Ovarian Cancer, Cancer Stem Cells and Current Treatment Strategies: A Potential Role of Magmas in the Current Treatment Methods

Abstract
:1. Introduction
Different Origins of OC
2. Heterogeneity in OC
2.1. Genetic Changes
2.2. Epigenetic Changes
3. Ovarian Cancer Stem Cells
4. Metastases and Tumour Microenvironment in OC
5. Magmas (Mitochondrial Associated Granulocyte Macrophage Colony Stimulating Factor Signaling Molecule)
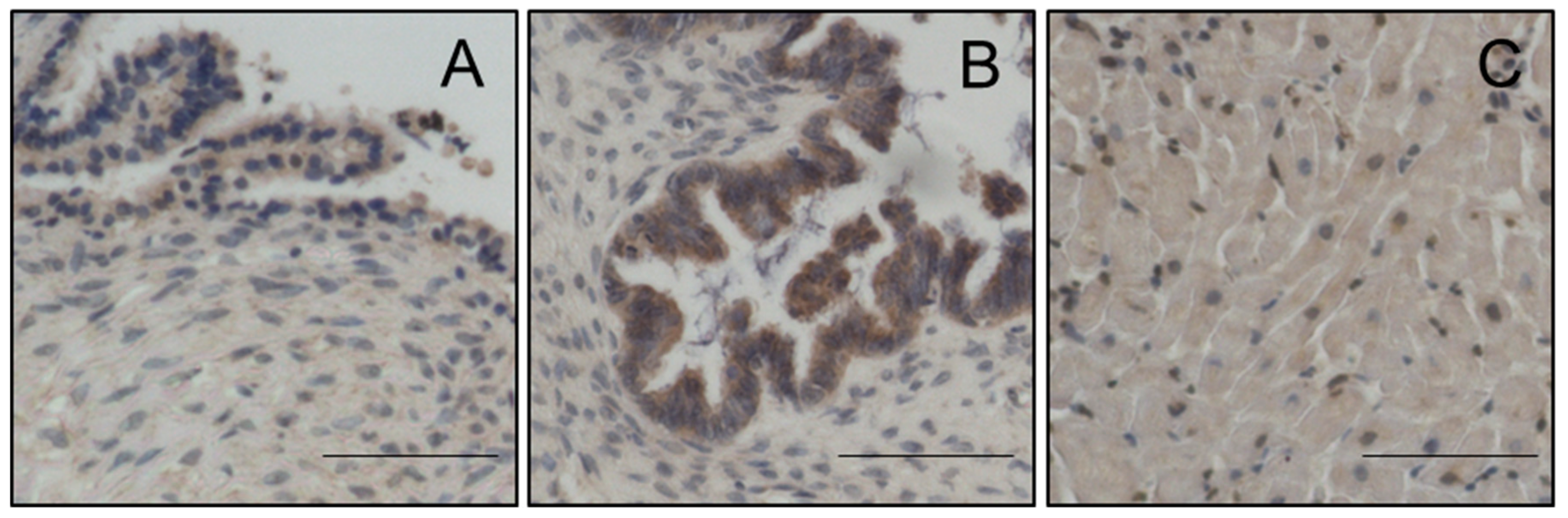
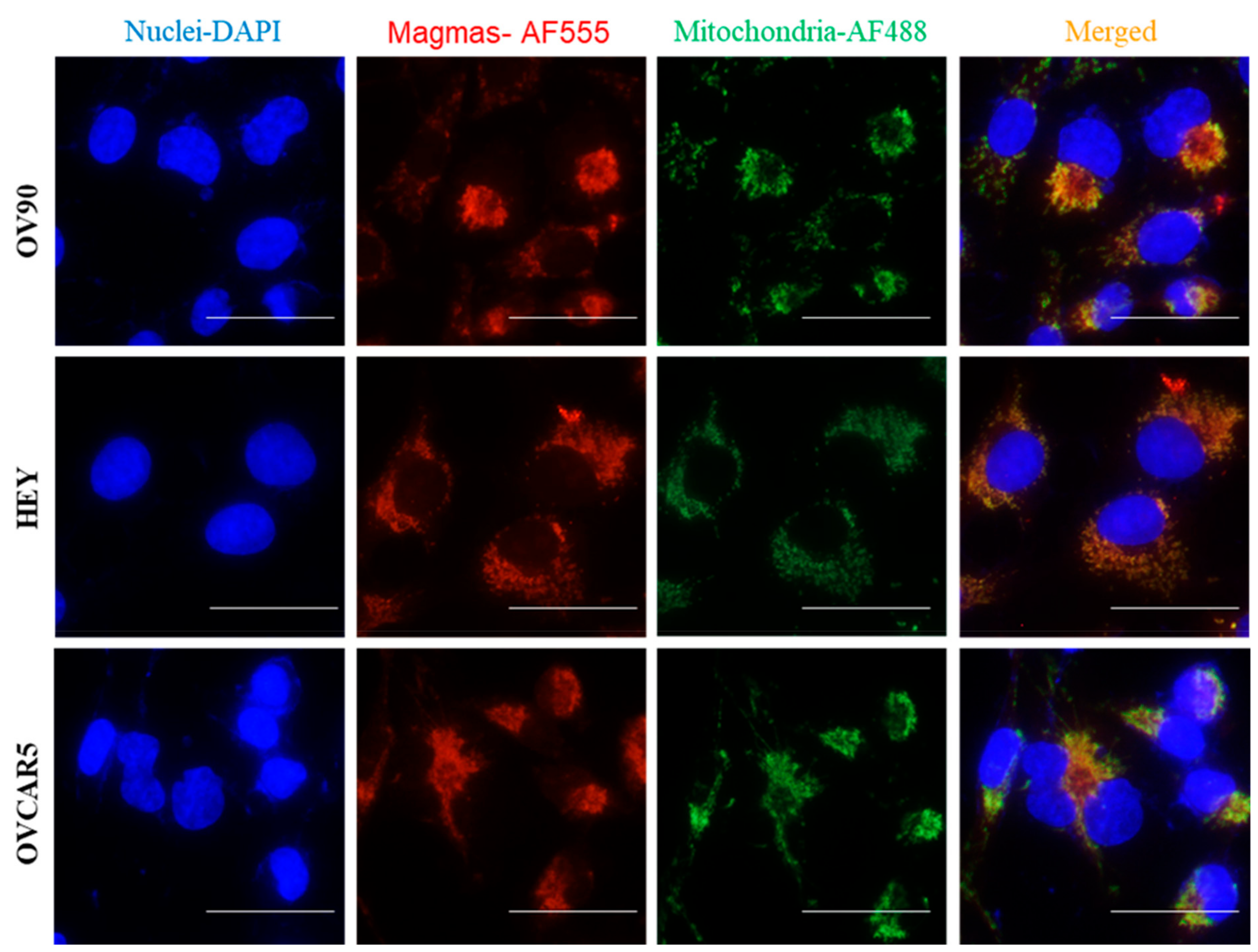
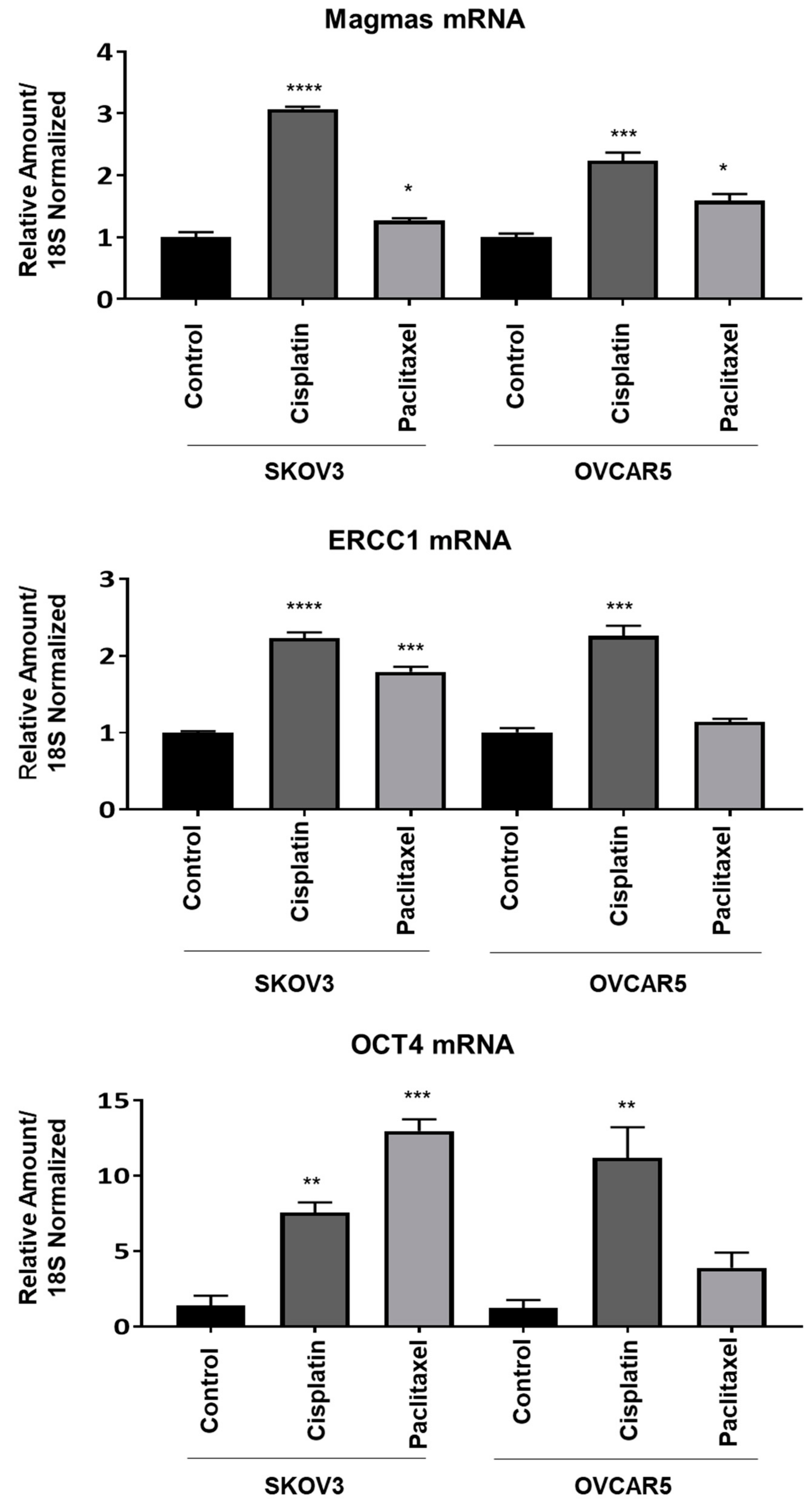
Magmas Expression in Ovarian Tumours, OC Cell Lines and in Mice Xenografts Before and after Chemotherapy Treatment, and the Effect of Magmas Inhibition on a Chemotherapy-Treated Ovarian Cancer Cell Line: A Proof of Concept Data
6. Ovarian Cancer Treatment Strategies
6.1. Current Treatment
6.2. Pathway to Precision Medicine
6.3. Targeting Folate Receptor (FR)
6.4. Immunotherapy
6.5. Metronomic Chemotherapy
6.6. Nanoparticle Drug Delivery
6.7. Patient- Derived Tumour Organoid Modelling
6.8. Ovarian Cancer Patient-Derived Organoid Model
6.9. CSC-Based Therapy
Signaling Pathways as Potential Targets for Ovarian CSCs
6.10. Metabolic Targeting of Ovarian CSCs
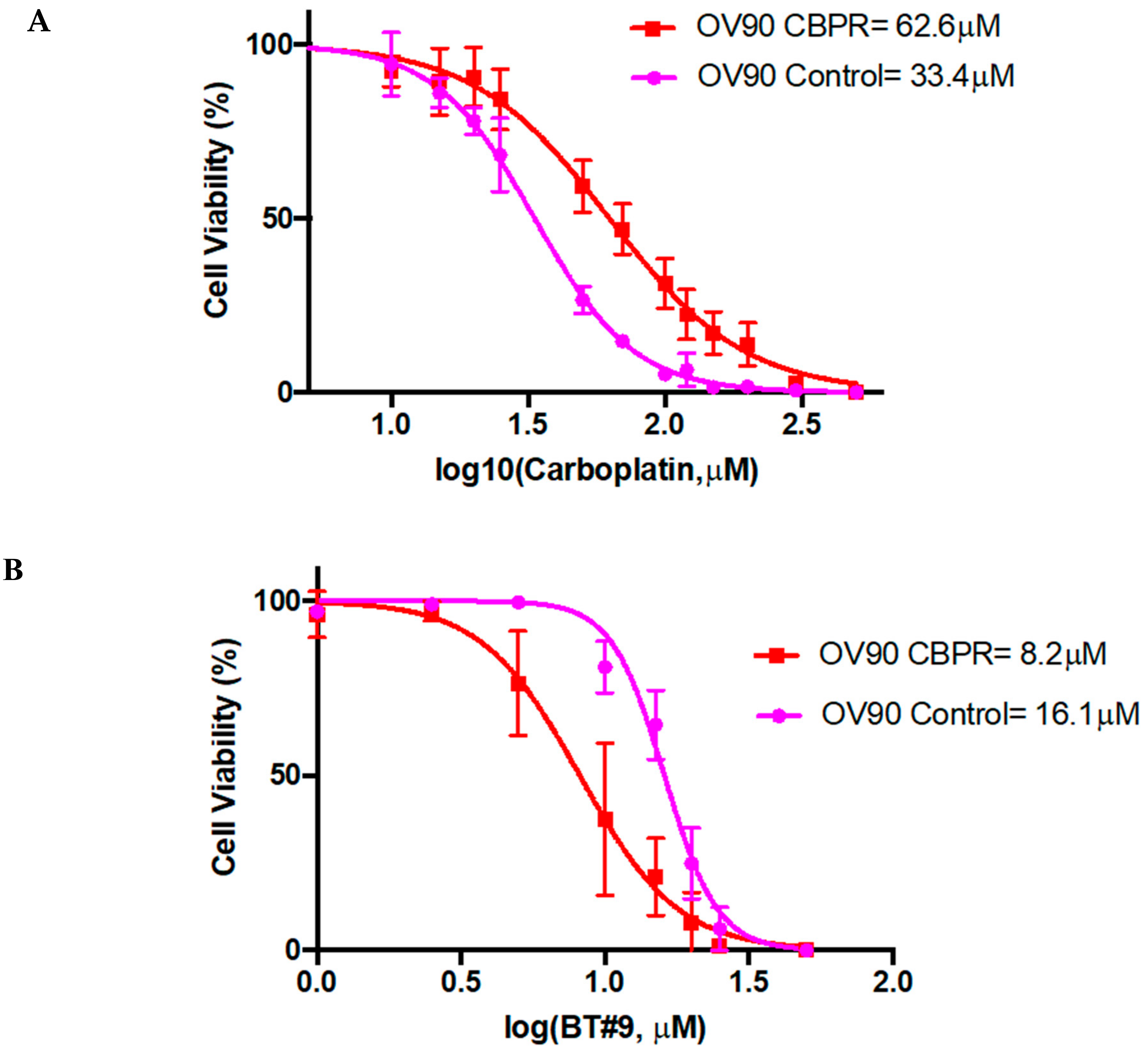
6.11. Role of Magmas Inhibitor in Ovarian Cancer Treatment
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Coburn, S.B.; Bray, F.; Sherman, M.E.; Trabert, B. International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Int. J. Cancer 2017, 140, 2451–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.C.; Huang, C.F.; Lai, M.S.; Shen, L.J.; Wu, F.L.; Cheng, W.F. Prognostic factors in epithelial ovarian cancer: A population-based study. PLoS ONE 2018, 13, e0194993. [Google Scholar] [CrossRef] [Green Version]
- Kipps, E.; Tan, D.S.; Kaye, S.B. Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Herzog, T.J. Recurrent ovarian cancer: How important is it to treat to disease progression? Clin. Cancer Res. 2004, 10, 7439–7449. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.M.; Cardenas, C.; Tedja, R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 2019, 11, 1083. [Google Scholar] [CrossRef] [Green Version]
- Nwani, N.G.; Sima, L.E.; Nieves-Neira, W.; Matei, D. Targeting the Microenvironment in High Grade Serous Ovarian Cancer. Cancers 2018, 10, 266. [Google Scholar] [CrossRef] [Green Version]
- Piek, J.M.; van Diest, P.J.; Zweemer, R.P.; Jansen, J.W.; Poort-Keesom, R.J.; Menko, F.H.; Gille, J.J.; Jongsma, A.P.; Pals, G.; Kenemans, P.; et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol. 2001, 195, 451–456. [Google Scholar] [CrossRef]
- Piek, J.M.; Torrenga, B.; Hermsen, B.; Verheijen, R.H.; Zweemer, R.P.; Gille, J.J.; Kenemans, P.; van Diest, P.J.; Menko, F.H. Histopathological characteristics of BRCA1- and BRCA2-associated intraperitoneal cancer: A clinic-based study. Fam. Cancer 2003, 2, 73–78. [Google Scholar] [CrossRef]
- Karst, A.M.; Drapkin, R. Ovarian cancer pathogenesis: A model in evolution. J. Oncol. 2010, 2010, 932371. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Miron, A.; Drapkin, R.; Nucci, M.R.; Medeiros, F.; Saleemuddin, A.; Garber, J.; Birch, C.; Mou, H.; Gordon, R.W.; et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 2007, 211, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Levanon, K.; Crum, C.; Drapkin, R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazaeri, A.A.; Bryant, J.L.; Park, H.; Li, H.; Dahiya, N.; Stoler, M.H.; Ferriss, J.S.; Dutta, A. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia 2011, 13, 899–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Ning, G.; Howitt, B.E.; Mehra, K.; Wu, L.; Wang, X.; Hong, Y.; Kern, F.; Wei, T.S.; Zhang, T.; et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. J. Pathol. 2016, 238, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Marquez, R.T.; Baggerly, K.A.; Patterson, A.P.; Liu, J.; Broaddus, R.; Frumovitz, M.; Atkinson, E.N.; Smith, D.I.; Hartmann, L.; Fishman, D.; et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin. Cancer Res. 2005, 11, 6116–6126. [Google Scholar] [CrossRef] [Green Version]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Jin, V.; Bergsten, T.M.; Austin, J.R.; Lantvit, D.D.; Russo, A.; Burdette, J.E. Loss of PTEN in Fallopian Tube Epithelium Results in Multicellular Tumor Spheroid Formation and Metastasis to the Ovary. Cancers 2019, 11, 884. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Czarnecki, A.A.; Dean, M.; Modi, D.A.; Lantvit, D.D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.J.; Burdette, J.E. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef]
- Medeiros, F.; Muto, M.G.; Lee, Y.; Elvin, J.A.; Callahan, M.J.; Feltmate, C.; Garber, J.E.; Cramer, D.W.; Crum, C.P. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am. J. Surg. Pathol. 2006, 30, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Bahar-Shany, K.; Brand, H.; Sapoznik, S.; Jacob-Hirsch, J.; Yung, Y.; Korach, J.; Perri, T.; Cohen, Y.; Hourvitz, A.; Levanon, K. Exposure of fallopian tube epithelium to follicular fluid mimics carcinogenic changes in precursor lesions of serous papillary carcinoma. Gynecol. Oncol. 2014, 132, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Wong, A.S.; Choi, K.C.; Kang, S.K.; Leung, P.C. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.F.; Hu, W.; Sood, A.K. Microenvironment and pathogenesis of epithelial ovarian cancer. Horm Cancer 2010, 1, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, W.J.; Martinchick, J.F. Oxidative damage to DNA of ovarian surface epithelial cells affected by ovulation: Carcinogenic implication and chemoprevention. Exp. Biol. Med. 2004, 229, 546–552. [Google Scholar] [CrossRef]
- Folkins, A.K.; Saleemuddin, A.; Garrett, L.A.; Garber, J.E.; Muto, M.G.; Tworoger, S.S.; Crum, C.P. Epidemiologic correlates of ovarian cortical inclusion cysts (CICs) support a dual precursor pathway to pelvic epithelial cancer. Gynecol. Oncol. 2009, 115, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.E. Pathology of ovarian cancer precursors. J. Cell. Biochem. Suppl. 1995, 23, 208–218. [Google Scholar] [CrossRef]
- Banet, N.; Kurman, R.J. Two types of ovarian cortical inclusion cysts: Proposed origin and possible role in ovarian serous carcinogenesis. Int. J. Gynecol. Pathol. 2015, 34, 3–8. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Shih, I.M. Ovarian Cancer Is an Imported Disease: Fact or Fiction? Curr. Obstet. Gynecol. Rep. 2012, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.H.; Bast, R.C., Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef] [Green Version]
- Kurman, R.J.; Shih Ie, M. Pathogenesis of ovarian cancer: Lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalwani, N.; Prasad, S.R.; Vikram, R.; Shanbhogue, A.K.; Huettner, P.C.; Fasih, N. Histologic, molecular, and cytogenetic features of ovarian cancers: Implications for diagnosis and treatment. Radiographics 2011, 31, 625–646. [Google Scholar] [CrossRef] [PubMed]
- Koshiyama, M.; Matsumura, N.; Konishi, I. Recent concepts of ovarian carcinogenesis: Type I and type II. Biomed. Res. Int. 2014, 2014, 934261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Pan, S.; Hernandez, K.M.; Loth, R.M.; Andrade, J.; Volchenboum, S.L.; Faber, P.; Montag, A.; Lastra, R.; Peter, M.E.; et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discov. 2016, 6, 1342–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayani, J.; Paderova, J.; Murphy, J.; Rosen, B.; Zielenska, M.; Squire, J.A. Distinct patterns of structural and numerical chromosomal instability characterize sporadic ovarian cancer. Neoplasia 2008, 10, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Natanzon, Y.; Goode, E.L.; Cunningham, J.M. Epigenetics in ovarian cancer. Semin. Cancer Biol. 2018, 51, 160–169. [Google Scholar] [CrossRef]
- Hyler, A.R.; Baudoin, N.C.; Brown, M.S.; Stremler, M.A.; Cimini, D.; Davalos, R.V.; Schmelz, E.M. Fluid shear stress impacts ovarian cancer cell viability, subcellular organization, and promotes genomic instability. PLoS ONE 2018, 13, e0194170. [Google Scholar] [CrossRef]
- Pejovic, T.; Yates, J.E.; Liu, H.Y.; Hays, L.E.; Akkari, Y.; Torimaru, Y.; Keeble, W.; Rathbun, R.K.; Rodgers, W.H.; Bale, A.E.; et al. Cytogenetic instability in ovarian epithelial cells from women at risk of ovarian cancer. Cancer Res. 2006, 66, 9017–9025. [Google Scholar] [CrossRef] [Green Version]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Klymenko, Y.; Nephew, K.P. Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled. Cancers 2018, 10, 295. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef] [PubMed]
- Kautiainen, T.L.; Jones, P.A. DNA methyltransferase levels in tumorigenic and nontumorigenic cells in culture. J. Biol. Chem. 1986, 261, 1594–1598. [Google Scholar] [PubMed]
- Teodoridis, J.M.; Hall, J.; Marsh, S.; Kannall, H.D.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.L.; Strathdee, G.; et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005, 65, 8961–8967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.J.; Straughn, J.M.; Buchsbaum, D.J.; Arend, R.C. Epigenetic therapy for the treatment of epithelial ovarian cancer: A clinical review. Gynecol. Oncol. Rep. 2017, 20, 81–86. [Google Scholar] [CrossRef]
- Schondorf, T.; Ebert, M.P.; Hoffmann, J.; Becker, M.; Moser, N.; Pur, S.; Gohring, U.J.; Weisshaar, M.P. Hypermethylation of the PTEN gene in ovarian cancer cell lines. Cancer Lett. 2004, 207, 215–220. [Google Scholar] [CrossRef]
- Watts, G.S.; Futscher, B.W.; Holtan, N.; Degeest, K.; Domann, F.E.; Rose, S.L. DNA methylation changes in ovarian cancer are cumulative with disease progression and identify tumor stage. BMC Med. Genom. 2008, 1, 47. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Klinkebiel, D.; Barger, C.J.; Pandey, S.; Guda, C.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. Global DNA hypomethylation in epithelial ovarian cancer: Passive demethylation and association with genomic instability. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Widschwendter, M.; Jiang, G.; Woods, C.; Muller, H.M.; Fiegl, H.; Goebel, G.; Marth, C.; Muller-Holzner, E.; Zeimet, A.G.; Laird, P.W.; et al. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004, 64, 4472–4480. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Gayther, S.A.; Apostolidou, S.; Jones, A.; Lechner, M.; Beck, S.; Jacobs, I.J.; et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS ONE 2009, 4, e8274. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Cancerous ovarian stem cells: Obscure targets for therapy but relevant to chemoresistance. J. Cell Biochem. 2013, 114, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Abubaker, K.; Findlay, J.K. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol. Asp. Med. 2014, 39, 110–125. [Google Scholar] [CrossRef]
- Iyer, D.N.; Sin, W.Y.; Ng, L. Linking stemness with colorectal cancer initiation, progression, and therapy. World J. Stem Cells 2019, 11, 519–534. [Google Scholar] [CrossRef]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef] [Green Version]
- Virant-Klun, I.; Zech, N.; Rozman, P.; Vogler, A.; Cvjeticanin, B.; Klemenc, P.; Malicev, E.; Meden-Vrtovec, H. Putative stem cells with an embryonic character isolated from the ovarian surface epithelium of women with no naturally present follicles and oocytes. Differentiation 2008, 76, 843–856. [Google Scholar] [CrossRef]
- Parte, S.; Bhartiya, D.; Telang, J.; Daithankar, V.; Salvi, V.; Zaveri, K.; Hinduja, I. Detection, characterization, and spontaneous differentiation in vitro of very small embryonic-like putative stem cells in adult mammalian ovary. Stem Cells Dev. 2011, 20, 1451–1464. [Google Scholar] [CrossRef]
- Virant-Klun, I.; Rozman, P.; Cvjeticanin, B.; Vrtacnik-Bokal, E.; Novakovic, S.; Rulicke, T.; Dovc, P.; Meden-Vrtovec, H. Parthenogenetic embryo-like structures in the human ovarian surface epithelium cell culture in postmenopausal women with no naturally present follicles and oocytes. Stem Cells Dev. 2009, 18, 137–149. [Google Scholar] [CrossRef]
- Auersperg, N. The stem-cell profile of ovarian surface epithelium is reproduced in the oviductal fimbriae, with increased stem-cell marker density in distal parts of the fimbriae. Int. J. Gynecol. Pathol. 2013, 32, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N. The origin of ovarian carcinomas: A unifying hypothesis. Int. J. Gynecol. Pathol. 2011, 30, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Szotek, P.P.; Chang, H.L.; Brennand, K.; Fujino, A.; Pieretti-Vanmarcke, R.; Lo Celso, C.; Dombkowski, D.; Preffer, F.; Cohen, K.S.; Teixeira, J.; et al. Normal ovarian surface epithelial label-retaining cells exhibit stem/progenitor cell characteristics. Proc. Natl. Acad. Sci. USA 2008, 105, 12469–12473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flesken-Nikitin, A.; Hwang, C.I.; Cheng, C.Y.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [Green Version]
- Ferrandina, G.; Bonanno, G.; Pierelli, L.; Perillo, A.; Procoli, A.; Mariotti, A.; Corallo, M.; Martinelli, E.; Rutella, S.; Paglia, A.; et al. Expression of CD133–1 and CD133–2 in ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.Q.; Choi, Y.P.; Kang, S.; Youn, J.H.; Cho, N.H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010, 29, 2672–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landen, C.N.; Goodman, B., Jr.; Katre, A.A.; Steg, A.D.; Nick, A.M.; Stone, R.L.; Miller, L.D.; Mejia, P.V.; Jennings, N.B.; Gershenson, D.M.; et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther. 2010, 9, 3186–3199. [Google Scholar] [CrossRef] [Green Version]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [Green Version]
- Vathipadiekal, V.; Saxena, D.; Mok, S.C.; Hauschka, P.V.; Ozbun, L.; Birrer, M.J. Identification of a potential ovarian cancer stem cell gene expression profile from advanced stage papillary serous ovarian cancer. PLoS ONE 2012, 7, e29079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.M.; Joseph, S.; Sudhahar, C.G.; Cowden Dahl, K.D. Enrichment for chemoresistant ovarian cancer stem cells from human cell lines. J. Vis. Exp. 2014, 91, e51891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, T.J.; Vanderhyden, B.C. AKT mediates the pro-survival effects of KIT in ovarian cancer cells and is a determinant of sensitivity to imatinib mesylate. Gynecol. Oncol. 2007, 105, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.; Bobbs, A.; Sattler, R.; Kurkewich, J.L.; Dausinas, P.B.; Nallathamby, P.; Cowden Dahl, K.D. CD133 Promotes Adhesion to the Ovarian Cancer Metastatic Niche. Cancer Growth Metastasis 2018, 11, 1179064418767882. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr. Cancer Drug Targets 2010, 10, 268–278. [Google Scholar] [CrossRef]
- Ahmed, N.; Thompson, E.W.; Quinn, M.A. Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: An exception to the norm. J. Cell Physiol. 2007, 213, 581–588. [Google Scholar] [CrossRef]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef]
- Roy, L.; Cowden Dahl, K.D. Can Stemness and Chemoresistance Be Therapeutically Targeted via Signaling Pathways in Ovarian Cancer? Cancers 2018, 10, 241. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Xu, L.; Tang, H.; Yang, Q.; Yi, X.; Fang, Y.; Zhu, Y.; Wang, Z. The role of the PTEN/PI3K/Akt pathway on prognosis in epithelial ovarian cancer: A meta-analysis. Oncologist 2014, 19, 528–535. [Google Scholar] [CrossRef] [Green Version]
- Abubaker, K.; Luwor, R.B.; Escalona, R.; McNally, O.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Targeted Disruption of the JAK2/STAT3 Pathway in Combination with Systemic Administration of Paclitaxel Inhibits the Priming of Ovarian Cancer Stem Cells Leading to a Reduced Tumor Burden. Front. Oncol. 2014, 4, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abubaker, K.; Luwor, R.B.; Zhu, H.; McNally, O.; Quinn, M.A.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 2014, 14, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.; Luwor, R.; Burns, C.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Momelotinib decreased cancer stem cell associated tumor burden and prolonged disease-free remission period in a mouse model of human ovarian cancer. Oncotarget 2018, 9, 16599–16618. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Torres, C.; Gaytan-Cervantes, J.; Vazquez-Santillan, K.; Mandujano-Tinoco, E.A.; Ceballos-Cancino, G.; Garcia-Venzor, A.; Zampedri, C.; Sanchez-Maldonado, P.; Mojica-Espinosa, R.; Jimenez-Hernandez, L.E.; et al. NF-kappaB Participates in the Stem Cell Phenotype of Ovarian Cancer Cells. Arch. Med. Res. 2017, 48, 343–351. [Google Scholar] [CrossRef]
- Leizer, A.L.; Alvero, A.B.; Fu, H.H.; Holmberg, J.C.; Cheng, Y.C.; Silasi, D.A.; Rutherford, T.; Mor, G. Regulation of inflammation by the NF-kappaB pathway in ovarian cancer stem cells. Am. J. Reprod. Immunol. 2011, 65, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog--a cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [Green Version]
- Barbolina, M.V. Molecular Mechanisms Regulating Organ-Specific Metastases in Epithelial Ovarian Carcinoma. Cancers 2018, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.G.; Piver, M.S.; Tsukada, Y.; Lau, T.S. Metastatic patterns in histologic variants of ovarian cancer. An autopsy study. Cancer 1989, 64, 1508–1513. [Google Scholar]
- Reed, E.; Zerbe, C.S.; Brawley, O.W.; Bicher, A.; Steinberg, S.M. Analysis of autopsy evaluations of ovarian cancer patients treated at the National Cancer Institute, 1972–1988. Am. J. Clin. Oncol. 2000, 23, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Drakes, M.L.; Stiff, P.J. Regulation of Ovarian Cancer Prognosis by Immune Cells in the Tumor Microenvironment. Cancers 2018, 10, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, N.M.; Barbolina, M.V.; Liu, Y.; Sun, L.; Munshi, H.G.; Stack, M.S. Ovarian cancer cell detachment and multicellular aggregate formation are regulated by membrane type 1 matrix metalloproteinase: A potential role in I.p. metastatic dissemination. Cancer Res. 2009, 69, 7121–7129. [Google Scholar] [CrossRef] [Green Version]
- Steinkamp, M.P.; Winner, K.K.; Davies, S.; Muller, C.; Zhang, Y.; Hoffman, R.M.; Shirinifard, A.; Moses, M.; Jiang, Y.; Wilson, B.S. Ovarian tumor attachment, invasion, and vascularization reflect unique microenvironments in the peritoneum: Insights from xenograft and mathematical models. Front. Oncol. 2013, 3, 97. [Google Scholar] [CrossRef] [Green Version]
- Ellerbroek, S.M.; Wu, Y.I.; Overall, C.M.; Stack, M.S. Functional interplay between type I collagen and cell surface matrix metalloproteinase activity. J. Biol. Chem. 2001, 276, 24833–24842. [Google Scholar] [CrossRef] [Green Version]
- Moser, T.L.; Pizzo, S.V.; Bafetti, L.M.; Fishman, D.A.; Stack, M.S. Evidence for preferential adhesion of ovarian epithelial carcinoma cells to type I collagen mediated by the alpha2beta1 integrin. Int. J. Cancer 1996, 67, 695–701. [Google Scholar] [CrossRef]
- Bilandzic, M.; Rainczuk, A.; Green, E.; Fairweather, N.; Jobling, T.W.; Plebanski, M.; Stephens, A.N. Keratin-14 (KRT14) Positive Leader Cells Mediate Mesothelial Clearance and Invasion by Ovarian Cancer Cells. Cancers 2019, 11, 1228. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, L.; Karimnia, N.; Stephens, A.; Bilandzic, M. Therapeutic Targeting of Collective Invasion in Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 1466. [Google Scholar] [CrossRef] [Green Version]
- Platell, C.; Cooper, D.; Papadimitriou, J.M.; Hall, J.C. The omentum. World J. Gastroenterol 2000, 6, 169–176. [Google Scholar]
- Yoo, E.; Kim, J.H.; Kim, M.J.; Yu, J.S.; Chung, J.J.; Yoo, H.S.; Kim, K.W. Greater and lesser omenta: Normal anatomy and pathologic processes. Radiographics 2007, 27, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.M.; Funk, H.M.; Thiolloy, S.; Lotan, T.L.; Hickson, J.; Prins, G.S.; Drew, A.F.; Rinker-Schaeffer, C.W. In vitro metastatic colonization of human ovarian cancer cells to the omentum. Clin. Exp. Metastasis 2010, 27, 185–196. [Google Scholar] [CrossRef]
- Shimotsuma, M.; Shields, J.W.; Simpson-Morgan, M.W.; Sakuyama, A.; Shirasu, M.; Hagiwara, A.; Takahashi, T. Morpho-physiological function and role of omental milky spots as omentum-associated lymphoid tissue (OALT) in the peritoneal cavity. Lymphology 1993, 26, 90–101. [Google Scholar]
- Clark, R.; Krishnan, V.; Schoof, M.; Rodriguez, I.; Theriault, B.; Chekmareva, M.; Rinker-Schaeffer, C. Milky spots promote ovarian cancer metastatic colonization of peritoneal adipose in experimental models. Am. J. Pathol. 2013, 183, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Gusky, H.C.; Diedrich, J.; MacDougald, O.A.; Podgorski, I. Omentum and bone marrow: How adipocyte-rich organs create tumour microenvironments conducive for metastatic progression. Obes. Rev. 2016, 17, 1015–1029. [Google Scholar] [CrossRef]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res. 2016, 175, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormio, G.; Rossi, C.; Cazzolla, A.; Resta, L.; Loverro, G.; Greco, P.; Selvaggi, L. Distant metastases in ovarian carcinoma. Int. J. Gynecol. Cancer 2003, 13, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Sorosky, J.I.; Dolan, M.; Anderson, B.; Buller, R.E. Distant metastases in ovarian cancer: Association with p53 mutations. Clin. Cancer Res. 1999, 5, 2485–2490. [Google Scholar] [PubMed]
- Jubinsky, P.T.; Messer, A.; Bender, J.; Morris, R.E.; Ciraolo, G.M.; Witte, D.P.; Hawley, R.G.; Short, M.K. Identification and characterization of Magmas, a novel mitochondria-associated protein involved in granulocyte-macrophage colony-stimulating factor signal transduction. Exp. Hematol. 2001, 29, 1392–1402. [Google Scholar] [CrossRef]
- Jubinsky, P.T.; Short, M.K.; Mutema, G.; Witte, D.P. Developmental expression of Magmas in murine tissues and its co-expression with the GM-CSF receptor. J. Histochem. Cytochem. 2003, 51, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Huang, C.H.; Short, M.K.; Jubinsky, P.T. Magmas gene structure and evolution. Silico Biol. 2005, 5, 251–263. [Google Scholar]
- Sinha, D.; Joshi, N.; Chittoor, B.; Samji, P.; D’Silva, P. Role of Magmas in protein transport and human mitochondria biogenesis. Hum. Mol. Genet. 2010, 19, 1248–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozany, C.; Mokranjac, D.; Sichting, M.; Neupert, W.; Hell, K. The J domain-related cochaperone Tim16 is a constituent of the mitochondrial TIM23 preprotein translocase. Nat. Struct. Mol. Biol. 2004, 11, 234–241. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, P.R.; Schilke, B.; Walter, W.; Craig, E.A. Role of Pam16’s degenerate J domain in protein import across the mitochondrial inner membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 12419–12424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliati, F.; Gentilin, E.; Buratto, M.; Mole, D.; degli Uberti, E.C.; Zatelli, M.C. Magmas, a gene newly identified as overexpressed in human and mouse ACTH-secreting pituitary adenomas, protects pituitary cells from apoptotic stimuli. Endocrinology 2010, 151, 4635–4642. [Google Scholar] [CrossRef] [Green Version]
- Tagliati, F.; Gagliano, T.; Gentilin, E.; Minoia, M.; Mole, D.; Delgi Uberti, E.C.; Zatelli, M.C. Magmas overexpression inhibits staurosporine induced apoptosis in rat pituitary adenoma cell lines. PLoS ONE 2013, 8, e75194. [Google Scholar] [CrossRef]
- Jubinsky, P.T.; Short, M.K.; Mutema, G.; Morris, R.E.; Ciraolo, G.M.; Li, M. Magmas expression in neoplastic human prostate. J. Mol. Histol. 2005, 36, 69–75. [Google Scholar] [CrossRef]
- Srivastava, S.; Sinha, D.; Saha, P.P.; Marthala, H.; D’Silva, P. Magmas functions as a ROS regulator and provides cytoprotection against oxidative stress-mediated damages. Cell Death Dis. 2014, 5, e1394. [Google Scholar] [CrossRef] [Green Version]
- Di, K.; Lomeli, N.; Bota, D.A.; Das, B.C. Magmas inhibition as a potential treatment strategy in malignant glioma. J. Neurooncol. 2019, 141, 267–276. [Google Scholar] [CrossRef]
- Jubinsky, P.T.; Short, M.K.; Ghanem, M.; Das, B.C. Design, synthesis, and biological activity of novel Magmas inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3479–3482. [Google Scholar] [CrossRef] [PubMed]
- Mehawej, C.; Delahodde, A.; Legeai-Mallet, L.; Delague, V.; Kaci, N.; Desvignes, J.P.; Kibar, Z.; Capo-Chichi, J.M.; Chouery, E.; Munnich, A.; et al. The impairment of MAGMAS function in human is responsible for a severe skeletal dysplasia. PLoS Genet. 2014, 10, e1004311. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Kaye, S.B. New strategies in the treatment of ovarian cancer: Current clinical perspectives and future potential. Clin. Cancer Res. 2013, 19, 961–968. [Google Scholar] [CrossRef] [Green Version]
- Ozols, R.F. Paclitaxel plus carboplatin in the treatment of ovarian cancer. Semin. Oncol. 1999, 26 (Suppl. 2), 84–89. [Google Scholar]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R.; et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef]
- Karam, A.; Ledermann, J.A.; Kim, J.W.; Sehouli, J.; Lu, K.; Gourley, C.; Katsumata, N.; Burger, R.A.; Nam, B.H.; Bacon, M.; et al. Fifth Ovarian Cancer Consensus Conference of the Gynecologic Cancer InterGroup: First-line interventions. Ann. Oncol. 2017, 28, 711–717. [Google Scholar] [CrossRef]
- Wilson, M.K.; Pujade-Lauraine, E.; Aoki, D.; Mirza, M.R.; Lorusso, D.; Oza, A.M.; du Bois, A.; Vergote, I.; Reuss, A.; Bacon, M.; et al. Fifth Ovarian Cancer Consensus Conference of the Gynecologic Cancer InterGroup: Recurrent disease. Ann. Oncol. 2017, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- McGee, J.; Bookman, M.; Harter, P.; Marth, C.; McNeish, I.; Moore, K.N.; Poveda, A.; Hilpert, F.; Hasegawa, K.; Bacon, M.; et al. Fifth Ovarian Cancer Consensus Conference: Individualized therapy and patient factors. Ann. Oncol. 2017, 28, 702–710. [Google Scholar] [CrossRef] [PubMed]
- du Bois, A.; Reuss, A.; Pujade-Lauraine, E.; Harter, P.; Ray-Coquard, I.; Pfisterer, J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: A combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: By the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer 2009, 115, 1234–1244. [Google Scholar]
- Ozols, R.F. Future directions in the treatment of ovarian cancer. Semin. Oncol. 2002, 29 (Suppl. 1), 32–42. [Google Scholar] [CrossRef] [PubMed]
- Katopodis, P.; Chudasama, D.; Wander, G.; Sales, L.; Kumar, J.; Pandhal, M.; Anikin, V.; Chatterjee, J.; Hall, M.; Karteris, E. Kinase Inhibitors and Ovarian Cancer. Cancers 2019, 11, 1357. [Google Scholar] [CrossRef] [Green Version]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Oza, A.M.; Matulonis, U.A.; Malander, S.; Hudgens, S.; Sehouli, J.; Del Campo, J.M.; Berton-Rigaud, D.; Banerjee, S.; Scambia, G.; Berek, J.S.; et al. Quality of life in patients with recurrent ovarian cancer treated with niraparib versus placebo (ENGOT-OV16/NOVA): Results from a double-blind, phase 3, randomised controlled trial. Lancet Oncol. 2018, 19, 1117–1125. [Google Scholar] [CrossRef]
- Friedlander, M.; Gebski, V.; Gibbs, E.; Davies, L.; Bloomfield, R.; Hilpert, F.; Wenzel, L.B.; Eek, D.; Rodrigues, M.; Clamp, A.; et al. Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): A placebo-controlled, phase 3 randomised trial. Lancet Oncol. 2018, 19, 1126–1134. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: An updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017, 17, 1579–1589. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Ben-Baruch, N.E.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 389, 2403–2415. [Google Scholar] [CrossRef]
- Mirza, M.R.; Avall-Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.-B.; Malander, S.; Anttila, M.; Louise Werner, T.; Lund, B.; Lindahl, G.; et al. Combination of niraparib and bevacizumab versus niraparib alone as treatment of recurrent platinum-sensitive ovarian cancer. A randomized controlled chemotherapy-free study-NSGO-AVANOVA2/ENGOT-OV24. J. Clin. Oncol. 2019, 37, 5505. [Google Scholar] [CrossRef]
- Bellio, C.; DiGloria, C.; Foster, R.; James, K.; Konstantinopoulos, P.A.; Growdon, W.B.; Rueda, B.R. PARP Inhibition Induces Enrichment of DNA Repair-Proficient CD133 and CD117 Positive Ovarian Cancer Stem Cells. Mol. Cancer. Res. 2019, 17, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Jarrar, A.; Lotti, F.; De Vecchio, J.; Ferrandon, S.; Gantt, G.; Mace, A.; Karagkounis, G.; Orloff, M.; Venere, M.; Hitomi, M.; et al. Poly(ADP-Ribose) Polymerase Inhibition Sensitizes Colorectal Cancer-Initiating Cells to Chemotherapy. Stem Cells 2019, 37, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Saunders, C.J.; Miller, N.A.; Soden, S.E.; Dinwiddie, D.L.; Noll, A.; Alnadi, N.A.; Andraws, N.; Patterson, M.L.; Krivohlavek, L.A.; Fellis, J.; et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 2012, 4, 154ra135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Lee, J.; Zhang, S.; Benyshek, C.; Dokmeci, M.R.; Khademhosseini, A. Engineering Precision Medicine. Adv. Sci. 2019, 6, 1801039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A collagen-remodeling gene signature regulated by TGF-beta signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Kalli, K.R.; Oberg, A.L.; Keeney, G.L.; Christianson, T.J.; Low, P.S.; Knutson, K.L.; Hartmann, L.C. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol. Oncol. 2008, 108, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blattler, W.A.; Goldmacher, V.S. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Martin, L.P.; O’Malley, D.M.; Matulonis, U.A.; Konner, J.A.; Perez, R.P.; Bauer, T.M.; Ruiz-Soto, R.; Birrer, M.J. Safety and Activity of Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha-Targeting Antibody-Drug Conjugate, in Platinum-Resistant Ovarian, Fallopian Tube, or Primary Peritoneal Cancer: A Phase I Expansion Study. J. Clin. Oncol. 2017, 35, 1112–1118. [Google Scholar] [CrossRef]
- Moore, K.N.; Vergote, I.; Oaknin, A.; Colombo, N.; Banerjee, S.; Oza, A.; Pautier, P.; Malek, K.; Birrer, M.J. FORWARD, I: A Phase III study of mirvetuximab soravtansine versus chemotherapy in platinum-resistant ovarian cancer. Future Oncol. 2018, 14, 1669–1678. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Ovarian Tumor Tissue Analysis, C.; Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.A.; Chen, W.; Fogarty, Z.C.; Gentry-Maharaj, A.; Toloczko, A.; Hein, A.; et al. Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [CrossRef] [Green Version]
- Santoiemma, P.P.; Reyes, C.; Wang, L.P.; McLane, M.W.; Feldman, M.D.; Tanyi, J.L.; Powell, D.J., Jr. Systematic evaluation of multiple immune markers reveals prognostic factors in ovarian cancer. Gynecol. Oncol. 2016, 143, 120–127. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Huang, R.Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef] [PubMed]
- Ino, K. Indoleamine 2, 3-dioxygenase and immune tolerance in ovarian cancer. Curr. Opin. Obstet. Gynecol. 2011, 23, 13–18. [Google Scholar] [CrossRef]
- Okla, K.; Wertel, I.; Polak, G.; Surowka, J.; Wawruszak, A.; Kotarski, J. Tumor-Associated Macrophages and Myeloid-Derived Suppressor Cells as Immunosuppressive Mechanism in Ovarian Cancer Patients: Progress and Challenges. Int. Rev. Immunol. 2016, 35, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Aptsiauri, N.; Cabrera, T.; Mendez, R.; Garcia-Lora, A.; Ruiz-Cabello, F.; Garrido, F. Role of altered expression of HLA class I molecules in cancer progression. Adv. Exp. Med. Biol. 2007, 601, 123–131. [Google Scholar] [PubMed]
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. 2017, 28 (Suppl. 8), viii1–viii7. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.; Sill, M.; Zamarin, D. NRG Oncology phase II trial of nivolumab with or without ipilimumab in patients with persistent or recurrent ovarian cancer. In Proceedings of the 17th Biennial meeting of the International Gynecologic Cancer Society, Kyoto, Japan, 14–16 September 2018. [Google Scholar]
- Wenham, R.M.; Fridley, B.; Boulware, D. Phase 2 trial of weekly paclitaxel with pembrolizumab in platinum recurrent ovarian cancer. In Proceedings of the 17th Biennial meeting of the International Gynecologic Cancer Society, Kyoto, Japan, 14–16 September 2018. [Google Scholar]
- Grunewald, C.M.; Schulz, W.A.; Skowron, M.A.; Hoffmann, M.J.; Niegisch, G. Tumor immunotherapy—The potential of epigenetic drugs to overcome resistance. Transl. Cancer Res. 2018, 7, 1151–1156. [Google Scholar] [CrossRef]
- Kareva, I.; Waxman, D.J.; Lakka Klement, G. Metronomic chemotherapy: An attractive alternative to maximum tolerated dose therapy that can activate anti-tumor immunity and minimize therapeutic resistance. Cancer Lett. 2015, 358, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerbel, R.S.; Kamen, B.A. The anti-angiogenic basis of metronomic chemotherapy. Nat. Rev. Cancer 2004, 4, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.S.; Hsu, C.C.; Pai, V.C.; Liao, W.Y.; Huang, S.S.; Tan, K.T.; Yen, C.J.; Hsu, S.C.; Chen, W.Y.; Shan, Y.S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef]
- Kareva, I. A Combination of Immune Checkpoint Inhibition with Metronomic Chemotherapy as a Way of Targeting Therapy-Resistant Cancer Cells. Int. J. Mol. Sci. 2017, 18, 2134. [Google Scholar] [CrossRef] [Green Version]
- Villa, M.; Attianese, D.; Petracchini, M.; Ferrero, A. Oral metronomic cyclophosphamide in advanced ovarian cancer: Long-lasting clinical response in an elderly frailty patient. Anticancer Drugs 2019, 30, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Perroud, H.A.; Scharovsky, O.G.; Rozados, V.R.; Alasino, C.M. Clinical response in patients with ovarian cancer treated with metronomic chemotherapy. Ecancermedicalscience 2017, 11, 723. [Google Scholar] [CrossRef] [Green Version]
- Rivkin, S.E.; Moon, J.; Iriarte, D.S.; Bailey, E.; Sloan, H.L.; Goodman, G.E.; BonDurant, A.E.; Velijovich, D.; Wahl, T.; Jiang, P.; et al. Phase Ib with expansion study of olaparib plus weekly (Metronomic) carboplatin and paclitaxel in relapsed ovarian cancer patients. Int. J. Gynecol. Cancer 2019, 29, 325–333. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Nichols, J.W.; Toh, K.; Nomoto, T.; Cabral, H.; Miura, Y.; Christie, R.J.; Yamada, N.; Ogura, T.; Kano, M.R.; et al. Vascular bursts enhance permeability of tumour blood vessels and improve nanoparticle delivery. Nat. Nanotechnol. 2016, 11, 533–538. [Google Scholar] [CrossRef]
- Lee, M.S.; Dees, E.C.; Wang, A.Z. Nanoparticle-Delivered Chemotherapy: Old Drugs in New Packages. Oncology 2017, 31, 198–208. [Google Scholar]
- Lee, J.K.; Bangayan, N.J.; Chai, T.; Smith, B.A.; Pariva, T.E.; Yun, S.; Vashisht, A.; Zhang, Q.; Park, J.W.; Corey, E.; et al. Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E4473–E4482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, J.; Luwor, R.B.; Ahmed, N.; Escalona, R.; Tan, F.H.; Fong, C.; Ratcliffe, J.; Scoble, J.A.; Drummond, C.J.; Tran, N. Paclitaxel-Loaded Self-Assembled Lipid Nanoparticles as Targeted Drug Delivery Systems for the Treatment of Aggressive Ovarian Cancer. ACS Appl. Mater. Interfaces 2018, 10, 25174–25185. [Google Scholar] [CrossRef] [PubMed]
- Byeon, Y.; Lee, J.W.; Choi, W.S.; Won, J.E.; Kim, G.H.; Kim, M.G.; Wi, T.I.; Lee, J.M.; Kang, T.H.; Jung, I.D.; et al. CD44-Targeting PLGA Nanoparticles Incorporating Paclitaxel and FAK siRNA Overcome Chemoresistance in Epithelial Ovarian Cancer. Cancer Res. 2018, 78, 6247–6256. [Google Scholar] [PubMed] [Green Version]
- Diaz Osterman, C.J.; Ozmadenci, D.; Kleinschmidt, E.G.; Taylor, K.N.; Barrie, A.M.; Jiang, S.; Bean, L.M.; Sulzmaier, F.J.; Jean, C.; Tancioni, I.; et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife 2019, 8. [Google Scholar] [CrossRef]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol. Cancer Res. 2016, 14, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Wilding, J.L.; Bodmer, W.F. Cancer cell lines for drug discovery and development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Nagle, P.W.; Plukker, J.T.M.; Muijs, C.T.; van Luijk, P.; Coppes, R.P. Patient-derived tumor organoids for prediction of cancer treatment response. Semin. Cancer Biol. 2018, 53, 258–264. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praharaj, P.P.; Bhutia, S.K.; Nagrath, S.; Bitting, R.L.; Deep, G. Circulating tumor cell-derived organoids: Current challenges and promises in medical research and precision medicine. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Sun, L.; Liu, M.; Mao, Y. Patient-derived organoids: A promising model for personalized cancer treatment. Gastroenterol. Rep. 2018, 6, 243–245. [Google Scholar] [CrossRef] [Green Version]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Lannagan, T.R.M.; Lee, Y.K.; Wang, T.; Roper, J.; Bettington, M.L.; Fennell, L.; Vrbanac, L.; Jonavicius, L.; Somashekar, R.; Gieniec, K.; et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut 2019, 68, 684–692. [Google Scholar] [CrossRef]
- Kopper, O.; de Witte, C.J.; Lohmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Jabs, J.; Zickgraf, F.M.; Park, J.; Wagner, S.; Jiang, X.; Jechow, K.; Kleinheinz, K.; Toprak, U.H.; Schneider, M.A.; Meister, M.; et al. Screening drug effects in patient-derived cancer cells links organoid responses to genome alterations. Mol. Syst. Biol. 2017, 13, 955. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J., Jr.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, N.; Hong, J.J.; Tofig, B.; Mapua, M.; Elashoff, D.; Moatamed, N.A.; Huang, J.; Memarzadeh, S.; Damoiseaux, R.; Soragni, A. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Invest. 2005, 115, 1503–1521. [Google Scholar] [CrossRef] [Green Version]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front. Oncol. 2018, 8, 203. [Google Scholar] [CrossRef]
- Marhaba, R.; Klingbeil, P.; Nuebel, T.; Nazarenko, I.; Buechler, M.W.; Zoeller, M. CD44 and EpCAM: Cancer-initiating cell markers. Curr. Mol. Med. 2008, 8, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Serafino, A.; Zonfrillo, M.; Andreola, F.; Psaila, R.; Mercuri, L.; Moroni, N.; Renier, D.; Campisi, M.; Secchieri, C.; Pierimarchi, P. CD44-targeting for antitumor drug delivery: A new SN-38-hyaluronan bioconjugate for locoregional treatment of peritoneal carcinomatosis. Curr. Cancer Drug Targets 2011, 11, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Yo, Y.T.; Lee, H.Y.; Liao, Y.P.; Chao, T.K.; Su, P.H.; Lai, H.C. ALDH1-bright epithelial ovarian cancer cells are associated with CD44 expression, drug resistance, and poor clinical outcome. Am. J. Pathol. 2012, 180, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 2012, 130, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: Potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518. [Google Scholar] [CrossRef]
- Khandekar, D.; Amara, S.; Tiriveedhi, V. Immunogenicity of Tumor Initiating Stem Cells: Potential Applications in Novel Anticancer Therapy. Front. Oncol. 2019, 9, 315. [Google Scholar] [CrossRef] [Green Version]
- Visus, C.; Ito, D.; Amoscato, A.; Maciejewska-Franczak, M.; Abdelsalem, A.; Dhir, R.; Shin, D.M.; Donnenberg, V.S.; Whiteside, T.L.; DeLeo, A.B. Identification of human aldehyde dehydrogenase 1 family member A1 as a novel CD8+ T-cell-defined tumor antigen in squamous cell carcinoma of the head and neck. Cancer Res. 2007, 67, 10538–10545. [Google Scholar] [CrossRef] [Green Version]
- Visus, C.; Wang, Y.; Lozano-Leon, A.; Ferris, R.L.; Silver, S.; Szczepanski, M.J.; Brand, R.E.; Ferrone, C.R.; Whiteside, T.L.; Ferrone, S.; et al. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8(+) T cells. Clin. Cancer Res. 2011, 17, 6174–6184. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, K.M.; Feldman, D.; Matthews, P.; Radfar, S.; Pickering, R.; Turkula, S.; Zebroski, H.; Dhodapkar, M.V. Natural immunity to pluripotency antigen OCT4 in humans. Proc. Natl. Acad. Sci. USA 2010, 107, 8718–8723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wefers, C.; Schreibelt, G.; Massuger, L.; de Vries, I.J.M.; Torensma, R. Immune Curbing of Cancer Stem Cells by CTLs Directed to NANOG. Front. Immunol. 2018, 9, 1412. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/mTOR signaling pathway as a therapeutic target for ovarian cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Greening, D.; Samardzija, C.; Escalona, R.M.; Chen, M.; Findlay, J.K.; Kannourakis, G. Unique proteome signature of post-chemotherapy ovarian cancer ascites-derived tumor cells. Sci. Rep. 2016, 6, 30061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.G.; Mercado-Uribe, I.; Yang, G.; Bast, R.C., Jr.; Amin, H.M.; Lai, R.; Liu, J. The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer 2006, 107, 2730–2740. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Foster, R.; Bell, D.A.; Mahoney, J.; Wolak, K.; Vaidya, A.; Hampel, C.; Lee, H.; Seiden, M.V. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin. Cancer Res. 2006, 12, 5055–5063. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.; Peyrollier, K.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Biol. Chem. 2008, 283, 17635–17651. [Google Scholar] [CrossRef] [Green Version]
- Steg, A.D.; Katre, A.A.; Bevis, K.S.; Ziebarth, A.; Dobbin, Z.C.; Shah, M.M.; Alvarez, R.D.; Landen, C.N. Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol. Cancer Ther. 2012, 11, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; Hulver, M.W.; Schmelz, E.M. Ovarian tumor-initiating cells display a flexible metabolism. Exp. Cell Res. 2014, 328, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Ogishima, J.; et al. Detachment from the primary site and suspension in ascites as the initial step in metabolic reprogramming and metastasis to the omentum in ovarian cancer. Oncol. Lett. 2018, 15, 1357–1361. [Google Scholar] [CrossRef]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.R.; Yung, M.M.H.; Xuan, Y.; Zhan, S.; Leung, L.L.; Liang, R.R.; Leung, T.H.Y.; Yang, H.; Xu, D.; Sharma, R.; et al. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun. Biol. 2019, 2, 281. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Trifanescu, O.; Gruia, M.I.; Gales, L.; Trifanescu, R.; Anghel, R. Tumor is an Oxidative Stress Factor in Ovarian Cancer Patients. Chirurgia 2018, 113, 687–694. [Google Scholar] [CrossRef]
- Mutch, D.G.; Williams, S. Biology of epithelial ovarian cancer. Clin. Obstet. Gynecol. 1994, 37, 406–422. [Google Scholar] [CrossRef]
- Runyon, B.A.; Hoefs, J.C.; Morgan, T.R. Ascitic fluid analysis in malignancy-related ascites. Hepatology 1988, 8, 1104–1109. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Osada, R.; Horiuchi, A.; Kikuchi, N.; Yoshida, J.; Hayashi, A.; Ota, M.; Katsuyama, Y.; Melillo, G.; Konishi, I. Expression of hypoxia-inducible factor 1alpha, hypoxia-inducible factor 2alpha, and von Hippel-Lindau protein in epithelial ovarian neoplasms and allelic loss of von Hippel-Lindau gene: Nuclear expression of hypoxia-inducible factor 1alpha is an independent prognostic factor in ovarian carcinoma. Hum. Pathol. 2007, 38, 1310–1320. [Google Scholar] [PubMed]
- Cohen, S.; Mehrabi, S.; Yao, X.; Millingen, S.; Aikhionbare, F.O. Reactive Oxygen Species and Serous Epithelial Ovarian Adenocarcinoma. Cancer Res. J. 2016, 4, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugazhendhi, A.; Edison, T.; Velmurugan, B.K.; Jacob, J.A.; Karuppusamy, I. Toxicity of Doxorubicin (Dox) to different experimental organ systems. Life Sci. 2018, 200, 26–30. [Google Scholar] [CrossRef]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PARP Inhibitors | PARP Inhibitors + Antiangiogenic Agents/Chemotherapy | Immune Checkpoint Inhibitors in Combination with Other Anti-Cancer Agents |
|---|---|---|
| Gelmon KA et al.: Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study [136]. | Coleman RL et al.: Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer [147]. | Burger R et al.: NRG Oncology phase II trial of nivolumab with or without ipilimumab in patients with persistent or recurrent ovarian cancer. 17thBiennial meeting of the International Gynecologic Cancer Society, September 14–16, 2018, Kyoto, Japan [171]. |
| Ledermann J et al.: Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer [137]. | Mirza MR, et al.: Combination of niraparib and bevacizumab versus niraparib alone as treatment of recurrent platinum-sensitive ovarian cancer. A randomized controlled chemotherapy-free study-NSGO-AVANOVA2/ENGOT-OV24 [148]. | Wenham RM etal: Phase 2 trial of weekly paclitaxel with pembrolizumab in platinum recurrent ovarian cancer. 17thBiennial meeting of the International Gynecologic Cancer Society, September 14–16, 2018, Kyoto, Japan [172]. |
| Swisher EM et al.: Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial [138]. | Grunewald CM et al: Tumor immunotherapy-the potential of epigenetic drugs to overcome resistance. Translational Cancer Research 2018, 1151–1156. [173]. | |
| Mirza MR, et al.: Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer [139]. | ||
| Oza AM et al.: Quality of life in patients with recurrent ovarian cancer treated with niraparib versus placebo (ENGOT-OV16/NOVA): results from a double-blind, phase 3, randomised controlled trial [140]. | ||
| Friedlander M et al.: Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): a placebo-controlled, phase 3 randomised trial [141]. | ||
| Ledermann J et al.: Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial [142]. | ||
| Pujade-Lauraine E et al.: Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial [143]. | ||
| Coleman RL et al.: Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial [144]. | ||
| Liu JF et al.: Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study [145]. | ||
| Ledermann JA et al.: Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial [146]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, N.; Kadife, E.; Raza, A.; Short, M.; Jubinsky, P.T.; Kannourakis, G. Ovarian Cancer, Cancer Stem Cells and Current Treatment Strategies: A Potential Role of Magmas in the Current Treatment Methods. Cells 2020, 9, 719. https://doi.org/10.3390/cells9030719
Ahmed N, Kadife E, Raza A, Short M, Jubinsky PT, Kannourakis G. Ovarian Cancer, Cancer Stem Cells and Current Treatment Strategies: A Potential Role of Magmas in the Current Treatment Methods. Cells. 2020; 9(3):719. https://doi.org/10.3390/cells9030719
Chicago/Turabian StyleAhmed, Nuzhat, Elif Kadife, Ali Raza, Mary Short, Paul T. Jubinsky, and George Kannourakis. 2020. "Ovarian Cancer, Cancer Stem Cells and Current Treatment Strategies: A Potential Role of Magmas in the Current Treatment Methods" Cells 9, no. 3: 719. https://doi.org/10.3390/cells9030719