NRF2 Is an Upstream Regulator of MYC-Mediated Osteoclastogenesis and Pathological Bone Erosion

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Mouse Osteoclastogenesis
2.3. RNA Isolation and Quantitative-PCR
2.4. Chromatin Immunoprecipitation (ChIP) Assay
2.5. Reagents
2.6. Bone In Vivo Phenotype Analysis
2.7. K/BxN Serum Transfer Arthritis Mouse Model
2.8. RNA Interference
2.9. Serum CTX Assay
2.10. Statistical Analysis and Graphs
3. Results
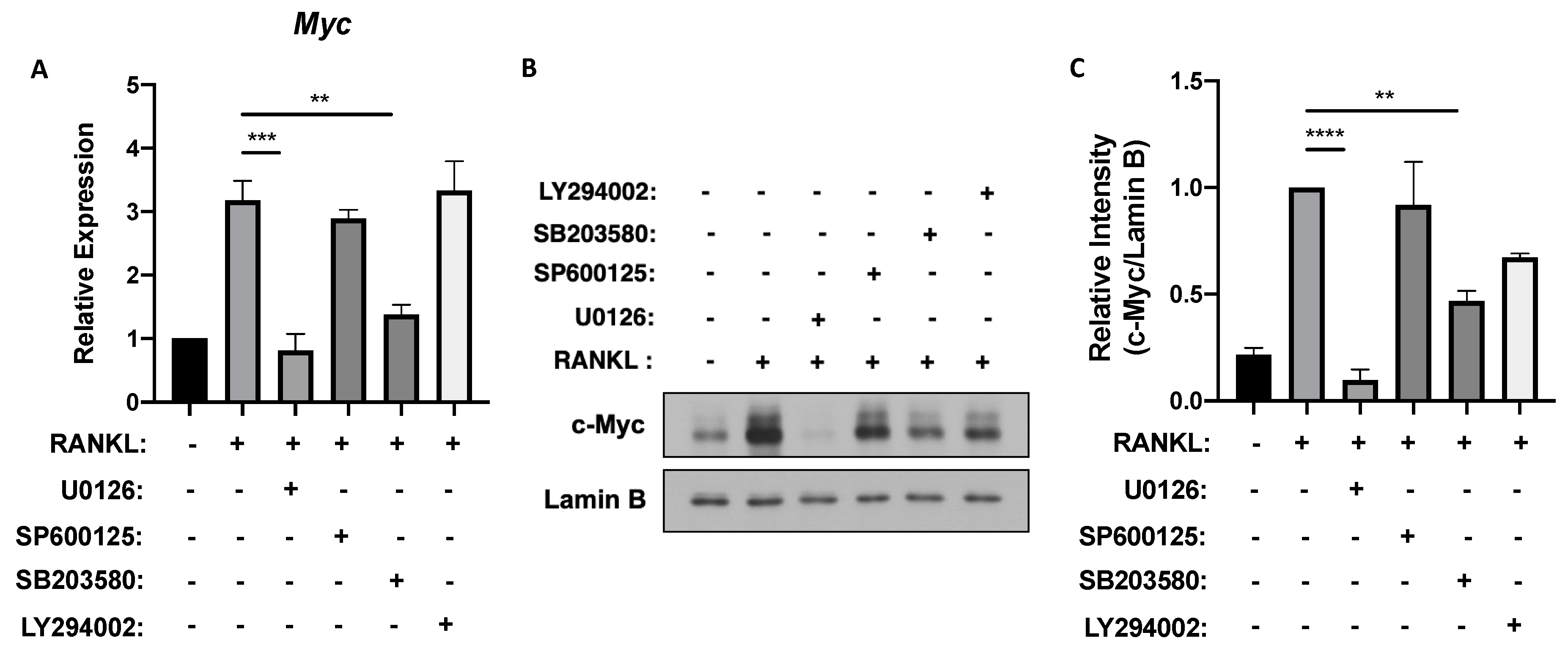
3.1. ERK and p38 Activation Is Required for MYC Expression in Osteoclastogenesis
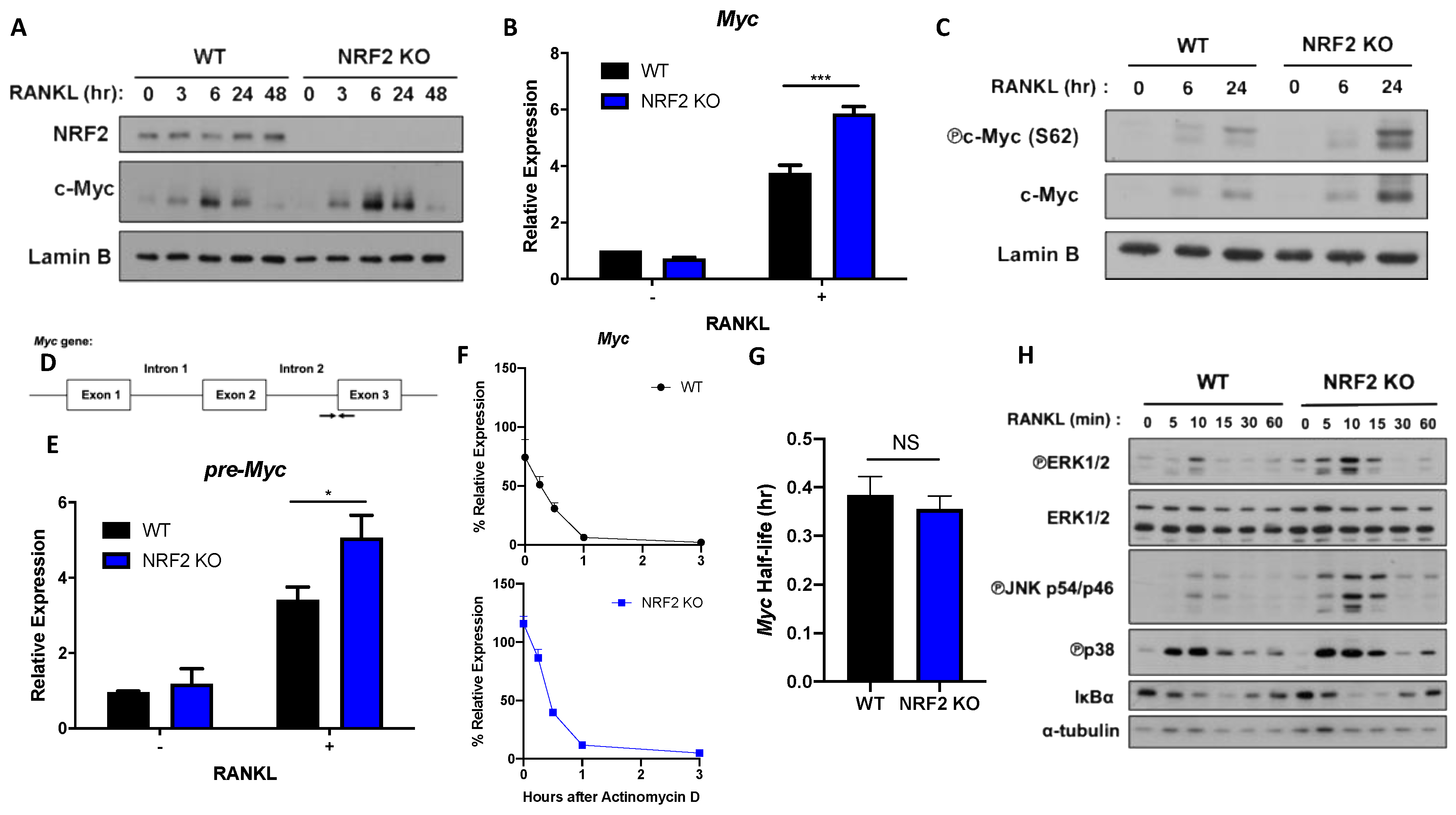
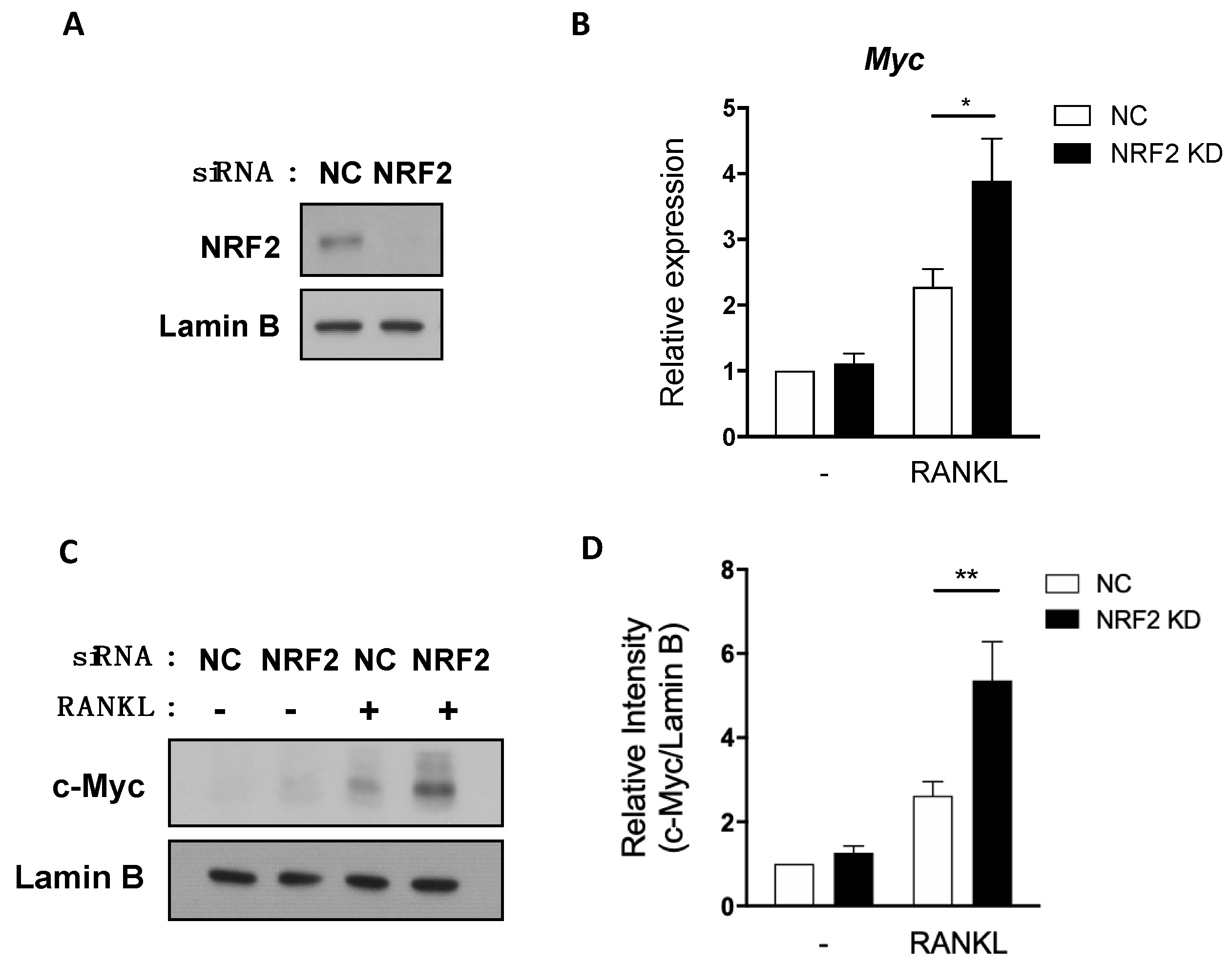
3.2. NRF2 Deficiency Enhances MYC Expression by Promoting ERK and p38 Phosphorylation
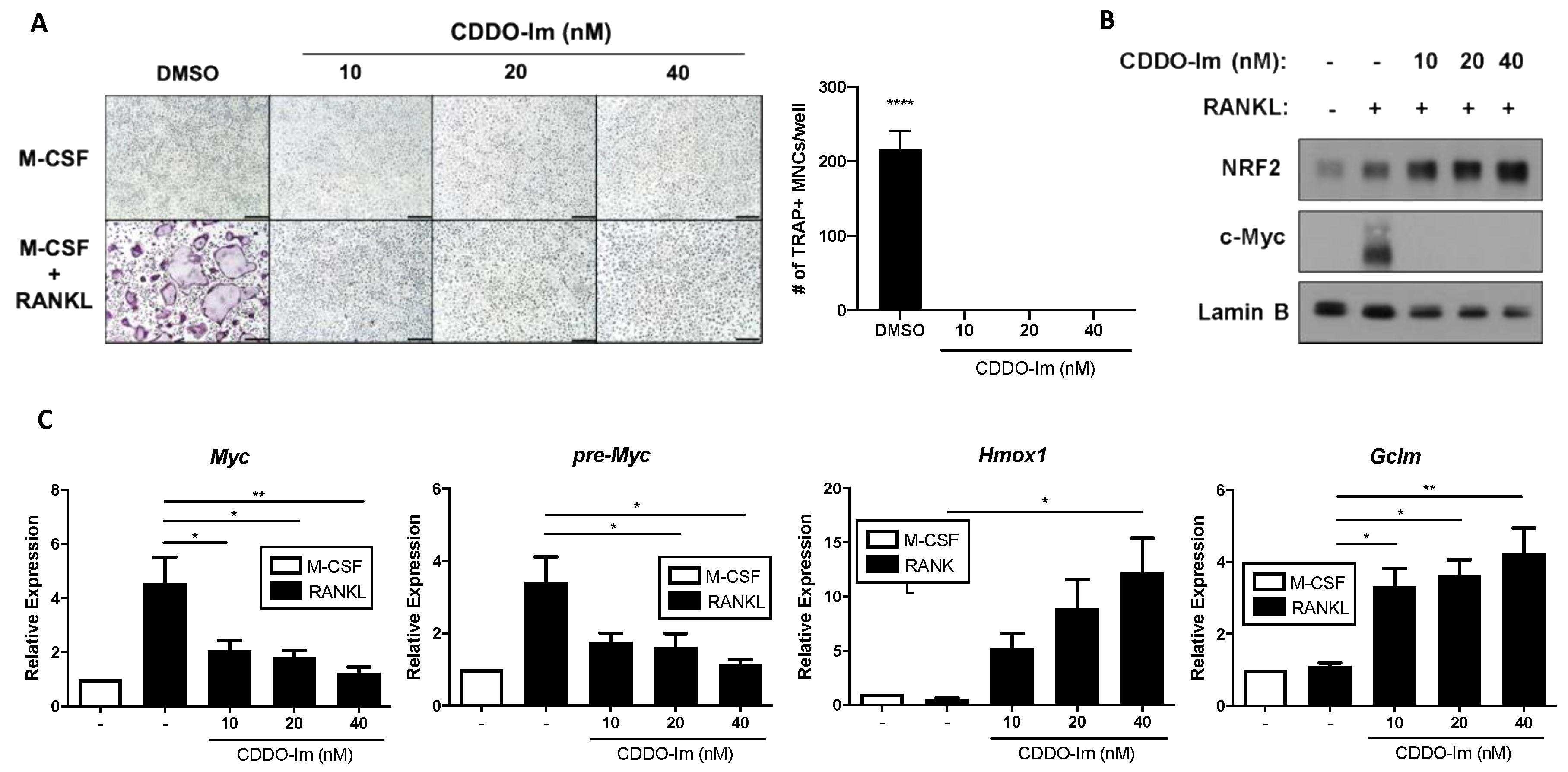
3.3. Hyperactivation of NRF2 Suppresses MYC Expression
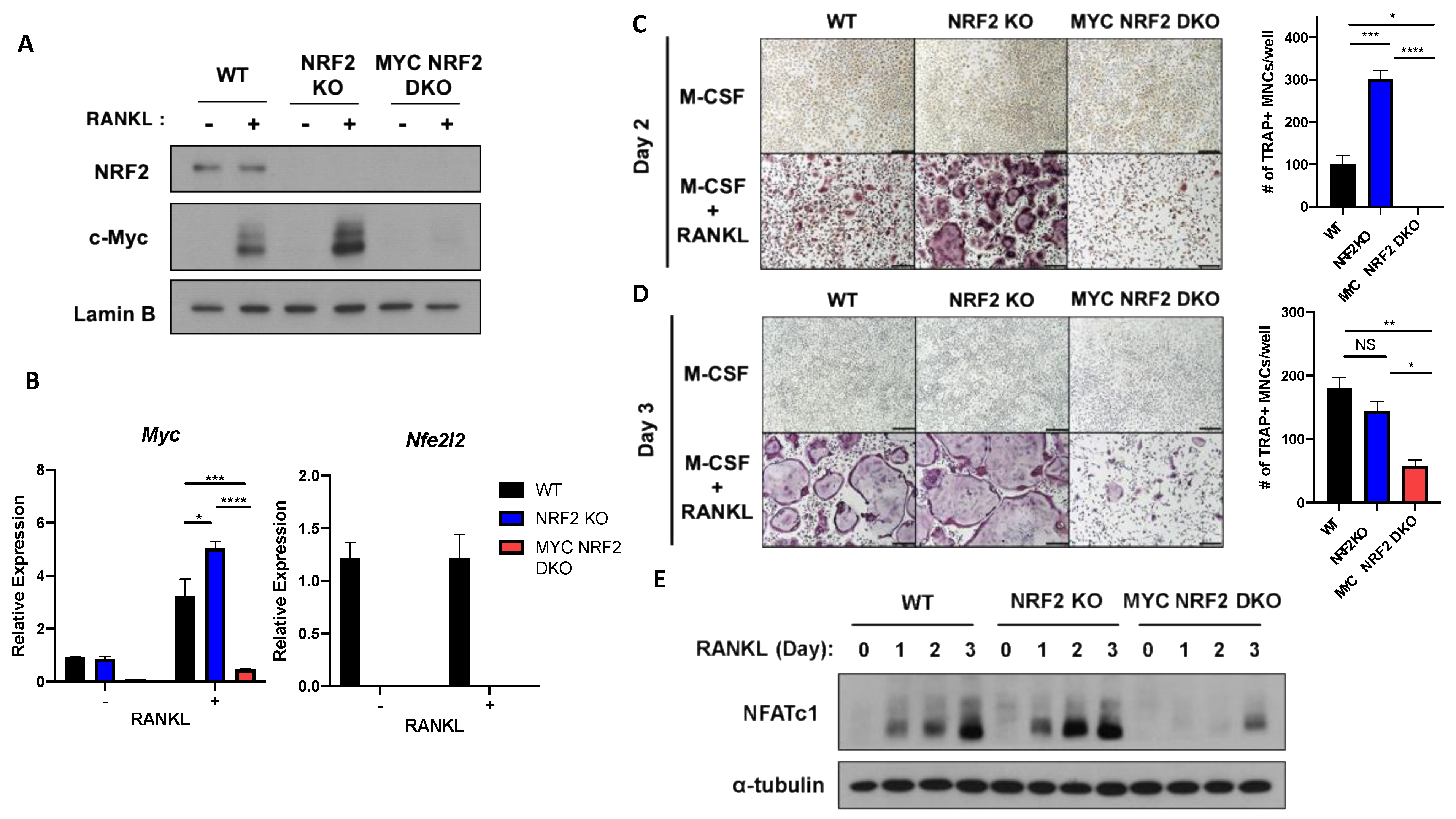
3.4. MYC Is Essential for NRF2 Deficiency-Induced Osteoclastogenesis
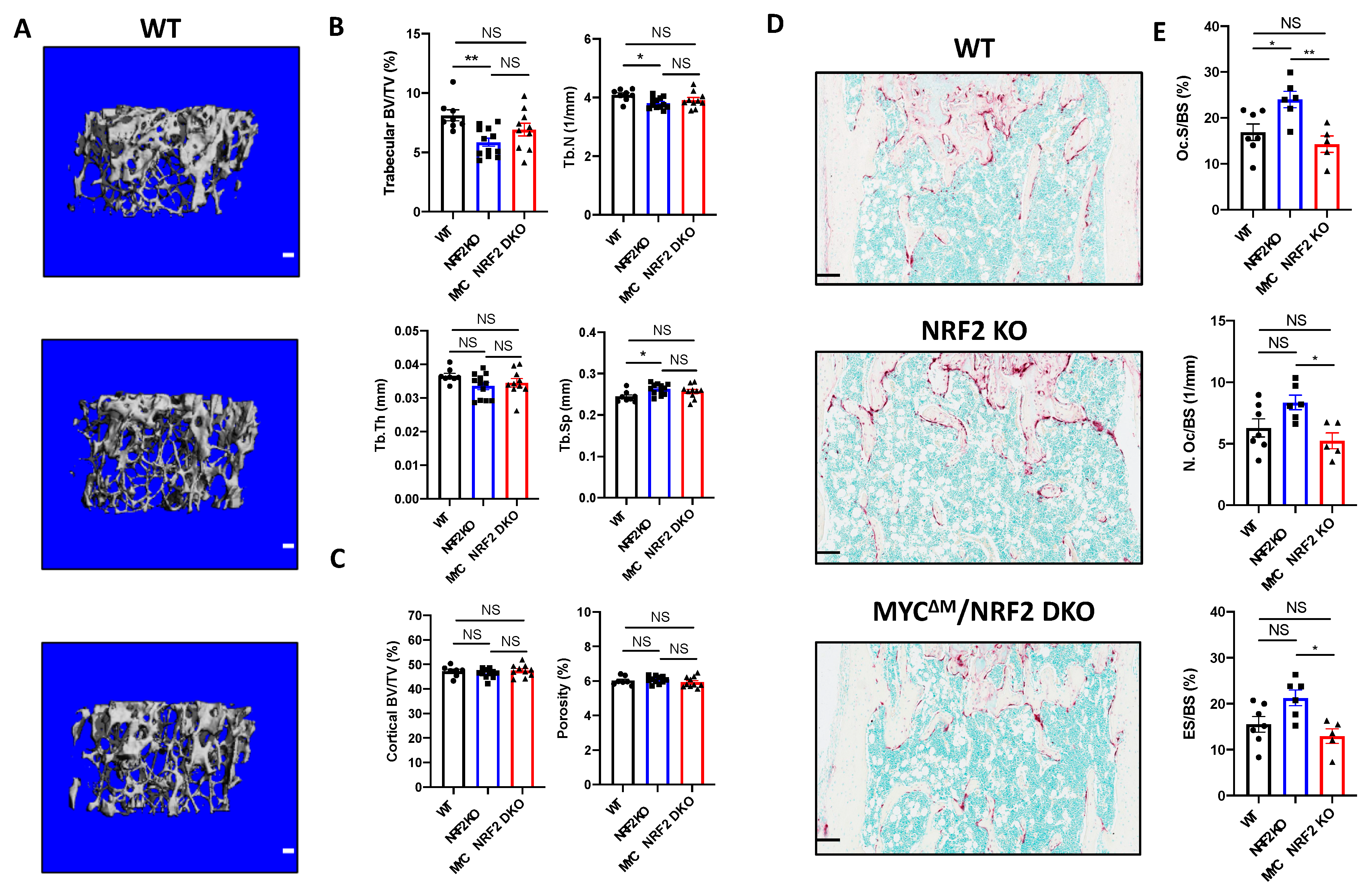
3.5. Myeloid-Specific Deletion of MYC Attenuates Osteoclast-Mediated Bone Loss Induced by NRF2 Deficiency
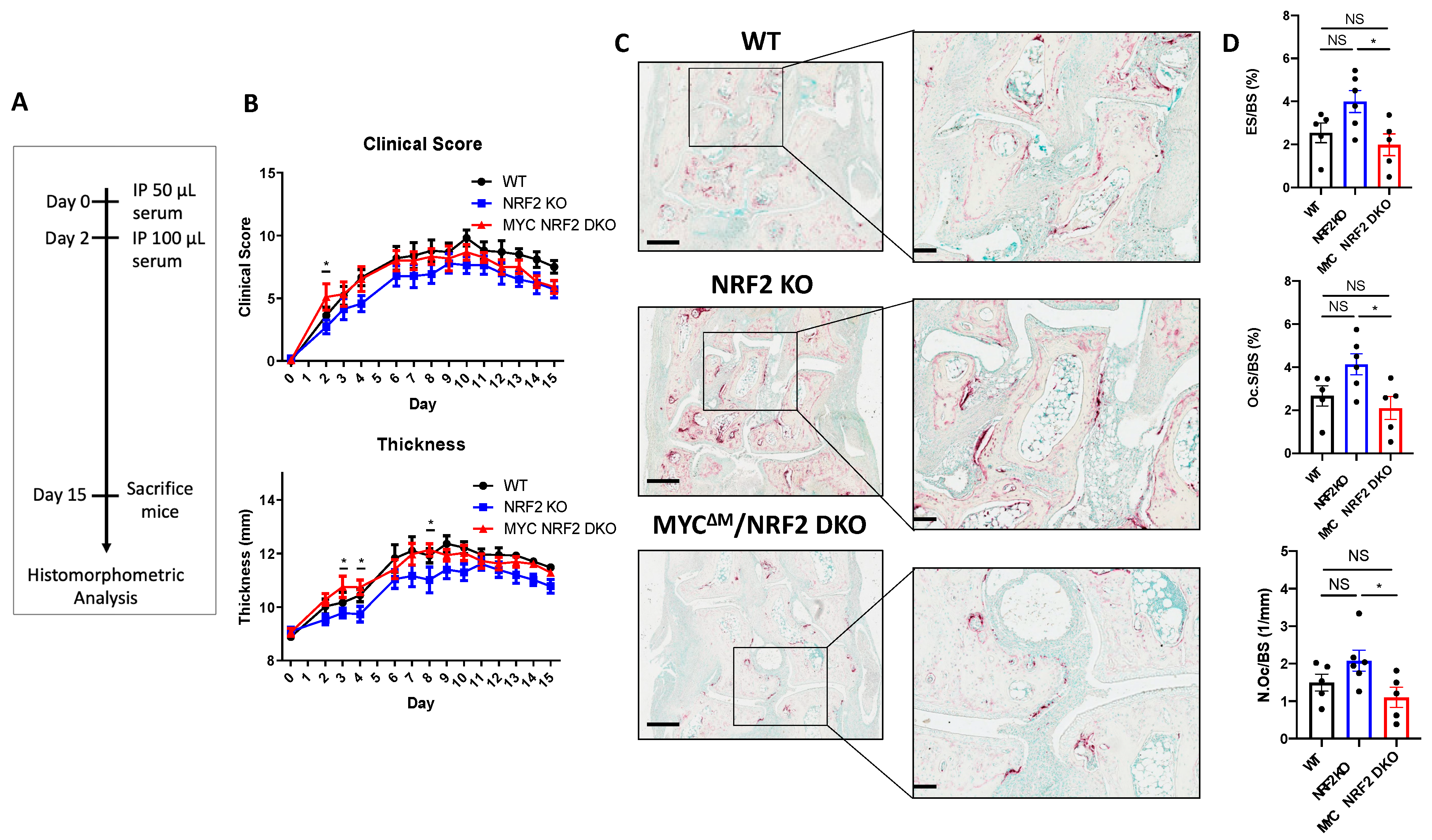
3.6. Myeloid-Specific MYC Deficiency Alleviates the Bone Loss in Serum Transfer-Induced Inflammatory Arthritis in NRF2-Deficient Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sato, K.; Takayanagi, H. Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr. Opin. Rheumatol. 2006, 18, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar]
- Park-Min, K.H. Mechanisms involved in normal and pathological osteoclastogenesis. Cell. Mol. Life Sci. 2018, 75, 2519–2528. [Google Scholar] [CrossRef]
- Novack, D.V.; Teitelbaum, S.L. The osteoclast: Friend or foe? Annu. Rev. Pathol. 2008, 3, 457–484. [Google Scholar] [CrossRef]
- Lee, K.; Seo, I.; Choi, M.H.; Jeong, D. Roles of Mitogen-Activated Protein Kinases in Osteoclast Biology. Int. J. Mol. Sci. 2018, 19, 3004. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, R.; Ferrari, S.; Russell, R.G. Denosumab and bisphosphonates: Different mechanisms of action and effects. Bone 2011, 48, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Janovska, Z. Bisphosphonate-related osteonecrosis of the jaws. A severe side effect of bisphosphonate therapy. Acta Med. 2012, 55, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Goessl, C.; Katz, L.; Dougall, W.C.; Kostenuik, P.J.; Zoog, H.B.; Braun, A.; Dansey, R.; Wagman, R.B. The development of denosumab for the treatment of diseases of bone loss and cancer-induced bone destruction. Ann. N. Y. Acad. Sci. 2012, 1263, 29–40. [Google Scholar] [CrossRef]
- Bae, S.; Lee, M.J.; Mun, S.H.; Giannopoulou, E.G.; Yong-Gonzalez, V.; Cross, J.R.; Murata, K.; Giguere, V.; van der Meulen, M.; Park-Min, K.H. MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRalpha. J. Clin. Investig. 2017, 127, 2555–2568. [Google Scholar] [CrossRef] [Green Version]
- Park-Min, K.H.; Lim, E.; Lee, M.J.; Park, S.H.; Giannopoulou, E.; Yarilina, A.; van der Meulen, M.; Zhao, B.; Smithers, N.; Witherington, J.; et al. Inhibition of osteoclastogenesis and inflammatory bone resorption by targeting BET proteins and epigenetic regulation. Nat. Commun. 2014, 5, 5418. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, J. The many ways of osteoclast activation. J. Clin. Investig. 2017, 127, 2530–2532. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [Green Version]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.X.; Xu, A.H.; Yang, Y.; Li, J. Role of Nrf2 in bone metabolism. J. Biomed. Sci. 2015, 22, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.X.; Li, L.; Corry, K.A.; Zhang, P.; Yang, Y.; Himes, E.; Mihuti, C.L.; Nelson, C.; Dai, G.; Li, J. Deletion of Nrf2 reduces skeletal mechanical properties and decreases load-driven bone formation. Bone 2015, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, L.; Ferrandiz, M.L.; Brines, R.; Guede, D.; Cuadrado, A.; Alcaraz, M.J. Effects of Nrf2 deficiency on bone microarchitecture in an experimental model of osteoporosis. Oxid. Med. Cell. Longev. 2014, 2014, 726590. [Google Scholar] [CrossRef]
- Hyeon, S.; Lee, H.; Yang, Y.; Jeong, W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic. Biol. Med. 2013, 65, 789–799. [Google Scholar] [CrossRef]
- Rana, T.; Schultz, M.A.; Freeman, M.L.; Biswas, S. Loss of Nrf2 accelerates ionizing radiation-induced bone loss by upregulating RANKL. Free Radic. Biol. Med. 2012, 53, 2298–2307. [Google Scholar] [CrossRef] [Green Version]
- Park, C.K.; Lee, Y.; Kim, K.H.; Lee, Z.H.; Joo, M.; Kim, H.H. Nrf2 is a novel regulator of bone acquisition. Bone 2014, 63, 36–46. [Google Scholar] [CrossRef]
- Nakamura, H.; Hirata, A.; Tsuji, T.; Yamamoto, T. Role of osteoclast extracellular signal-regulated kinase (ERK) in cell survival and maintenance of cell polarity. J. Bone Miner. Res. 2003, 18, 1198–1205. [Google Scholar] [CrossRef]
- Chen, S.; Qiong, Y.; Gardner, D.G. A role for p38 mitogen-activated protein kinase and c-myc in endothelin-dependent rat aortic smooth muscle cell proliferation. Hypertension 2006, 47, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Zipper, L.M.; Mulcahy, R.T. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol. Sci. 2003, 73, 124–134. [Google Scholar] [CrossRef]
- Bai, X.C.; Lu, D.; Bai, J.; Zheng, H.; Ke, Z.Y.; Li, X.M.; Luo, S.Q. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem. Biophys. Res. Commun. 2004, 314, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Choi, Y.G.; Baik, J.Y.; Han, S.Y.; Jeong, D.W.; Bae, Y.S.; Kim, N.; Lee, S.Y. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005, 106, 852–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Englen, M.D.; Valdez, Y.E.; Lehnert, N.M.; Lehnert, B.E. Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. J. Immunol. Methods 1995, 184, 281–283. [Google Scholar] [CrossRef]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Muller, R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Greenblatt, M.B.; Zou, W.; Huang, Z.; Wein, M.N.; Brady, N.; Hu, D.; Charron, J.; Brodkin, H.R.; Petsko, G.A.; et al. Schnurri-3 regulates ERK downstream of WNT signaling in osteoblasts. J. Clin. Investig 2013, 123, 4010–4022. [Google Scholar] [CrossRef] [Green Version]
- Parfitt, A.; Drezner, M.K.; Vlorieux, F.H.; Kanis, J.A.; Malluche, H.; Meunier, P.J.; Ott, S.M.; Recker, R.R. Bone histomorphometry:stardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 1987, 2, 595–610. [Google Scholar] [CrossRef]
- Korganow, A.S.; Ji, H.; Mangialaio, S.; Duchatelle, V.; Pelanda, R.; Martin, T.; Degott, C.; Kikutani, H.; Rajewsky, K.; Pasquali, J.L.; et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 1999, 10, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Murata, K.; Fang, C.; Terao, C.; Giannopoulou, E.G.; Lee, Y.J.; Lee, M.J.; Mun, S.H.; Bae, S.; Qiao, Y.; Yuan, R.; et al. Hypoxia-Sensitive COMMD1 Integrates Signaling and Cellular Metabolism in Human Macrophages and Suppresses Osteoclastogenesis. Immunity 2017, 47, 66–79.e5. [Google Scholar] [CrossRef] [Green Version]
- Park-Min, K.H.; Serbina, N.V.; Yang, W.; Ma, X.; Krystal, G.; Neel, B.G.; Nutt, S.L.; Hu, X.; Ivashkiv, L.B. FcgammaRIII-dependent inhibition of interferon-gamma responses mediates suppressive effects of intravenous immune globulin. Immunity 2007, 26, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, H.; Shinohara, F.; Kajiya, M.; Kodama, T. The Keap1/Nrf2 protein axis plays a role in osteoclast differentiation by regulating intracellular reactive oxygen species signaling. J. Biol. Chem. 2013, 288, 23009–23020. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Singhal, V.; Biswal, S.; Thimmulappa, R.K.; DiGirolamo, D.J. Nrf2 is required for normal postnatal bone acquisition in mice. Bone Res. 2014, 2, 14033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liby, K.; Hock, T.; Yore, M.M.; Suh, N.; Place, A.E.; Risingsong, R.; Williams, C.R.; Royce, D.B.; Honda, T.; Honda, Y.; et al. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 2005, 65, 4789–4798. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Chung, J.; Sussman, D.J.; Zeller, R.; Leder, P. The c-myc gene encodes superimposed RNA polymerase II and III promoters. Cell 1987, 51, 1001–1008. [Google Scholar] [CrossRef]
- Maicas, N.; Ferrandiz, M.L.; Brines, R.; Ibanez, L.; Cuadrado, A.; Koenders, M.I.; van den Berg, W.B.; Alcaraz, M.J. Deficiency of Nrf2 accelerates the effector phase of arthritis and aggravates joint disease. Antioxid. Redox Signal. 2011, 15, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Park, S.H.; Chen, J.; Qiao, Y.; Giannopoulou, E.; Berg, K.; Hanidu, A.; Li, J.; Nabozny, G.; Kang, K.; et al. Interferon-gamma Represses M2 Gene Expression in Human Macrophages by Disassembling Enhancers Bound by the Transcription Factor MAF. Immunity 2017, 47, 235–250.e4. [Google Scholar] [CrossRef]
- Cong, Q.; Jia, H.; Li, P.; Qiu, S.; Yeh, J.; Wang, Y.; Zhang, Z.L.; Ao, J.; Li, B.; Liu, H. p38alpha MAPK regulates proliferation and differentiation of osteoclast progenitors and bone remodeling in an aging-dependent manner. Sci. Rep. 2017, 7, 45964. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Udagawa, N.; Itoh, K.; Suda, K.; Murase, Y.; Nishihara, T.; Suda, T.; Takahashi, N. p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology 2002, 143, 3105–3113. [Google Scholar] [CrossRef]
- Park-Min, K.H.; Ji, J.D.; Antoniv, T.; Reid, A.C.; Silver, R.B.; Humphrey, M.B.; Nakamura, M.; Ivashkiv, L.B. IL-10 suppresses calcium-mediated costimulation of receptor activator NF-kappa B signaling during human osteoclast differentiation by inhibiting TREM-2 expression. J. Immunol. 2009, 183, 2444–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Staser, K.; Rhodes, S.D.; Liu, Y.; Wu, X.; Park, S.J.; Yuan, J.; Yang, X.; Li, X.; Jiang, L.; et al. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS ONE 2011, 6, e24780. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, H.S.; Yeon, J.T.; Choi, S.W.; Chun, C.H.; Kwak, H.B.; Oh, J. GM-CSF regulates fusion of mononuclear osteoclasts into bone-resorbing osteoclasts by activating the Ras/ERK pathway. J. Immunol. 2009, 183, 3390–3399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkhoff, E.; Houben, R.; Loffler, S.; Troppmair, J.; Lee, J.E.; Rapp, U.R. Regulation of c-myc expression by Ras/Raf signalling. Oncogene 1998, 16, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcon-Vargas, D.; Ronai, Z. c-Jun-NH2 kinase (JNK) contributes to the regulation of c-Myc protein stability. J. Biol. Chem. 2004, 279, 5008–5016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Braun, S.; Hanselmann, C.; Gassmann, M.G.; auf dem Keller, U.; Born-Berclaz, C.; Chan, K.; Kan, Y.W.; Werner, S. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell. Biol. 2002, 22, 5492–5505. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar]
- Yamaguchi, Y.; Kanzaki, H.; Katsumata, Y.; Itohiya, K.; Fukaya, S.; Miyamoto, Y.; Narimiya, T.; Wada, S.; Nakamura, Y. Dimethyl fumarate inhibits osteoclasts via attenuation of reactive oxygen species signalling by augmented antioxidation. J. Cell. Mol. Med. 2018, 22, 1138–1147. [Google Scholar]
- Callaway, D.A.; Jiang, J.X. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J. Bone Miner. Metab. 2015, 33, 359–370. [Google Scholar] [CrossRef]
- Maggio, D.; Barabani, M.; Pierandrei, M.; Polidori, M.C.; Catani, M.; Mecocci, P.; Senin, U.; Pacifici, R.; Cherubini, A. Marked decrease in plasma antioxidants in aged osteoporotic women: Results of a cross-sectional study. J. Clin. Endocrinol. Metab. 2003, 88, 1523–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerhan, J.R.; Saag, K.G.; Merlino, L.A.; Mikuls, T.R.; Criswell, L.A. Antioxidant micronutrients and risk of rheumatoid arthritis in a cohort of older women. Am. J. Epidemiol. 2003, 157, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Morita, M.; Ohuchi, M.; Kido, M.A.; Fukuma, Y.; Nishishita, K.; Okamoto, K.; Itoh, K.; Yamamoto, M.; Tsukuba, T. Effects of deficiency of Kelch-like ECH-associated protein 1 on skeletal organization: A mechanism for diminished nuclear factor of activated T cells cytoplasmic 1 during osteoclastogenesis. FASEB J. 2017, 31, 4011–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Corry, K.A.; Loughran, J.P.; Li, J. Moderate Nrf2 Activation by Genetic Disruption of Keap1 Has Sex-Specific Effects on Bone Mass in Mice. Sci. Rep. 2020, 10, 348. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, P.S.U.; Mun, S.H.; Zeng, S.L.; Kim, H.; Bae, S.; Park-Min, K.-H. NRF2 Is an Upstream Regulator of MYC-Mediated Osteoclastogenesis and Pathological Bone Erosion. Cells 2020, 9, 2133. https://doi.org/10.3390/cells9092133
Park PSU, Mun SH, Zeng SL, Kim H, Bae S, Park-Min K-H. NRF2 Is an Upstream Regulator of MYC-Mediated Osteoclastogenesis and Pathological Bone Erosion. Cells. 2020; 9(9):2133. https://doi.org/10.3390/cells9092133
Chicago/Turabian StylePark, Peter Sang Uk, Se Hwan Mun, Steven L. Zeng, Haemin Kim, Seyeon Bae, and Kyung-Hyun Park-Min. 2020. "NRF2 Is an Upstream Regulator of MYC-Mediated Osteoclastogenesis and Pathological Bone Erosion" Cells 9, no. 9: 2133. https://doi.org/10.3390/cells9092133