.; Liu, M.; Li, J.; Andrew, N.R.; Wei, M. Apelin-13 protects the heart against ischemia-reperfusion injury through inhibition of ER-dependent apoptotic pathways in a time-dependent fashion. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1471–H1486. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- Boyce, M.; Yuan, J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ. 2006, 13, 363–373. [Google Scholar] [CrossRef]
- Liu, X.H.; Zhang, Z.Y.; Sun, S.; Wu, X.D. Ischemic postconditioning protects myocardium from ischemia/reperfusion injury through attenuating endoplasmic reticulum stress. Shock 2008, 30, 422–427. [Google Scholar] [CrossRef]
- Pisarenko, O.I.; Shulzhenko, V.S.; Studneva, I.M.; Serebryakova, L.I.; Pelogeykina, Y.A.; Veselova, O.M. Signaling pathways of a structural analogue of apelin-12 involved in myocardial protection against ischemia/reperfusion injury. Peptides 2015, 73, 67–76. [Google Scholar] [CrossRef]
- Wang, C.; Liu, N.; Luan, R.; Li, Y.; Wang, D.; Zou, W.; Xing, Y.; Tao, L.; Cao, F.; Wang, H. Apelin protects sarcoplasmic reticulum function and cardiac performance in ischaemia-reperfusion by attenuating oxidation of sarcoplasmic reticulum Ca2+ ATPase and ryanodine receptor. Cardiovasc. Res. 2013, 100, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Japp, A.G.; Cruden, N.L.; Barnes, G.; van Gemeren, N.; Mathews, J.; Adamson, J.; Johnston, N.R.; Denvir, M.A.; Megson, I.L.; Flapan, A.D.; et al. Acute cardiovascular effects of apelin in humans: Potential role in patients with chronic heart failure. Circulation 2010, 121, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Yu, D.; Yang, R.; Zhao, Q.; Wang, J.; Zhang, H.; Qian, K.; Shi, Z.; Wang, W.; Brown, R.; et al. Recombinant Fc-Elabela fusion protein has extended plasma half-life andmitigates post-infarct heart dysfunction in rats. Int. J. Cardiol. 2019, 292, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; McKinnie, S.M.; Farhan, M.; Paul, M.; McDonald, T.; McLean, B.; Llorens-Cortes, C.; Hazra, S.; Murray, A.G.; Vederas, J.C.; et al. Angiotensin-Converting Enzyme 2 Metabolizes and Partially Inactivates Pyr-Apelin-13 and Apelin-17: Physiological Effects in the Cardiovascular System. Hypertension 2016, 68, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, C.; Lamer, T.; Wang, W.; McKinnie, S.M.K.; Iturrioz, X.; Llorens-Cortes, C.; Oudit, G.Y.; Vederas, J.C. Plasma kallikrein cleaves and inactivates apelin-17: Palmitoyl- and PEG-extended apelin-17 analogs as metabolically stable blood pressure-lowering agents. Eur. J. Med. Chem. 2019, 166, 119–124. [Google Scholar] [CrossRef]
- Zubrzycka, M.; Janecka, A. Substance P: Transmitter of nociception (Minireview). Endocr. Regul. 2000, 34, 195–201. [Google Scholar]
- Datar, P.; Srivastava, S.; Coutinho, E.; Govil, G. Substance P: Structure, function, and therapeutics. Curr. Top. Med. Chem. 2004, 4, 75–103. [Google Scholar] [CrossRef]
- Bossaller, C.; Reither, K.; Hehlert-Friedrich, C.; Auch-Schwelk, W.; Graf, K.; Grafe, M.; Fleck, E. In vivo measurement of endothelium-dependent vasodilation with substance P in man. Herz 1992, 17, 284–290. [Google Scholar]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, E.; Koyanagi, M.; Dimmeler, S. Enhancing the outcome of cell therapy for cardiac repair: Progress from bench to bedside and back. Circulation 2010, 121, 325–335. [Google Scholar] [CrossRef]
- Dimmeler, S.; Burchfield, J.; Zeiher, A.M. Cell-based therapy of myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Kim, S.; Lee, S.; Woo, J.S.; Lee, K.H.; Cheng, X.W.; Son, Y.; Kim, W. Substance-P Prevents Cardiac Ischemia-Reperfusion Injury by Modulating Stem Cell Mobilization and Causing Early Suppression of Injury-Mediated Inflammation. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 52, 40–56. [Google Scholar] [CrossRef] [Green Version]
- Jubair, S.; Li, J.; Dehlin, H.M.; Manteufel, E.J.; Goldspink, P.H.; Levick, S.P.; Janicki, J.S. Substance P induces cardioprotection in ischemia-reperfusion via activation of AKT. American journal of physiology. Heart Circ. Physiol. 2015, 309, H676–H684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehlin, H.M.; Levick, S.P. Substance P in heart failure: The good and the bad. Int. J. Cardiol. 2014, 170, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Hinson, J.P.; Kapas, S.; Smith, D.M. Adrenomedullin, a multifunctional regulatory peptide. Endocr. Rev. 2000, 21, 138–167. [Google Scholar] [CrossRef]
- Hamid, S.A.; Totzeck, M.; Drexhage, C.; Thompson, I.; Fowkes, R.C.; Rassaf, T.; Baxter, G.F. Nitric oxide/cGMP signalling mediates the cardioprotective action of adrenomedullin in reperfused myocardium. Basic Res. Cardiol. 2010, 105, 257–266. [Google Scholar] [CrossRef]
- Kato, K.; Yin, H.; Agata, J.; Yoshida, H.; Chao, L.; Chao, J. Adrenomedullin gene delivery attenuates myocardial infarction and apoptosis after ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1506–H1514. [Google Scholar] [CrossRef] [Green Version]
- Okumura, H.; Nagaya, N.; Itoh, T.; Okano, I.; Hino, J.; Mori, K.; Tsukamoto, Y.; Ishibashi-Ueda, H.; Miwa, S.; Tambara, K.; et al. Adrenomedullin infusion attenuates myocardial ischemia/reperfusion injury through the phosphatidylinositol 3-kinase/Akt-dependent pathway. Circulation 2004, 109, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Nagaya, N.; Satoh, T.; Nishikimi, T.; Uematsu, M.; Furuichi, S.; Sakamaki, F.; Oya, H.; Kyotani, S.; Nakanishi, N.; Goto, Y.; et al. Hemodynamic, renal, and hormonal effects of adrenomedullin infusion in patients with congestive heart failure. Circulation 2000, 101, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.H.; Jia, Y.X.; Pan, C.S.; Zhao, J.; Ouyang, M.; Yang, J.; Chang, J.K.; Tang, C.S.; Qi, Y.F. Effects of intermedin(1-53) on cardiac function and ischemia/reperfusion injury in isolated rat hearts. Biochem. Biophys. Res. Commun. 2005, 327, 713–719. [Google Scholar] [CrossRef]
- Yang, J.H.; Qi, Y.F.; Jia, Y.X.; Pan, C.S.; Zhao, J.; Yang, J.; Chang, J.K.; Tang, C.S. Protective effects of intermedin/adrenomedullin2 on ischemia/reperfusion injury in isolated rat hearts. Peptides 2005, 26, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Heng, Y.; Zhang, W.; Jiang, J.Y.; Liu, Y.; Li, C.L.; Chen, H.B.; Xin, D.J. Huang. Intermedin is upregulated and has protective roles in a mouse ischemia/reperfusion model. Hypertens. Res. 2009, 32, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Song, J.Q.; Teng, X.; Cai, Y.; Tang, C.S.; Qi, Y.F. Activation of Akt/GSK-3beta signaling pathway is involved in intermedin(1-53) protection against myocardial apoptosis induced by ischemia/reperfusion. Apoptosis 2009, 14, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.; Satoh, F.; Murakami, O.; Totsune, K.; Suzuki, T.; Sasano, H.; Ito, S.; Takahashi, K. Expression of adrenomedullin2/intermedin in human brain, heart, and kidney. Peptides 2007, 28, 1095–1103. [Google Scholar] [CrossRef]
- Bell, D.; Campbell, M.; Ferguson, M.; Sayers, L.; Donaghy, L.; O’Regan, A.; Jewhurst, V.; Harbinson, M. AM(1)-receptor-dependent protection by intermedin of human vascular and cardiac non-vascular cells from ischaemia-reperfusion injury. J. Physiol. 2012, 590, 1181–1197. [Google Scholar] [CrossRef]
- Holmes, D.; Campbell, M.; Harbinson, M.; Bell, D. Protective effects of intermedin on cardiovascular, pulmonary and renal diseases: Comparison with adrenomedullin and CGRP. Curr. Protein Pept. Sci. 2013, 14, 294–329. [Google Scholar] [CrossRef]
- Lv, Z.; Wu, K.; Chen, X.; Zhang, X.; Hong, B. Plasma intermedin levels in patients with acute myocardial infarction. Peptides 2013, 43, 121–125. [Google Scholar] [CrossRef]
- Bell, D.; Campbell, M.; McAleer, S.F.; Ferguson, M.; Donaghy, L.; Harbinson, M.T. Endothelium-derived intermedin/adrenomedullin-2 protects human ventricular cardiomyocytes from ischaemia-reoxygenation injury predominantly via the AM(1) receptor. Peptides 2016, 76, 1–13. [Google Scholar] [CrossRef]
- Puymirat, E.; Simon, T.; Cayla, G.; Cottin, Y.; Elbaz, M.; Coste, P.; Lemesle, G.; Motreff, P.; Popovic, B.; Khalife, K.; et al. Acute Myocardial Infarction: Changes in Patient Characteristics, Management, and 6-Month Outcomes Over a Period of 20 Years in the FAST-MI Program (French Registry of Acute ST-Elevation or Non-ST-Elevation Myocardial Infarction) 1995 to 2015. Circulation 2017, 136, 1908–1919. [Google Scholar] [CrossRef]
- Szummer, K.; Wallentin, L.; Lindhagen, L.; Alfredsson, J.; Erlinge, D.; Held, C.; James, S.; Kellerth, T.; Lindahl, B.; Ravn-Fischer, A.; et al. Improved outcomes in patients with ST-elevation myocardial infarction during the last 20 years are related to implementation of evidence-based treatments: Experiences from the SWEDEHEART registry 1995–2014. Eur. Heart J. 2017, 38, 3056–3065. [Google Scholar] [CrossRef] [Green Version]
- Szummer, K.; Wallentin, L.; Lindhagen, L.; Alfredsson, J.; Erlinge, D.; Held, C.; James, S.; Kellerth, T.; Lindahl, B.; Ravn-Fischer, A.; et al. Relations between implementation of new treatments and improved outcomes in patients with non-ST-elevation myocardial infarction during the last 20 years: Experiences from SWEDEHEART registry 1995 to 2014. Eur. Heart J. 2018, 39, 3766–3776. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, G.M.; Meier, P.; White, S.K.; Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury: Looking beyond primary PCI. Eur. Heart J. 2013, 34, 1714–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Botker, H.E.; Engstrom, T.; Erlinge, D.; Heusch, G.; Ibanez, B.; Kloner, R.A.; Ovize, M.; Yellon, D.M.; Garcia-Dorado, D. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: Trials and tribulations. Eur. Heart J. 2017, 38, 935–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Ibanez, B.; Prat-González, S.; Speidl, W.S.; Vilahur, G.; Pinero, A.; Cimmino, G.; García, M.J.; Fuster, V.; Sanz, J.; Badimon, J.J. Early metoprolol administration before coronary reperfusion results in increased myocardial salvage: Analysis of ischemic myocardium at risk using cardiac magnetic resonance. Circulation 2007, 115, 2909–2916. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, J.M.; Fernández-Jiménez, R.; García-Alvarez, A.; Pizarro, G.; Galán-Arriola, C.; Fernández-Friera, L.; Mateos, A.; Nuno-Ayala, M.; Aguero, J.; Sánchez-González, J.; et al. Impact of the Timing of Metoprolol Administration During STEMI on Infarct Size and Ventricular Function. J. Am. Coll. Cardiol. 2016, 67, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Roolvink, V.; Ibáñez, B.; Ottervanger, J.P.; Pizarro, G.; van Royen, N.; Mateos, A.; Dambrink, J.E.; Escalera, N.; Lipsic, E.; Albarran, A.; et al. Early Intravenous Beta-Blockers in Patients with ST-Segment Elevation Myocardial Infarction Before Primary Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2016, 67, 2705–2715. [Google Scholar] [CrossRef]
- Sun, H.; Guo, T.; Liu, L.; Yu, Z.; Xu, W.; Chen, W.; Shen, L.; Wang, J.; Dou, X. Ischemic postconditioning inhibits apoptosis after acute myocardial infarction in pigs. Heart Surg. Forum 2010, 13, E305–E310. [Google Scholar] [CrossRef] [Green Version]
- Engstrøm, T.; Kelbæk, H.; Helqvist, S.; Høfsten, D.E.; Kløvgaard, L.; Clemmensen, P.; Holmvang, L.; Jørgensen, E.; Pedersen, F.; Saunamaki, K.; et al. Effect of Ischemic Postconditioning During Primary Percutaneous Coronary Intervention for Patients With ST-Segment Elevation Myocardial Infarction: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 490–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G.; Botker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote ischemic conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, A.; Lourenço, A.P. Randomized controlled trial of remote ischaemic conditioning in ST-elevation myocardial infarction as adjuvant to primary angioplasty (RIC-STEMI). Basic Res. Cardiol. 2018, 113, 14. [Google Scholar] [CrossRef] [PubMed]
- McLeod, S.L.; Iansavichene, A.; Cheskes, S. Remote Ischemic Perconditioning to Reduce Reperfusion Injury during Acute ST-Segment-Elevation Myocardial Infarction: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Eitel, I.; Stiermaier, T.; Rommel, K.P.; Fuernau, G.; Sandri, M.; Mangner, N.; Linke, A.; Erbs, S.; Lurz, P.; Boudriot, E.; et al. Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: The randomized LIPSIA CONDITIONING trial. Eur. Heart J. 2015, 36, 3049–3057. [Google Scholar] [CrossRef]
- Stiermaier, T.; Jensen, J.O.; Rommel, K.P.; de Waha-Thiele, S.; Fuernau, G.; Desch, S.; Thiele, H.; Eitel, I. Combined Intrahospital Remote Ischemic Perconditioning and Postconditioning Improves Clinical Outcome in ST-Elevation Myocardial Infarction. Circ. Res. 2019, 124, 1482–1491. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Packer, M.; McMurray, J.J. Importance of endogenous compensatory vasoactive peptides in broadening the effects of inhibitors of the renin-angiotensin system for the treatment of heart failure. Lancet 2017, 389, 1831–1840. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Velazquez, E.J.; Morrow, D.A.; DeVore, A.D.; Duffy, C.I.; Ambrosy, A.P.; McCague, K.; Rocha, R.; Braunwald, E.; Investigators, P.-H. Angiotensin-Neprilysin Inhibition in Acute Decompensated Heart Failure. N. Engl. J. Med. 2019, 380, 539–548. [Google Scholar] [CrossRef]
- Imran, M.; Hassan, M.Q.; Akhtar, M.S.; Rahman, O.; Akhtar, M.; Najmi, A.K. Sacubitril and valsartan protect from experimental myocardial infarction by ameliorating oxidative damage in Wistar rats. Clin Exp. Hypertens. 2019, 41, 62–69. [Google Scholar] [CrossRef]
- Nakano, A.; Miura, T.; Miki, T.; Nozawa, Y.; Ichikawa, Y.; Ura, N.; Shimamoto, K. Effects of neutral endopeptidase 24.11 inhibition on myocardial infarct size and ischemic preconditioning in rabbits. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2002, 366, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Liu, Y.H.; Peterson, E.; Carretero, O.A. Effect of neutral endopeptidase 24.11 inhibition on myocardial ischemia/reperfusion injury: The role of kinins. J. Cardiovasc. Pharmacol. 1997, 29, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Kaikita, K.; Sato, K.; Sueta, D.; Fujisue, K.; Arima, Y.; Oimatsu, Y.; Mitsuse, T.; Onoue, Y.; Araki, S.; et al. Cardioprotective Effects of LCZ696 (Sacubitril/Valsartan) After Experimental Acute Myocardial Infarction. Circ. Res. 2017, 2, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Torrado, J.; Cain, C.; Mauro, A.G.; Romeo, F.; Ockaili, R.; Chau, V.Q.; Nestler, J.A.; Devarakonda, T.; Ghosh, S.; Das, A.; et al. Sacubitril/Valsartan Averts Adverse Post-Infarction Ventricular Remodeling and Preserves Systolic Function in Rabbits. J. Am. Coll. Cardiol. 2018, 72, 2342–2356. [Google Scholar] [CrossRef]
- Morrow, D.A.; Velazquez, E.J.; DeVore, A.D.; Prescott, M.F.; Duffy, C.I.; Gurmu, Y.; McCague, K.; Rocha, R.; Braunwald, E. Cardiovascular biomarkers in patients with acute decompensated heart failure randomized to sacubitril-valsartan or enalapril in the PIONEER-HF trial. Eur. Heart J. 2019, 40, 3345–3352. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Senni, M.; McMurray, J.J.; Wachter, R.; McIntyre, H.F.; Reyes, A.; Majercak, I.; Andreka, P.; Shehova-Yankova, N.; Anand, I.; Yilmaz, M.B.; et al. Initiating sacubitril/valsartan (LCZ696) in heart failure: Results of TITRATION, a double-blind, randomized comparison of two uptitration regimens. Eur. J. Heart Fail. 2016, 18, 1193–1202. [Google Scholar] [CrossRef]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 119–177. [Google Scholar] [CrossRef] [Green Version]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; Antonio, R.S.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin Ameliorates Adverse Left Ventricular Remodeling in Nondiabetic Heart Failure by Enhancing Myocardial Energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellis, A.; Mauro, C.; Barbato, E.; Di Gioia, G.; Sorriento, D.; Trimarco, B.; Morisco, C. The Rationale of Neprilysin Inhibition in Prevention of Myocardial Ischemia-Reperfusion Injury during ST-Elevation Myocardial Infarction. Cells 2020, 9, 2134. https://doi.org/10.3390/cells9092134
Bellis A, Mauro C, Barbato E, Di Gioia G, Sorriento D, Trimarco B, Morisco C. The Rationale of Neprilysin Inhibition in Prevention of Myocardial Ischemia-Reperfusion Injury during ST-Elevation Myocardial Infarction. Cells. 2020; 9(9):2134. https://doi.org/10.3390/cells9092134
Chicago/Turabian StyleBellis, Alessandro, Ciro Mauro, Emanuele Barbato, Giuseppe Di Gioia, Daniela Sorriento, Bruno Trimarco, and Carmine Morisco. 2020. "The Rationale of Neprilysin Inhibition in Prevention of Myocardial Ischemia-Reperfusion Injury during ST-Elevation Myocardial Infarction" Cells 9, no. 9: 2134. https://doi.org/10.3390/cells9092134