Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Microglia in the Healthy Central Nervous System
3. Microglia in the Pathological CNS
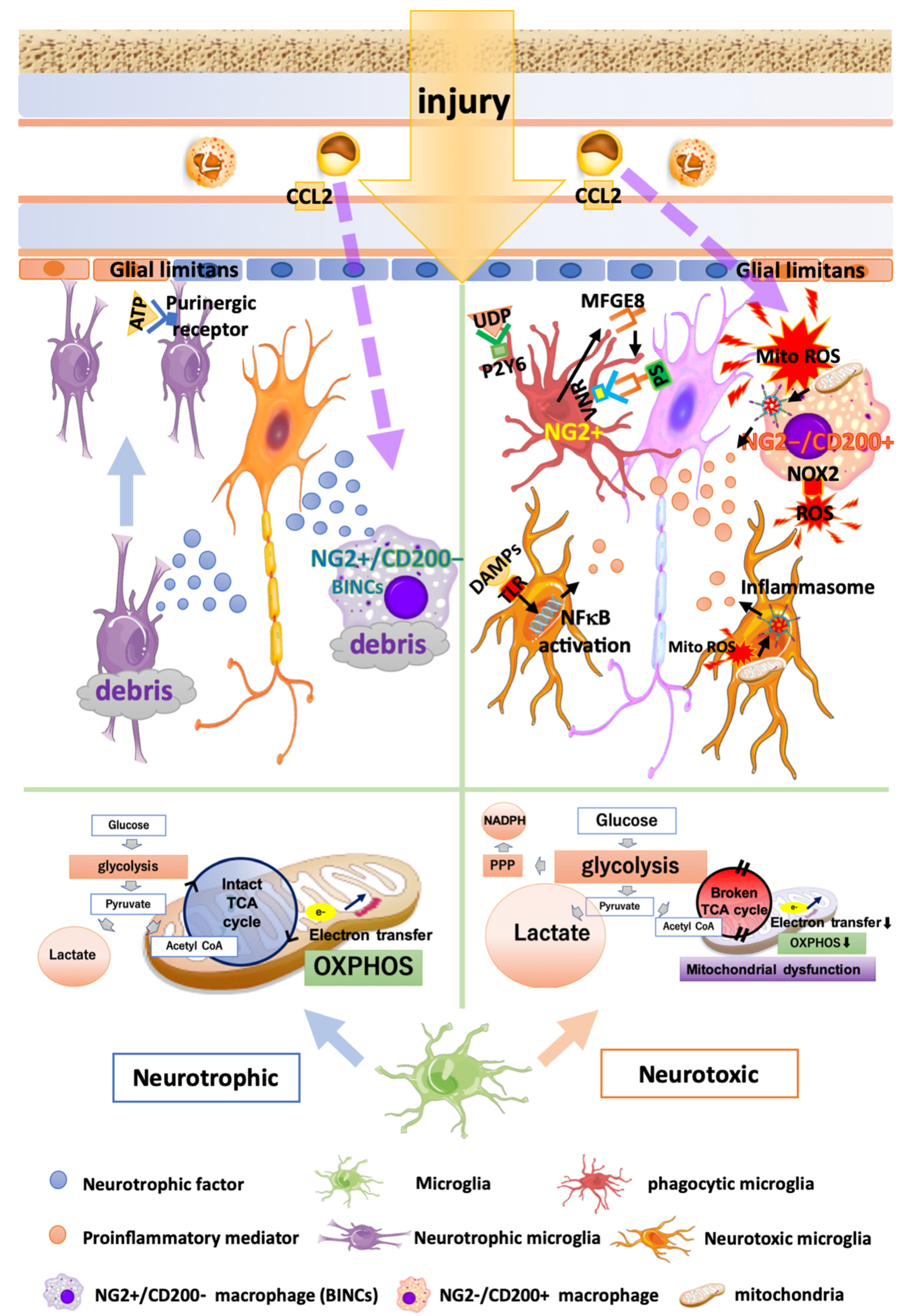
3.1. Heterogeneity of Microglia in Traumatic Brain Injury and Cerebral Infarction
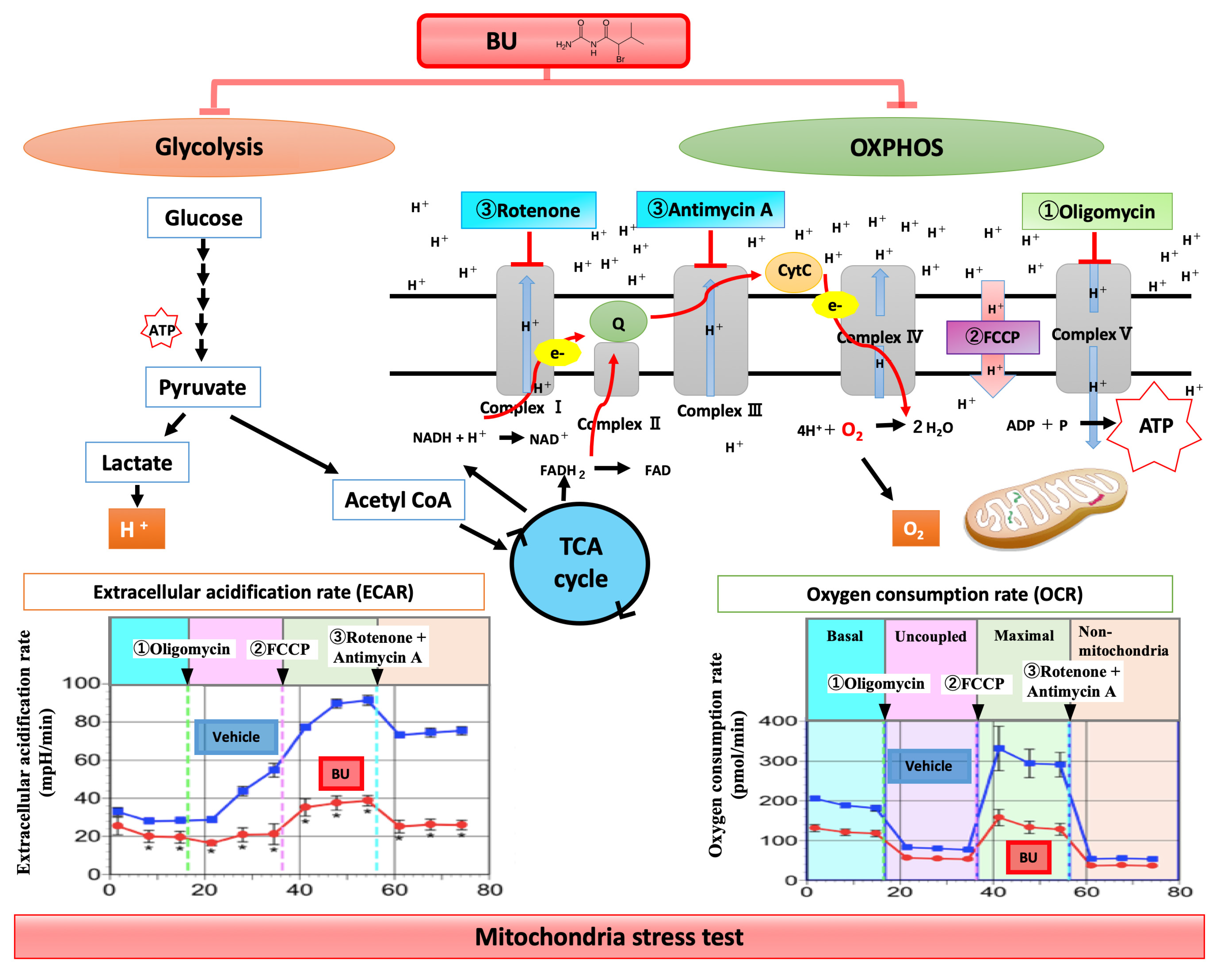
3.2. Metabolic Changes in Microglia in TBI and Infarction
3.3. Phagocytosis by Microglia and Find-Me/Eat-Me Signals in TBI and Infarction
3.4. Oxidative Stress Caused by Microglia and Macrophages in TBI and Infarction
3.5. Heterogeneity of Blood-Borne Macrophages in TBI and Infarction
3.6. Carbon Monoxide Poisoning
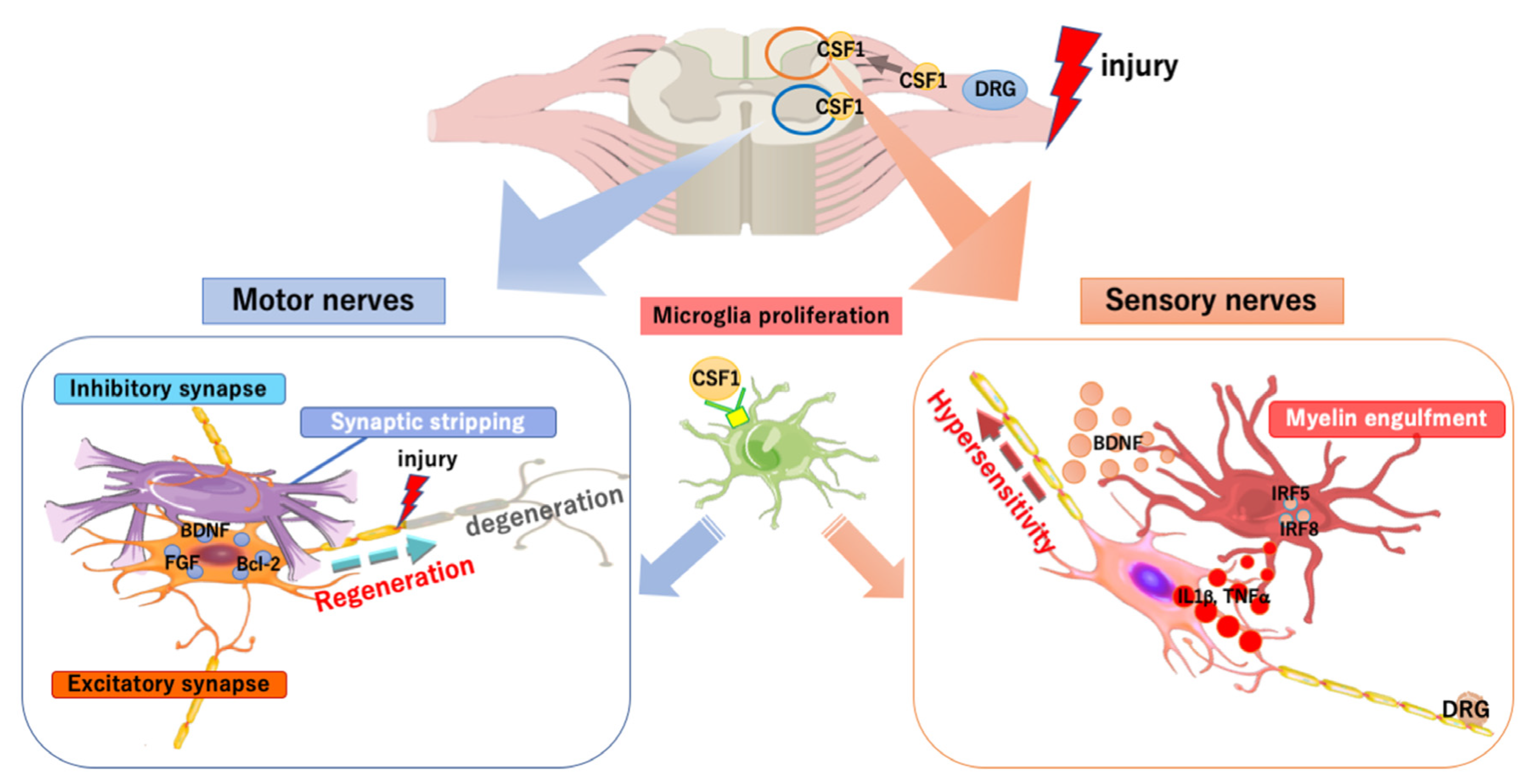
4. Microglia/Macrophages in the PNS Injury
5. Therapeutic Approaches Targeting Microglia
5.1. Immunomodulation
5.1.1. Suppressing the Proinflammatory Reaction of Microglia/Macrophages
5.1.2. Enhancing the Neuroprotective Function of Microglia/Macrophages
5.2. Controlling Metabolism of Microglia/Macrophages
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Dudvarski Stankovic, N.; Teodorczyk, M.; Ploen, R.; Zipp, F.; Schmidt, M.H.H. Microglia-blood vessel interactions: A double-edged sword in brain pathologies. Acta Neuropathol. 2016, 131, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Tsuda, M. Microglia in neuropathic pain: Cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018, 19, 138–152. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Moorhouse, A.J.; Miyamoto, A.; Nabekura, J. Microglia: Actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 2013, 36, 209–217. [Google Scholar] [CrossRef]
- Augusto-Oliveira, M.; Arrifano, G.P.; Lopes-Araujo, A.; Santos-Sacramento, L.; Takeda, P.Y.; Anthony, D.C.; Malva, J.O.; Crespo-Lopez, M.E. What Do Microglia Really Do in Healthy Adult Brain? Cells 2019, 8, 1293. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Walton, N.M.; Sutter, B.M.; Laywell, E.D.; Levkoff, L.H.; Kearns, S.M.; Marshall, G.P., 2nd; Scheffler, B.; Steindler, D.A. Microglia instruct subventricular zone neurogenesis. Glia 2006, 54, 815–825. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, M.E.; Miyanishi, K.; Takeda, H.; Islam, A.; Matsuoka, N.; Kubo, M.; Matsumoto, S.; Kunieda, T.; Nomoto, M.; Yano, H.; et al. Phagocytic elimination of synapses by microglia during sleep. Glia 2020, 68, 44–59. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Koyanagi, S.; Kusunose, N.; Okada, R.; Wu, Z.; Tozaki-Saitoh, H.; Ukai, K.; Kohsaka, S.; Inoue, K.; Ohdo, S.; et al. The intrinsic microglial molecular clock controls synaptic strength via the circadian expression of cathepsin S. Sci. Rep. 2013, 3, 2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peirano, P.D.; Algarin, C.R. Sleep in brain development. Biol. Res. 2007, 40, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M.G.; Issa, N.P.; Stryker, M.P. Sleep enhances plasticity in the developing visual cortex. Neuron 2001, 30, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Stickgold, R.; Walker, M.P. Sleep-dependent memory consolidation and reconsolidation. Sleep Med. 2007, 8, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275 Pt 3, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef]
- Lagraoui, M.; Latoche, J.R.; Cartwright, N.G.; Sukumar, G.; Dalgard, C.L.; Schaefer, B.C. Controlled cortical impact and craniotomy induce strikingly similar profiles of inflammatory gene expression, but with distinct kinetics. Front. Neurol. 2012, 3, 155. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.W.; Wang, J.; Dhandapani, K.M.; Wang, R.; Brann, D.W. NADPH oxidases in traumatic brain injury - Promising therapeutic targets? Redox Biol. 2018, 16, 285–293. [Google Scholar] [CrossRef]
- Hall, E.D.; Wang, J.A.; Miller, D.M. Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Exp. Neurol. 2012, 238, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Kobiyama, K.; Takeshita, F.; Ishii, K.J. Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front. Cell. Infect. Microbiol. 2012, 2, 168. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xing, H.; Wan, L.; Jiang, X.; Wang, C.; Wu, Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed. Pharmacother. 2018, 105, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciano, P.G.; Eberwine, J.H.; Ragupathi, R.; Saatman, K.E.; Meaney, D.F.; McIntosh, T.K. Expression profiling following traumatic brain injury: A review. Neurochem. Res. 2002, 27, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Biber, K.; Finsen, B. Inflammatory cytokines in experimental and human stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1677–1698. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Devanney, N.A.; Stewart, A.N.; Gensel, J.C. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp. Neurol. 2020, 329, 113310. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, T.; Tanaka, J.; Sekiya, K.; Nishikawa, Y.; Abe, N.; Hamada, T.; Kitamura, S.; Ikemune, K.; Ochi, S.; Choudhury, M.E.; et al. Chronic constriction injury of the sciatic nerve in rats causes different activation modes of microglia between the anterior and posterior horns of the spinal cord. Neurochem. Int. 2020, 134, 104672. [Google Scholar] [CrossRef]
- Gimeno-Bayon, J.; Lopez-Lopez, A.; Rodriguez, M.J.; Mahy, N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J. Neurosci. Res. 2014, 92, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Lauro, C.; Chece, G.; Monaco, L.; Antonangeli, F.; Peruzzi, G.; Rinaldo, S.; Paone, A.; Cutruzzola, F.; Limatola, C. Fractalkine Modulates Microglia Metabolism in Brain Ischemia. Front. Cell Neurosci. 2019, 13, 414. [Google Scholar] [CrossRef]
- Tu, D.; Gao, Y.; Yang, R.; Guan, T.; Hong, J.S.; Gao, H.M. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J. Neuroinflammation 2019, 16, 255. [Google Scholar] [CrossRef] [Green Version]
- Honda, S.; Sasaki, Y.; Ohsawa, K.; Imai, Y.; Nakamura, Y.; Inoue, K.; Kohsaka, S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J. Neurosci. 2001, 21, 1975–1982. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef]
- Fourgeaud, L.; Traves, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagorska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, N.; Takano, T.; Pei, Y.; Xavier, A.L.; Goldman, S.A.; Nedergaard, M. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc. Natl. Acad. Sci. USA 2016, 113, 1074–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Thery, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neher, J.J.; Neniskyte, U.; Zhao, J.W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef]
- Suzuki, J.; Fujii, T.; Imao, T.; Ishihara, K.; Kuba, H.; Nagata, S. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem. 2013, 288, 13305–13316. [Google Scholar] [CrossRef] [Green Version]
- Levano, K.; Punia, V.; Raghunath, M.; Debata, P.R.; Curcio, G.M.; Mogha, A.; Purkayastha, S.; McCloskey, D.; Fata, J.; Banerjee, P. Atp8a1 deficiency is associated with phosphatidylserine externalization in hippocampus and delayed hippocampus-dependent learning. J. Neurochem. 2012, 120, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Fricker, M.; Neher, J.J.; Zhao, J.W.; Thery, C.; Tolkovsky, A.M.; Brown, G.C. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J. Neurosci. 2012, 32, 2657–2666. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, K.; Nishioka, R.; Ikeda, A.; Mise, A.; Takahashi, H.; Yano, H.; Kumon, Y.; Ohnishi, T.; Tanaka, J. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-beta1. Glia 2014, 62, 185–198. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Ohsawa, K.; Inoue, K.; Kohsaka, S. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 2013, 61, 47–54. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Hornik, T.; Brown, G.C. Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 2014, 62, 1463–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Matozaki, T.; Murata, Y.; Okazawa, H.; Ohnishi, H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009, 19, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Lundgren, D.H.; Jaiswal, S.; Chao, M.; Graham, K.L.; Garris, C.S.; Axtell, R.C.; Ho, P.P.; Lock, C.B.; Woodard, J.I.; et al. Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J. Exp. Med. 2012, 209, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ma, M.W.; Dhandapani, K.M.; Brann, D.W. Regulatory role of NADPH oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free Radic Biol. Med. 2017, 113, 119–131. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Dhandapani, K.M.; Brann, D.W. NADPH Oxidase 2 Regulates NLRP3 Inflammasome Activation in the Brain after Traumatic Brain Injury. Oxid Med. Cell Longev. 2017, 2017, 6057609. [Google Scholar] [CrossRef]
- Gao, L.; Laude, K.; Cai, H. Mitochondrial pathophysiology, reactive oxygen species, and cardiovascular diseases. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 137–155. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Abe, N.; Choudhury, M.E.; Watanabe, M.; Kawasaki, S.; Nishihara, T.; Yano, H.; Matsumoto, S.; Kunieda, T.; Kumon, Y.; Yorozuya, T.; et al. Comparison of the detrimental features of microglia and infiltrated macrophages in traumatic brain injury: A study using a hypnotic bromovalerylurea. Glia 2018, 66, 2158–2173. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar Sahel, D.; Kaira, M.; Raj, K.; Sharma, S.; Singh, S. Mitochondrial dysfunctioning and neuroinflammation: Recent highlights on the possible mechanisms involved in Traumatic Brain Injury. Neurosci. Lett. 2019, 710, 134347. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.J.; Mena, N.P.; Nunez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Islam, A.; Choudhury, M.E.; Kigami, Y.; Utsunomiya, R.; Matsumoto, S.; Watanabe, H.; Kumon, Y.; Kunieda, T.; Yano, H.; Tanaka, J. Sustained anti-inflammatory effects of TGF-beta1 on microglia/macrophages. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 721–734. [Google Scholar] [CrossRef]
- Dhandapani, K.M.; Brann, D.W. Transforming growth factor-beta: A neuroprotective factor in cerebral ischemia. Cell Biochem. Biophys. 2003, 39, 13–22. [Google Scholar] [CrossRef]
- Szalay, G.; Martinecz, B.; Lenart, N.; Kornyei, Z.; Orsolits, B.; Judak, L.; Csaszar, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290 e1217. [Google Scholar] [CrossRef]
- DePaula-Silva, A.B.; Gorbea, C.; Doty, D.J.; Libbey, J.E.; Sanchez, J.M.S.; Hanak, T.J.; Cazalla, D.; Fujinami, R.S. Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J. Neuroinflammation 2019, 16, 152. [Google Scholar] [CrossRef] [Green Version]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, H.; Kumon, Y.; Watanabe, H.; Ohnishi, T.; Shudou, M.; Chuai, M.; Imai, Y.; Takahashi, H.; Tanaka, J. Accumulation of macrophage-like cells expressing NG2 proteoglycan and Iba1 in ischemic core of rat brain after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 2008, 28, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirkin, A.; Matsumoto, H.; Takahashi, H.; Inoue, A.; Tagawa, M.; Ohue, S.; Watanabe, H.; Yano, H.; Kumon, Y.; Ohnishi, T.; et al. Iba1(+)/NG2(+) macrophage-like cells expressing a variety of neuroprotective factors ameliorate ischemic damage of the brain. J. Cereb. Blood Flow Metab. 2010, 30, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, S.; Tanaka, J.; Yano, H.; Takahashi, H.; Sugimoto, K.; Ohue, S.; Inoue, A.; Aono, H.; Kusakawa, A.; Watanabe, H.; et al. CD200+ and CD200- macrophages accumulated in ischemic lesions of rat brain: The two populations cannot be classified as either M1 or M2 macrophages. J. Neuroimmunol. 2015, 282, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef]
- Costello, D.A.; Lyons, A.; Denieffe, S.; Browne, T.C.; Cox, F.F.; Lynch, M.A. Long term potentiation is impaired in membrane glycoprotein CD200-deficient mice: A role for Toll-like receptor activation. J. Biol. Chem. 2011, 286, 34722–34732. [Google Scholar] [CrossRef] [Green Version]
- Cox, F.F.; Carney, D.; Miller, A.M.; Lynch, M.A. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain Behav. Immun. 2012, 26, 789–796. [Google Scholar] [CrossRef]
- Nishihara, T.; Ochi, M.; Sugimoto, K.; Takahashi, H.; Yano, H.; Kumon, Y.; Ohnishi, T.; Tanaka, J. Subcutaneous injection containing IL-3 and GM-CSF ameliorates stab wound-induced brain injury in rats. Exp. Neurol. 2011, 229, 507–516. [Google Scholar] [CrossRef]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thom, S.R.; Bhopale, V.M.; Fisher, D.; Zhang, J.; Gimotty, P. Delayed neuropathology after carbon monoxide poisoning is immune-mediated. Proc. Natl. Acad. Sci. USA 2004, 101, 13660–13665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, G.; Ren, M.; Wang, X.; Jiang, H.; Yin, X.; Wang, S.; Wang, X.; Feng, H. Allopurinol reduces severity of delayed neurologic sequelae in experimental carbon monoxide toxicity in rats. Neurotoxicology 2015, 48, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, K.; Nishihara, T.; Abe, N.; Konishi, A.; Nandate, H.; Hamada, T.; Ikemune, K.; Takasaki, Y.; Tanaka, J.; Asano, M.; et al. Carbon monoxide poisoning-induced delayed encephalopathy accompanies decreased microglial cell numbers: Distinctive pathophysiological features from hypoxemia-induced brain damage. Brain Res. 2019, 1710, 22–32. [Google Scholar] [CrossRef]
- Lyons, S.A.; Kettenmann, H. Oligodendrocytes and microglia are selectively vulnerable to combined hypoxia and hypoglycemia injury in vitro. J. Cereb. Blood Flow Metab. 1998, 18, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Zheng, B.; Fan, L.W.; Rhodes, P.G.; Cai, Z. IGF-1 protects oligodendrocyte progenitors against TNFalpha-induced damage by activation of PI3K/Akt and interruption of the mitochondrial apoptotic pathway. Glia 2007, 55, 1099–1107. [Google Scholar] [CrossRef]
- Nicholas, R.S.; Stevens, S.; Wing, M.G.; Compston, D.A. Microglia-derived IGF-2 prevents TNFalpha induced death of mature oligodendrocytes in vitro. J. Neuroimmunol. 2002, 124, 36–44. [Google Scholar] [CrossRef]
- Ohya, W.; Funakoshi, H.; Kurosawa, T.; Nakamura, T. Hepatocyte growth factor (HGF) promotes oligodendrocyte progenitor cell proliferation and inhibits its differentiation during postnatal development in the rat. Brain Res. 2007, 1147, 51–65. [Google Scholar] [CrossRef]
- Reuss, B.; von Bohlen und Halbach, O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003, 313, 139–157. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Siebert, H.; Sachse, A.; Kuziel, W.A.; Maeda, N.; Bruck, W. The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system. J. Neuroimmunol. 2000, 110, 177–185. [Google Scholar] [CrossRef]
- Nishihara, T.; Remacle, A.G.; Angert, M.; Shubayev, I.; Shiryaev, S.A.; Liu, H.; Dolkas, J.; Chernov, A.V.; Strongin, A.Y.; Shubayev, V.I. Matrix metalloproteinase-14 both sheds cell surface neuronal glial antigen 2 (NG2) proteoglycan on macrophages and governs the response to peripheral nerve injury. J. Biol. Chem. 2015, 290, 3693–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Z.; Kuhn, J.A.; Wang, X.; Colquitt, B.; Solorzano, C.; Vaman, S.; Guan, A.K.; Evans-Reinsch, Z.; Braz, J.; Devor, M.; et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci. 2016, 19, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Tsuda, M.; Yoshinaga, R.; Tozaki-Saitoh, H.; Ozato, K.; Tamura, T.; Inoue, K. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep. 2012, 1, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalla, R.; Liu, Z.; Xu, S.; Koppius, A.; Imai, Y.; Kloss, C.U.; Kohsaka, S.; Gschwendtner, A.; Moller, J.C.; Werner, A.; et al. Microglia and the early phase of immune surveillance in the axotomized facial motor nucleus: Impaired microglial activation and lymphocyte recruitment but no effect on neuronal survival or axonal regeneration in macrophage-colony stimulating factor-deficient mice. J. Comp. Neurol. 2001, 436, 182–201. [Google Scholar] [PubMed]
- Blinzinger, K.; Kreutzberg, G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat 1968, 85, 145–157. [Google Scholar] [CrossRef]
- Moran, L.B.; Graeber, M.B. The facial nerve axotomy model. Brain Res. Brain Res. Rev. 2004, 44, 154–178. [Google Scholar] [CrossRef]
- Yamada, J.; Hayashi, Y.; Jinno, S.; Wu, Z.; Inoue, K.; Kohsaka, S.; Nakanishi, H. Reduced synaptic activity precedes synaptic stripping in vagal motoneurons after axotomy. Glia 2008, 56, 1448–1462. [Google Scholar] [CrossRef]
- Nakajima, K.; Tohyama, Y.; Maeda, S.; Kohsaka, S.; Kurihara, T. Neuronal regulation by which microglia enhance the production of neurotrophic factors for GABAergic, catecholaminergic, and cholinergic neurons. Neurochem. Int. 2007, 50, 807–820. [Google Scholar] [CrossRef]
- Chen, Z.; Jalabi, W.; Hu, W.; Park, H.J.; Gale, J.T.; Kidd, G.J.; Bernatowicz, R.; Gossman, Z.C.; Chen, J.T.; Dutta, R.; et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat. Commun. 2014, 5, 4486. [Google Scholar] [CrossRef] [Green Version]
- Higaki, H.; Choudhury, M.E.; Kawamoto, C.; Miyamoto, K.; Islam, A.; Ishii, Y.; Miyanishi, K.; Takeda, H.; Seo, N.; Sugimoto, K.; et al. The hypnotic bromovalerylurea ameliorates 6-hydroxydopamine-induced dopaminergic neuron loss while suppressing expression of interferon regulatory factors by microglia. Neurochem. Int. 2016, 99, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J. Favorable and unfavorable roles of microglia and macrophages in the pathologic central nervous system. Neuroimmunol. Neuroinflammation 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, L.; Ma, Q.; Concepcion, K.R.; Song, M.A.; Zhang, P.; Fu, Y.; Xiao, D.; Zhang, L. Repression of the Glucocorticoid Receptor Aggravates Acute Ischemic Brain Injuries in Adult Mice. Int. J. Mol. Sci. 2018, 19, 2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campolo, M.; Ahmad, A.; Crupi, R.; Impellizzeri, D.; Morabito, R.; Esposito, E.; Cuzzocrea, S. Combination therapy with melatonin and dexamethasone in a mouse model of traumatic brain injury. J. Endocrinol. 2013, 217, 291–301. [Google Scholar] [CrossRef]
- Edwards, P.; Arango, M.; Balica, L.; Cottingham, R.; El-Sayed, H.; Farrell, B.; Fernandes, J.; Gogichaisvili, T.; Golden, N.; Hartzenberg, B.; et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet 2005, 365, 1957–1959. [Google Scholar] [CrossRef]
- Sandercock, P.A.; Soane, T. Corticosteroids for acute ischaemic stroke. Cochrane Database Syst. Rev. 2011, CD000064. [Google Scholar] [CrossRef]
- Thal, S.C.; Schaible, E.V.; Neuhaus, W.; Scheffer, D.; Brandstetter, M.; Engelhard, K.; Wunder, C.; Forster, C.Y. Inhibition of proteasomal glucocorticoid receptor degradation restores dexamethasone-mediated stabilization of the blood-brain barrier after traumatic brain injury. Crit. Care Med. 2013, 41, 1305–1315. [Google Scholar] [CrossRef]
- Feng, X.; Yuan, W. Dexamethasone enhanced functional recovery after sciatic nerve crush injury in rats. Biomed. Res. Int. 2015, 2015, 627923. [Google Scholar] [CrossRef]
- Bastos, L.F.; Medeiros, D.C.; Vieira, R.P.; Watkins, L.R.; Coelho, M.M.; Moraes, M.F. Intraneural dexamethasone applied simultaneously to rat sciatic nerve constriction delays the development of hyperalgesia and allodynia. Neurosci. Lett. 2012, 510, 20–23. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Lim, G.; Zeng, Q.; Sung, B.; Ai, Y.; Guo, G.; Yang, L.; Mao, J. Expression of central glucocorticoid receptors after peripheral nerve injury contributes to neuropathic pain behaviors in rats. J. Neurosci. 2004, 24, 8595–8605. [Google Scholar] [CrossRef]
- Hayashi, K.; Ohta, S.; Kawakami, Y.; Toda, M. Activation of dendritic-like cells and neural stem/progenitor cells in injured spinal cord by GM-CSF. Neurosci. Res. 2009, 64, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Kim, J.M.; Kong, T.H.; Park, S.R.; Ha, Y.; Kim, M.H.; Park, H.; Yoon, S.H.; Park, H.C.; Park, J.O.; et al. GM-CSF inhibits glial scar formation and shows long-term protective effect after spinal cord injury. J. Neurol. Sci. 2009, 277, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.E.; Sugimoto, K.; Kubo, M.; Nagai, M.; Nomoto, M.; Takahashi, H.; Yano, H.; Tanaka, J. A cytokine mixture of GM-CSF and IL-3 that induces a neuroprotective phenotype of microglia leading to amelioration of (6-OHDA)-induced Parkinsonism of rats. Brain Behav. 2011, 1, 26–43. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.C.; Tanaka, J.; Peng, H.; Desaki, J.; Matsuda, S.; Maeda, N.; Fujita, H.; Sato, K.; Sakanaka, M. Interleukin 3 prevents delayed neuronal death in the hippocampal CA1 field. J. Exp. Med. 1998, 188, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schabitz, W.R.; Kruger, C.; Pitzer, C.; Weber, D.; Laage, R.; Gassler, N.; Aronowski, J.; Mier, W.; Kirsch, F.; Dittgen, T.; et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J. Cereb. Blood Flow Metab. 2008, 28, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, C.; Sriram, S.; Muthian, G.; Bright, J.J. Signaling through JAK2-STAT5 pathway is essential for IL-3-induced activation of microglia. Glia 2004, 45, 188–196. [Google Scholar] [CrossRef]
- Kamigaki, M.; Hide, I.; Yanase, Y.; Shiraki, H.; Harada, K.; Tanaka, Y.; Seki, T.; Shirafuji, T.; Tanaka, S.; Hide, M.; et al. The Toll-like receptor 4-activated neuroprotective microglia subpopulation survives via granulocyte macrophage colony-stimulating factor and JAK2/STAT5 signaling. Neurochem. Int. 2016, 93, 82–94. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.; Ikeda, K.; Ichikawa, Y.; Igarashi, O.; Iwamoto, K.; Kinoshita, M. Protective effect of interleukin-3 and erythropoietin on motor neuron death after neonatal axotomy. Neurol. Res. 2002, 24, 643–646. [Google Scholar] [CrossRef]
- Bombeiro, A.L.; Pereira, B.T.N.; de Oliveira, A.L.R. Granulocyte-macrophage colony-stimulating factor improves mouse peripheral nerve regeneration following sciatic nerve crush. Eur. J. Neurosci. 2018, 48, 2152–2164. [Google Scholar] [CrossRef]
- Cook, A.D.; Pobjoy, J.; Steidl, S.; Durr, M.; Braine, E.L.; Turner, A.L.; Lacey, D.C.; Hamilton, J.A. Granulocyte-macrophage colony-stimulating factor is a key mediator in experimental osteoarthritis pain and disease development. Arthritis. Res. Ther. 2012, 14, R199. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.D.; Pobjoy, J.; Sarros, S.; Steidl, S.; Durr, M.; Lacey, D.C.; Hamilton, J.A. Granulocyte-macrophage colony-stimulating factor is a key mediator in inflammatory and arthritic pain. Ann. Rheum. Dis. 2013, 72, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Nicol, L.S.C.; Thornton, P.; Hatcher, J.P.; Glover, C.P.; Webster, C.I.; Burrell, M.; Hammett, K.; Jones, C.A.; Sleeman, M.A.; Billinton, A.; et al. Central inhibition of granulocyte-macrophage colony-stimulating factor is analgesic in experimental neuropathic pain. Pain 2018, 159, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donatien, P.; Anand, U.; Yiangou, Y.; Sinisi, M.; Fox, M.; MacQuillan, A.; Quick, T.; Korchev, Y.E.; Anand, P. Granulocyte-macrophage colony-stimulating factor receptor expression in clinical pain disorder tissues and role in neuronal sensitization. Pain Rep. 2018, 3, e676. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Ransohoff, R.M. Inflammatory reaction after traumatic brain injury: Therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol. Sci. 2015, 36, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, S.; Nishihara, T.; Kawasaki, S.; Abe, N.; Kuwabara, J.; Choudhury, M.E.; Takahashi, H.; Yano, H.; Nagaro, T.; Watanabe, Y.; et al. The ameliorative effects of a hypnotic bromvalerylurea in sepsis. Biochem. Biophys. Res. Commun. 2015, 459, 319–326. [Google Scholar] [CrossRef]
- Kawasaki, S.; Abe, N.; Ohtake, F.; Islam, A.; Choudhury, M.E.; Utsunomiya, R.; Kikuchi, S.; Nishihara, T.; Kuwabara, J.; Yano, H.; et al. Effects of hypnotic bromovalerylurea on microglial BV2 cells. J. Pharmacol. Sci. 2017, 134, 116–123. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Katoh, M.; Wu, B.; Nguyen, H.B.; Thai, T.Q.; Yamasaki, R.; Lu, H.; Rietsch, A.M.; Zorlu, M.M.; Shinozaki, Y.; Saitoh, Y.; et al. Polymorphic regulation of mitochondrial fission and fusion modifies phenotypes of microglia in neuroinflammation. Sci. Rep. 2017, 7, 4942. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Xia, S.X.; Li, Q.Q.; Gao, Y.; Shen, X.; Ma, L.; Zhang, M.Y.; Wang, T.; Li, Y.S.; Wang, Z.F.; et al. Mitochondrial division inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing cell death and improving functional outcome in a mouse model of traumatic brain injury. Brain Res. 2016, 1630, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Gomi, M.; Takagi, Y.; Morizane, A.; Doi, D.; Nishimura, M.; Miyamoto, S.; Takahashi, J. Functional recovery of the murine brain ischemia model using human induced pluripotent stem cell-derived telencephalic progenitors. Brain Res. 2012, 1459, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, O.; Drury-Stewart, D.; Song, M.; Faulkner, B.; Chen, D.; Yu, S.P.; Wei, L. Vector-free and transgene-free human iPS cells differentiate into functional neurons and enhance functional recovery after ischemic stroke in mice. PLoS ONE 2013, 8, e64160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.B.; Yan, H.; Krishnaney-Davison, R.; Al Sawaf, A.; Zhang, S.C. Survival and differentiation of transplanted neural stem cells derived from human induced pluripotent stem cells in a rat stroke model. J. Stroke Cerebrovasc. Dis. 2013, 22, 304–308. [Google Scholar] [CrossRef] [Green Version]
- Oki, K.; Tatarishvili, J.; Wood, J.; Koch, P.; Wattananit, S.; Mine, Y.; Monni, E.; Tornero, D.; Ahlenius, H.; Ladewig, J.; et al. Human-induced pluripotent stem cells form functional neurons and improve recovery after grafting in stroke-damaged brain. Stem. Cells 2012, 30, 1120–1133. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, N.; Nishihara, T.; Yorozuya, T.; Tanaka, J. Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems. Cells 2020, 9, 2132. https://doi.org/10.3390/cells9092132
Abe N, Nishihara T, Yorozuya T, Tanaka J. Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems. Cells. 2020; 9(9):2132. https://doi.org/10.3390/cells9092132
Chicago/Turabian StyleAbe, Naoki, Tasuku Nishihara, Toshihiro Yorozuya, and Junya Tanaka. 2020. "Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems" Cells 9, no. 9: 2132. https://doi.org/10.3390/cells9092132