Phosphorylation of PLK3 Is Controlled by Protein Phosphatase 6


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cells
2.3. Plasmids
2.4. Real-Time Quantitative Reverse Transcriptase PCR (qRT-PCR)
2.5. Immunofluorescence
2.6. Immunoprecipitation
2.7. Cell Fractionation
2.8. Statistical Analysis
3. Results
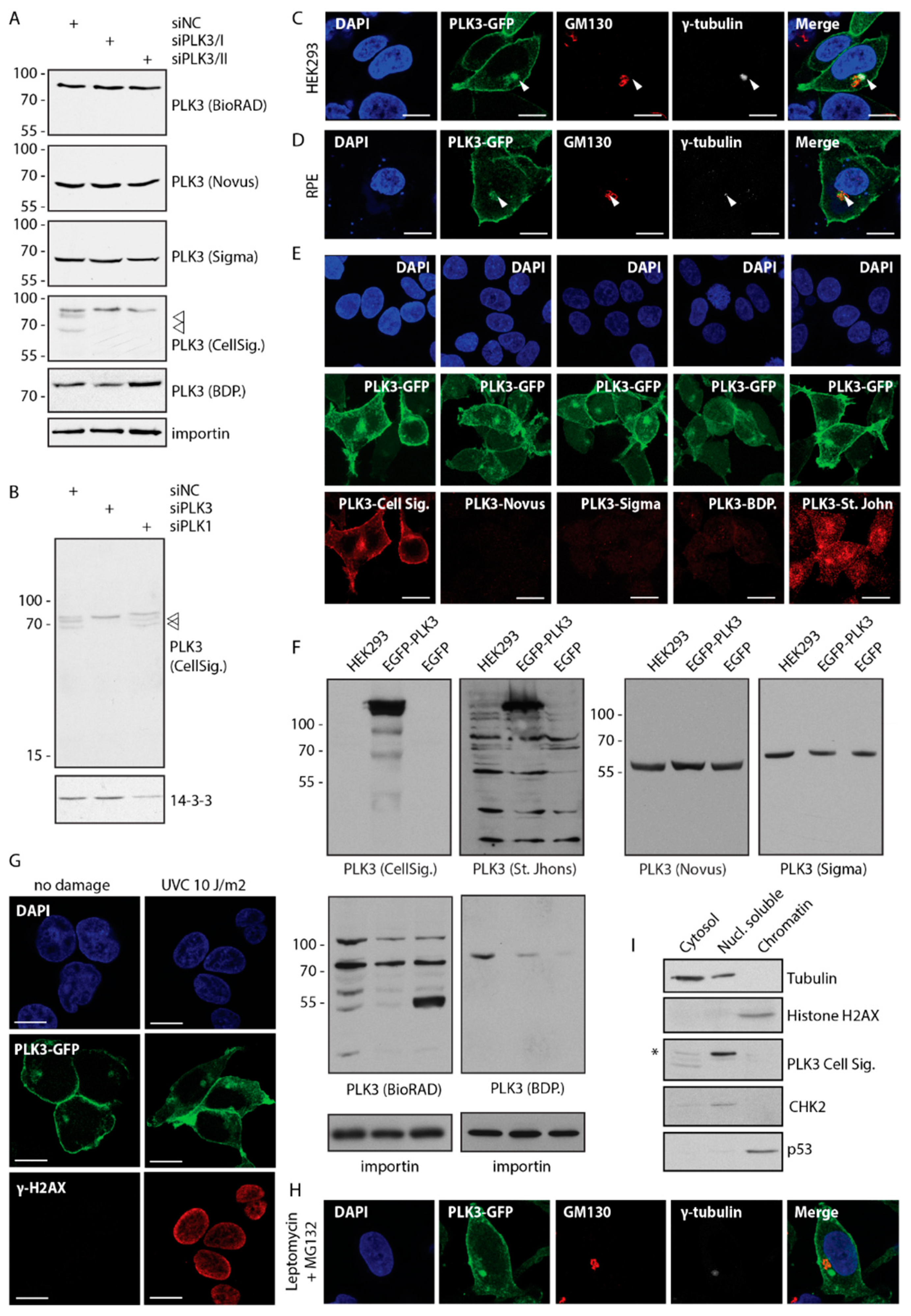
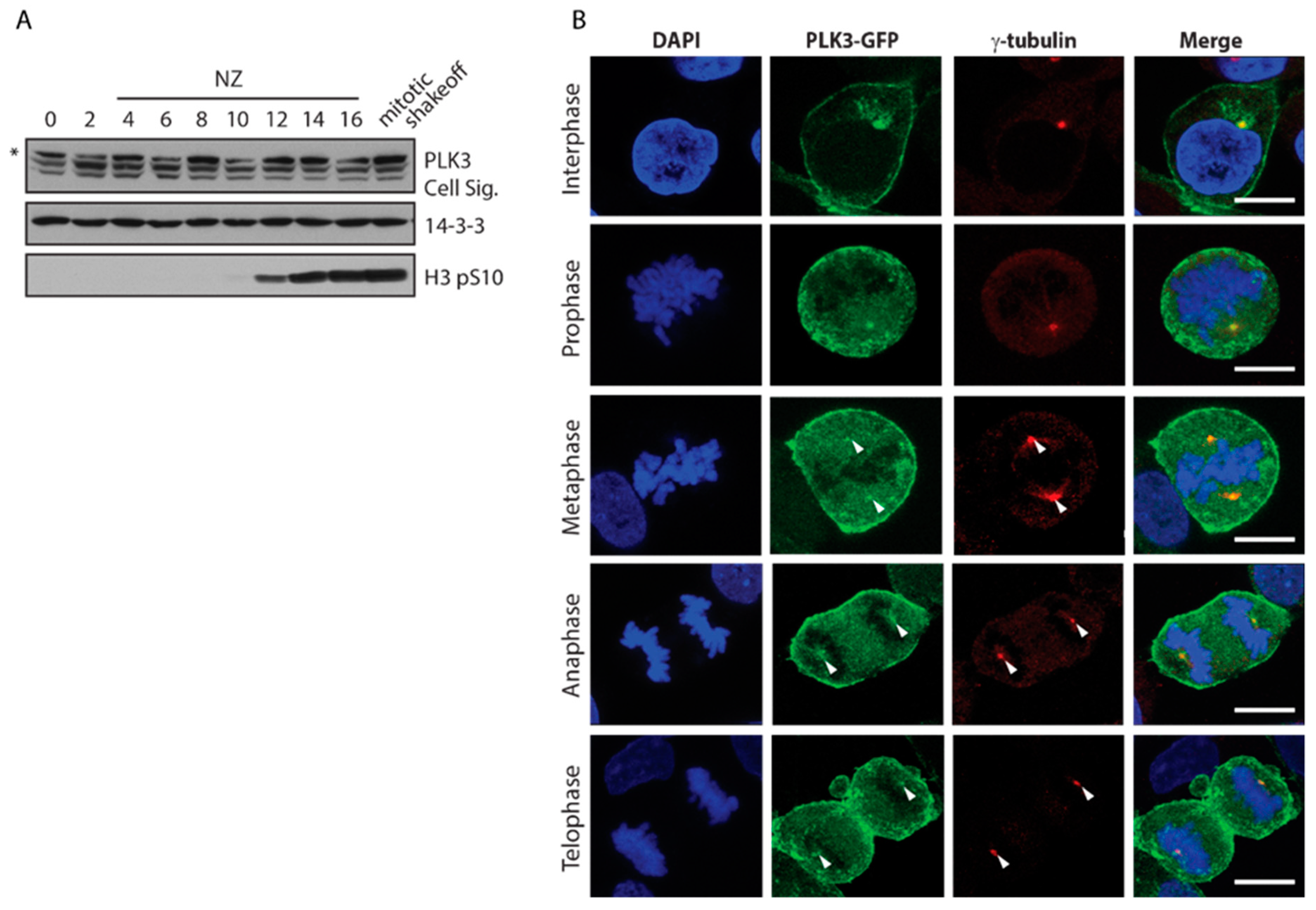
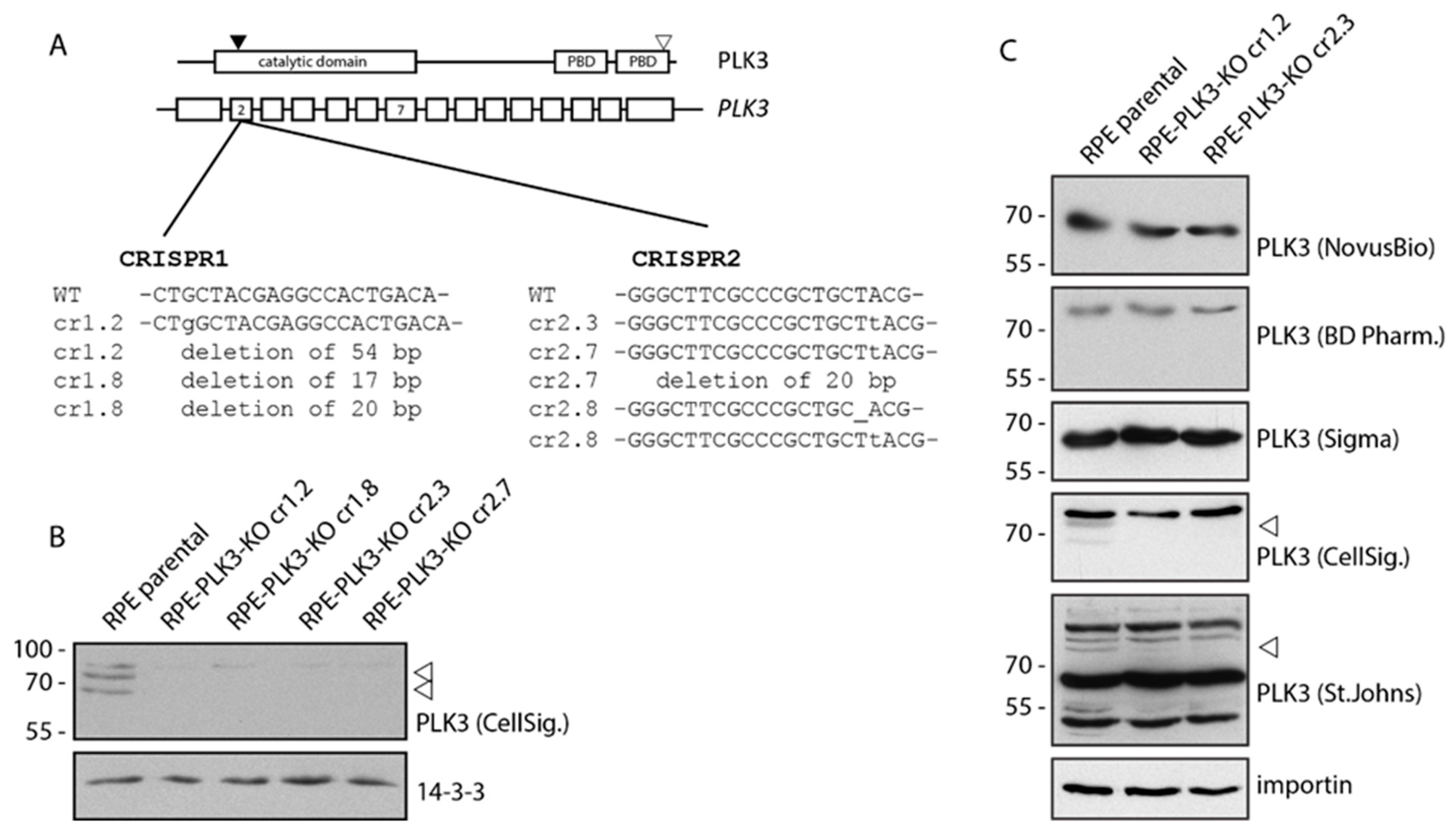
3.1. PLK3 Localizes to Plasma Membrane, Golgi and Centrosome
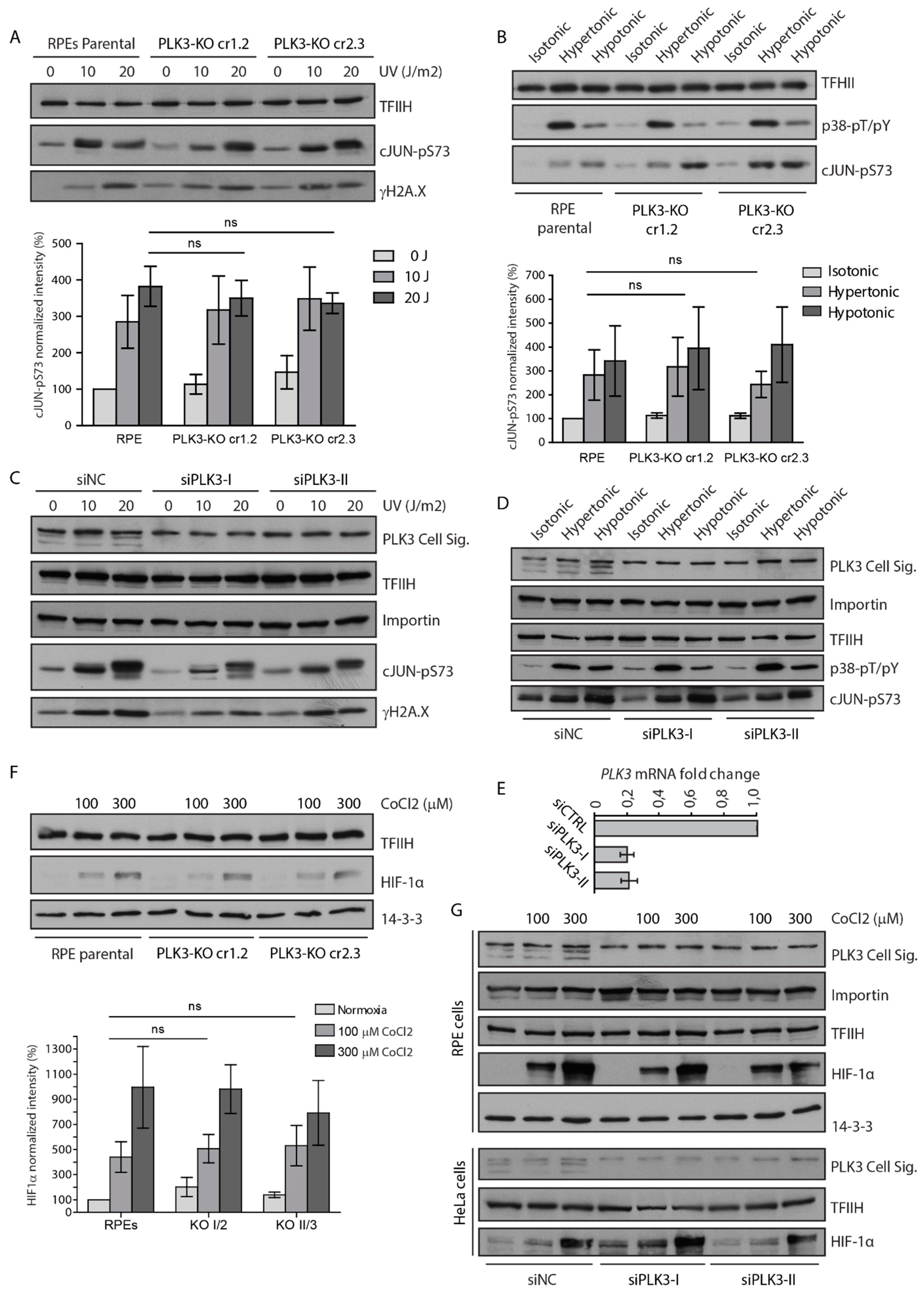
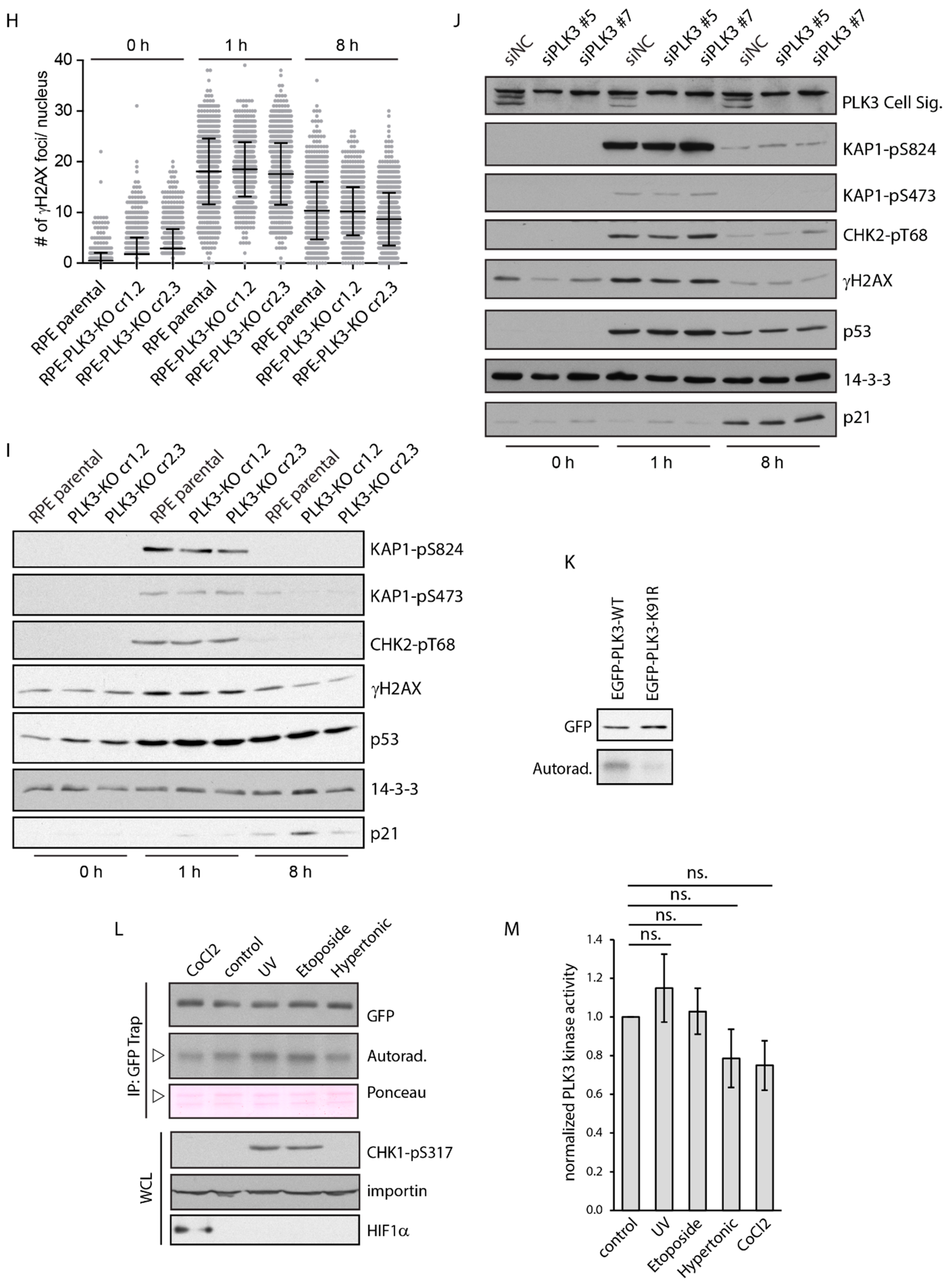
3.2. PLK3 is Disposable for Cell Response to Genotoxic Stress and Osmotic Stress
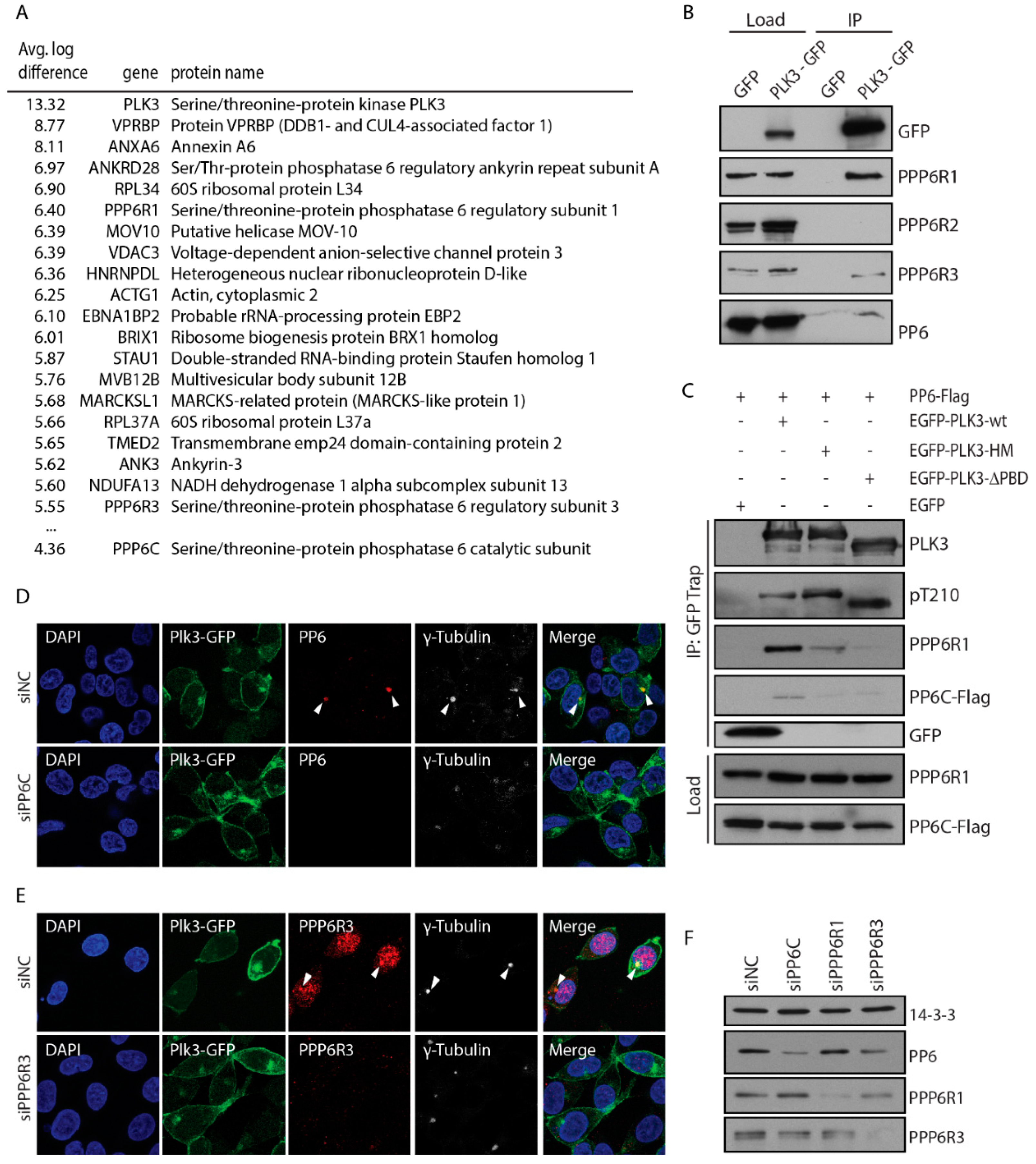
3.3. PLK3 Interacts with PP6 Phosphatase Through the PBD Domain
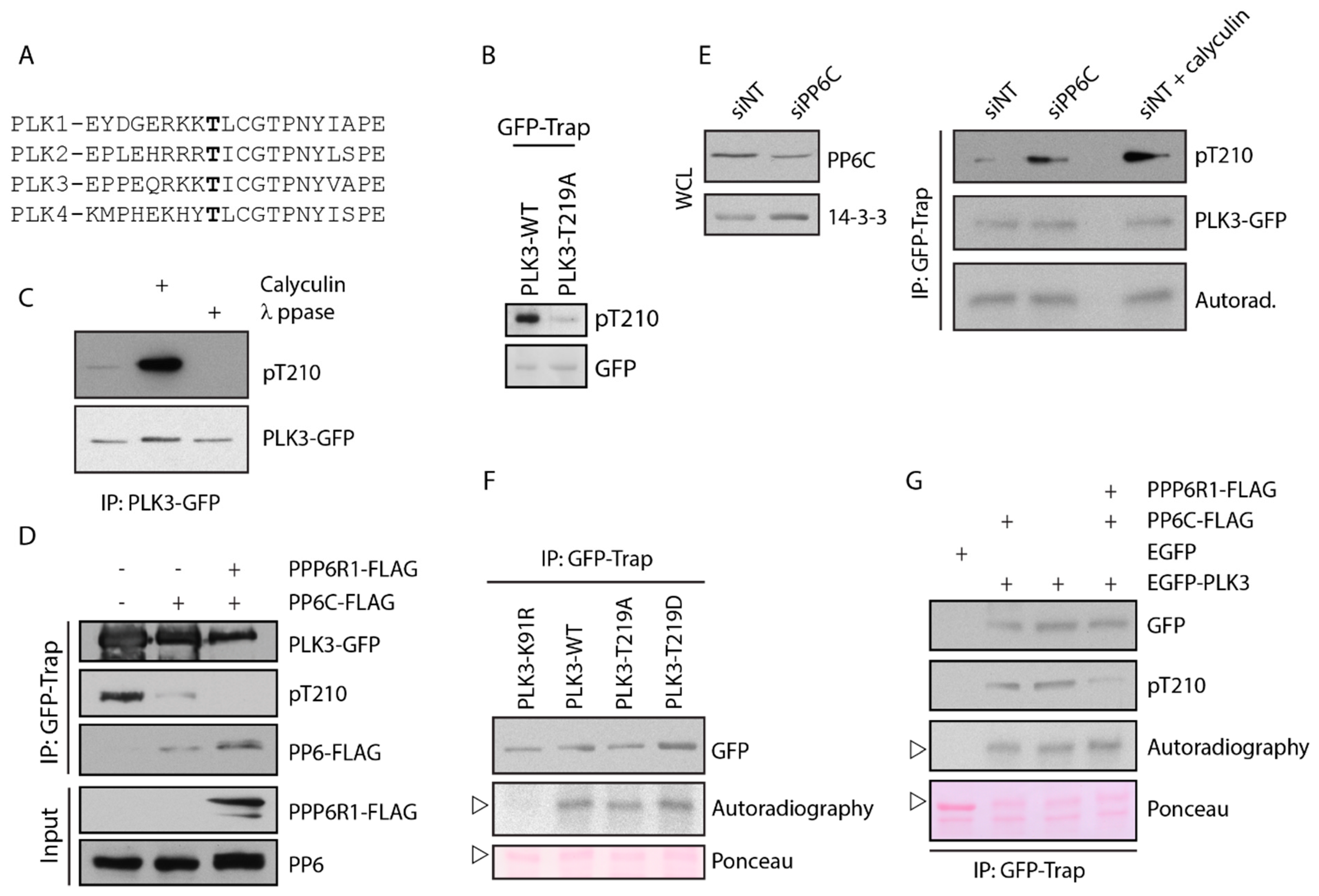
3.4. PLK3 Is Phosphorylated in the T-Loop but This Does Not Affect Its Kinase Activity
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef]
- Elia, A.E.H.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.C.; Smerdon, S.J.; Yaffe, M.B. The Molecular Basis for Phosphodependent Substrate Targeting and Regulation of Plks by the Polo-Box Domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Voest, E.E.; Medema, R.H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 2010, 10, 825–841. [Google Scholar] [CrossRef]
- Petronczki, M.; Lénárt, P.; Peters, J.-M. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev. Cell 2008, 14, 646–659. [Google Scholar] [CrossRef] [Green Version]
- Habedanck, R.; Stierhof, Y.-D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef]
- De Cárcer, G.; Escobar, B.; Higuero, A.M.; García, L.; Ansón, A.; Pérez, G.; Mollejo, M.; Manning, G.; Meléndez, B.; Abad-Rodríguez, J.; et al. Plk5, a Polo Box Domain-Only Protein with Specific Roles in Neuron Differentiation and Glioblastoma Suppression. Mol. Cell. Biol. 2011, 31, 1225–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtrich, U.; Wolf, G.; Yuan, J.; Bereiter-Hahn, J.; Karn, T.; Weiler, M.; Kauselmann, G.; Rehli, M.; Andreesen, R.; Kaufmann, M.; et al. Adhesion induced expression of the serine/threonine kinase Fnk in human macrophages. Oncogene 2000, 19, 4832–4839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Liu, M.-A.; Yuan, Y.-L.O.; Erikson, R.L. The serum-inducible protein kinase Snk is a G1 phase polo-like kinase that is inhibited by the calcium- and integrin-binding protein CIB. Mol. Cancer Res. 2003, 1, 376–384. [Google Scholar] [PubMed]
- Zimmerman, W.C.; Erikson, R.L. Polo-like kinase 3 is required for entry into S phase. Proc. Natl. Acad. Sci. USA 2007, 104, 1847–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahassi, E.L.M.; Hennigan, R.F.; Myer, D.L.; Stambrook, P.J. Cdc25C phosphorylation on serine 191 by Plk3 promotes its nuclear translocation. Oncogene 2004, 23, 2658–2663. [Google Scholar] [CrossRef] [Green Version]
- Myer, D.L.; Robbins, S.B.; Yin, M.; Boivin, G.P.; Liu, Y.; Greis, K.D.; Bahassi, E.M.; Stambrook, P.J. Absence of polo-like kinase 3 in mice stabilizes Cdc25A after DNA damage but is not sufficient to produce tumors. Mutat. Res. 2011, 714, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bahassi, E.M.; Conn, C.W.; Myer, D.L.; Hennigan, R.F.; McGowan, C.H.; Sanchez, Y.; Stambrook, P.J. Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene 2002, 21, 6633–6640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahassi, E.M.; Myer, D.L.; McKenney, R.J.; Hennigan, R.F.; Stambrook, P.J. Priming phosphorylation of Chk2 by polo-like kinase 3 (Plk3) mediates its full activation by ATM and a downstream checkpoint in response to DNA damage. Mutat. Res. 2006, 596, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Wu, H.; Wang, Q.; Cogswell, J.P.; Husain, I.; Conn, C.; Stambrook, P.; Jhanwar-Uniyal, M.; Dai, W. Plk3 Functionally Links DNA Damage to Cell Cycle Arrest and Apoptosis at Least in Part via the p53 Pathway. J. Biol. Chem. 2001, 276, 43305–43312. [Google Scholar] [CrossRef] [Green Version]
- Barton, O.; Naumann, S.C.; Diemer-Biehs, R.; Künzel, J.; Steinlage, M.; Conrad, S.; Makharashvili, N.; Wang, J.; Feng, L.; Lopez, B.S.; et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell Biol. 2014, 206, 877–894. [Google Scholar] [CrossRef]
- Wang, L.; Gao, J.; Dai, W.; Lu, L. Activation of Polo-like Kinase 3 by Hypoxic Stresses. J. Biol. Chem. 2008, 283, 25928–25935. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Yao, Y.; Lu, L.; Costa, M.; Dai, W. Plk3 Functions as an Essential Component of the Hypoxia Regulatory Pathway by Direct Phosphorylation of HIF-1α. J. Biol. Chem. 2010, 285, 38944–38950. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bai, J.; Shen, R.; Brown, S.A.N.; Komissarova, E.; Huang, Y.; Jiang, N.; Alberts, G.F.; Costa, M.; Lu, L.; et al. Polo-like Kinase 3 Functions as a Tumor Suppressor and Is a Negative Regulator of Hypoxia-Inducible Factor-1α under Hypoxic Conditions. Cancer Res. 2008, 68, 4077–4085. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Dai, W.; Lu, L. Osmotic Stress-induced Phosphorylation of H2AX by Polo-like Kinase 3 Affects Cell Cycle Progression in Human Corneal Epithelial Cells. J. Biol. Chem. 2014, 289, 29827–29835. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Payton, R.; Dai, W.; Lu, L. Hyperosmotic Stress-induced ATF-2 Activation through Polo-like Kinase 3 in Human Corneal Epithelial Cells. J. Biol. Chem. 2011, 286, 1951–1958. [Google Scholar] [CrossRef] [Green Version]
- Colanzi, A.; Sutterlin, C.; Malhotra, V. RAF1-activated MEK1 is found on the Golgi apparatus in late prophase and is required for Golgi complex fragmentation in mitosis. J. Cell Biol. 2003, 161, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Wang, Q.; Ruan, Q.; Liu, T.; Jhanwar-Uniyal, M.; Guan, K.; Dai, W. MEK1-induced Golgi dynamics during cell cycle progression is partly mediated by Polo-like kinase-3. Oncogene 2004, 23, 3822–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Q.; Wang, Q.; Xie, S.; Fang, Y.; Darzynkiewicz, Z.; Guan, K.; Jhanwar-Uniyal, M.; Dai, W. Polo-like kinase 3 is Golgi localized and involved in regulating Golgi fragmentation during the cell cycle. Exp. Cell Res. 2004, 294, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, B.; Ohama, T.; Daugherty, A.E.; Brautigan, D.L. Protein Phosphatase 6 Regulatory Subunits Composed of Ankyrin Repeat Domains. Biochemistry 2008, 47, 1442–1451. [Google Scholar] [CrossRef]
- Ohama, T. The multiple functions of protein phosphatase 6. Biochimi. Biophys. Acta (BBA) Mol. Cell Res. 2019, 1866, 74–82. [Google Scholar] [CrossRef]
- Rusin, S.F.; Schlosser, K.A.; Adamo, M.E.; Kettenbach, A.N. Quantitative phosphoproteomics reveals new roles for the protein phosphatase PP6 in mitotic cells. Sci. Signal. 2015, 8, rs12. [Google Scholar] [CrossRef] [Green Version]
- Zeng, K.; Bastos, R.N.; Barr, F.A.; Gruneberg, U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J. Cell Biol. 2010, 191, 1315–1332. [Google Scholar] [CrossRef] [Green Version]
- Mi, J.; Dziegielewski, J.; Bolesta, E.; Brautigan, D.L.; Larner, J.M. Activation of DNA-PK by Ionizing Radiation Is Mediated by Protein Phosphatase 6. PLoS ONE 2009, 4, e4395. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Wang, Y.; Sheng, K.; Fei, X.; Guo, Q.; Larner, J.; Kong, X.; Qiu, Y.; Mi, J. Serine/threonine protein phosphatase 6 modulates the radiation sensitivity of glioblastoma. Cell Death Dis. 2011, 2, e241. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Momoi, Y.; Tanuma, N.; Kishimoto, A.; Ogoh, H.; Kato, H.; Suzuki, M.; Sakamoto, Y.; Inoue, Y.; Nomura, M.; et al. Abrogation of protein phosphatase 6 promotes skin carcinogenesis induced by DMBA. Oncogene 2015, 34, 4647–4655. [Google Scholar] [CrossRef] [Green Version]
- Hammond, D.; Zeng, K.; Espert, A.; Bastos, R.N.; Baron, R.D.; Gruneberg, U.; Barr, F.A. Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J. Cell Sci. 2013, 126, 3429–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdova, K.; Storchova, R.; Palek, M.; Macurek, L. WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors. Cells 2019, 8, 1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Stern, D.F. NFBD1/KIAA0170 Is a Chromatin-associated Protein Involved in DNA Damage Signaling Pathways. J. Biol. Chem. 2003, 278, 8795–8803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macůrek, L.; Lindqvist, A.; Voets, O.; Kool, J.; Vos, H.R.; Medema, R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates γH2AX to promote checkpoint inhibition. Oncogene 2010, 29, 2281–2291. [Google Scholar] [CrossRef] [Green Version]
- Alberts, G.; Winkles, J. Murine FGF-inducible kinase is rapidly degraded via the nuclear ubiquitin-proteosome system when overexpressed in NIH 3T3 cells. Cell Cycle 2004, 3, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Helmke, C.; Raab, M.; Rödel, F.; Matthess, Y.; Oellerich, T.; Mandal, R.; Sanhaji, M.; Urlaub, H.; Rödel, C.; Becker, S.; et al. Ligand stimulation of CD95 induces activation of Plk3 followed by phosphorylation of caspase-8. Cell Res. 2016, 26, 914–934. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Wang, X.; Jhanwar-Uniyal, M.; Darzynkiewicz, Z.; Dai, W. Polo Box Domain of Plk3 Functions as a Centrosome Localization Signal, Overexpression of Which Causes Mitotic Arrest, Cytokinesis Defects, and Apoptosis. J. Biolo. Chem. 2006, 281, 10577–10582. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Dai, W.; Lu, L. Stress-induced c-Jun Activation Mediated by Polo-like Kinase 3 in Corneal Epithelial Cells. J. Biol. Chem. 2007, 282, 32121–32127. [Google Scholar] [CrossRef] [Green Version]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef]
- Kleiblova, P.; Stolarova, L.; Krizova, K.; Lhota, F.; Hojny, J.; Zemankova, P.; Havranek, O.; Vocka, M.; Cerna, M.; Lhotova, K.; et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int. J. Cancer 2019, 145, 1782–1797. [Google Scholar] [CrossRef]
- Van Vugt, M.A.T.M.; Smits, V.A.J.; Klompmaker, R.; Medema, R.H. Inhibition of Polo-like Kinase-1 by DNA Damage Occurs in an ATM- or ATR-dependent Fashion. J. Biol. Chem. 2001, 276, 41656–41660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenbach, A.N.; Schlosser, K.A.; Lyons, S.P.; Nasa, I.; Gui, J.; Adamo, M.E.; Gerber, S.A. Global assessment of its network dynamics reveals that the kinase Plk1 inhibits the phosphatase PP6 to promote Aurora A activity. Sci. Signal. 2018, 11, eaaq1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macurek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, W.; Macůrek, L.; Freire, R.; Lindqvist, A.; Medema, R.H. Bora and Aurora-A continue to activate Plk1 in mitosis. J. Cell Sci. 2014, 127, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.H.Y.; Santamaria, A.; Silljé, H.W.; Nigg, E.A. Plk1 regulates mitotic Aurora A function through βTrCP-dependent degradation of hBora. Chromosom. Biol. Nucl. 2008, 117, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Swingle, M.; Ni, L.; Honkanen, R.E. Small-molecule inhibitors of ser/thr protein phosphatases: Specificity, use and common forms of abuse. Methods Mol. Biol. 2007, 365, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Umeda, T.; Niwa, K.; Naguro, I.; Ichijo, H. A PP6-ASK3 Module Coordinates the Bidirectional Cell Volume Regulation under Osmotic Stress. Cell Rep. 2018, 22, 2809–2817. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, D.; Zhang, J.; Menon, S.; Lord, C.; Chen, S.; Helm, J.; Thorsen, K.; Corbett, K.; Hay, J.; Ferro-Novick, S. Sit4p/PP6 Regulates ER-to-Golgi Traffic by Controlling the Dephosphorylation of COPII Coat Subunits. Mol. Biol. Cell 2013, 24, 2727–2738. [Google Scholar] [CrossRef]
- Ohama, T.; Wang, L.; Griner, E.; Brautigan, D. Protein Ser/Thr phosphatase-6 is required for maintenance of E-cadherin at adherens junctions. BMC Cell Biol. 2013, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Bai, S.; Du, T.; Lai, Y.; Chen, X.; Peng, S.; Ma, X.; Wu, W.; Guo, Z.; Huang, H. Polo-like kinase 3 is associated with poor prognosis and regulates proliferation and metastasis in prostate cancer. Cancer Manag. Res. 2019, 11, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Babagana, M.; Kichina, J.V.; Slabodkin, H.; Johnson, S.; Maslov, A.; Brown, L.; Attwood, K.; Nikiforov, M.A.; Kandel, E.S. The role of polo-like kinase 3 in the response of BRAF-mutant cells to targeted anticancer therapies. Mol. Carcinog. 2020, 59, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Sun, H.; Zhao, J.; Xu, Z.; Liu, Y.; Feng, H.; Peng, Z. Polo-like kinase 3 inhibits glucose metabolism in colorectal cancer by targeting HSP90/STAT3/HK2 signaling. J. Exp. Clin. Cancer Res. CR 2019, 38, 426. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aquino Perez, C.; Palek, M.; Stolarova, L.; von Morgen, P.; Macurek, L. Phosphorylation of PLK3 Is Controlled by Protein Phosphatase 6. Cells 2020, 9, 1506. https://doi.org/10.3390/cells9061506
Aquino Perez C, Palek M, Stolarova L, von Morgen P, Macurek L. Phosphorylation of PLK3 Is Controlled by Protein Phosphatase 6. Cells. 2020; 9(6):1506. https://doi.org/10.3390/cells9061506
Chicago/Turabian StyleAquino Perez, Cecilia, Matous Palek, Lenka Stolarova, Patrick von Morgen, and Libor Macurek. 2020. "Phosphorylation of PLK3 Is Controlled by Protein Phosphatase 6" Cells 9, no. 6: 1506. https://doi.org/10.3390/cells9061506