Relationship between Heart Disease and Liver Disease: A Two-Way Street


{kind=link}
Abstract
:1. Introduction
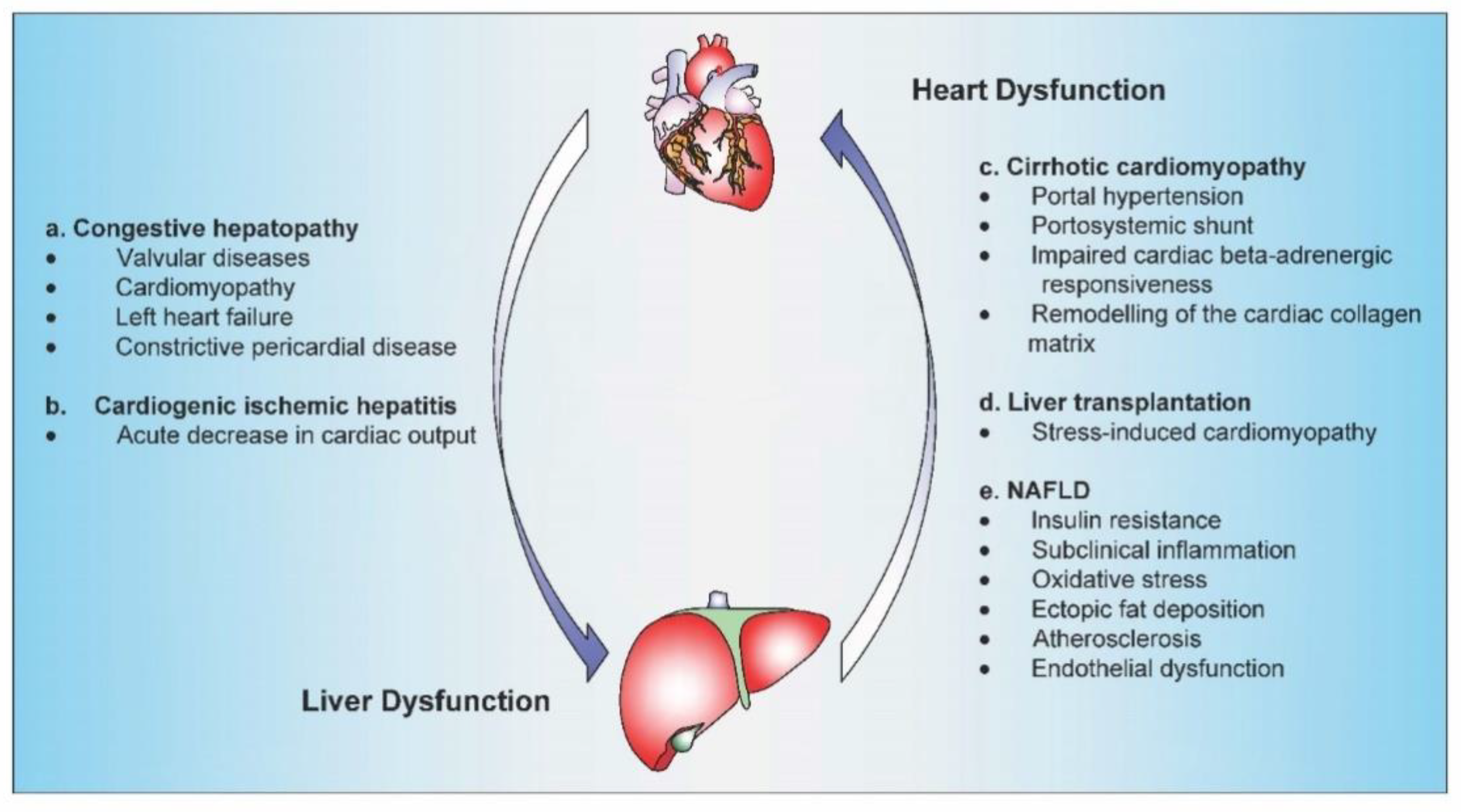
2. The heart as a Cause of Liver Disease
2.1. Congestive Hepatopathy
2.1.1. Pathophysiology and Presentation
2.1.2. Biochemical Pattern
2.1.3. Histopathology
2.1.4. Management
2.2. Cardiogenic Ischemic Hepatitis
2.2.1. Pathophysiology and Presentation
2.2.2. Biochemical Pattern
2.2.3. Histopathology
2.2.4. Management
3. The Liver as a Cause of Heart Disease
3.1. Cirrhotic Cardiomyopathy
3.1.1. Pathophysiology and Presentation
Hyperdynamic Circulation
Systolic Dysfunction
Diastolic Dysfunction
Electrophysiological Modifications
3.1.2. Diagnostic Approach
3.1.3. Management
3.2. Liver Transplantation
Stress-Induced Cardiomyopathy
Clinical Presentation
Pathophysiology
Predictors and Diagnostic Approach
Management
3.3. Nonalcoholic Fatty Liver Disease
3.3.1. Definition and Prevalence
3.3.2. Cardiac Disorders in NAFLD
3.3.3. Mechanisms Linking Cardiac Disorders to NAFLD
3.3.4. Predictors of Cardiac Events in NAFLD
3.3.5. Management
4. The Heart–Liver Axis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-AG | anandamide and 2-arachidonoylglycerol |
| ACE | angiotensin-converting enzyme |
| AEA | Arachidonoylethanolamide |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase |
| ANF | atrial natriuretic factor |
| ARBs | angiotensin receptor blockers |
| AST | aspartate aminotransferase |
| ATP | adenosine triphosphate |
| BMI | Body mass index |
| BNP | B-type natriuretic peptide |
| CB-1 | cannabinoid receptor type 1 |
| CBRs | cannabinoid receptors |
| CCM | cirrhotic cardiomyopathy |
| CCTA | coronary computed tomography angiography |
| CFR | coronary flow reserve |
| CRP | C-reactive protein |
| cTnC | troponin C |
| cTnI | troponin I |
| FetA | Fetuin-A |
| FGF21 | fibroblast growth factor 21 |
| GGT | gamma-glutamyltransferase |
| Il-1 β | interleukin 1 beta |
| iNOs | nitric oxide synthetase |
| INR | International Normalized Ratio |
| ITVR | isovolumetric relaxation time |
| LDH | lactate dehydrogenase |
| LDL-C | low-density lipoprotein cholesterol |
| LT | liver transplantation |
| LVAD | left ventricular assistive device support |
| MED13 | mediator complex subunit 13 |
| miR-208a | microRNA-208a |
| mtDNA | mitochondrial DNA |
| Myh6 | myosin Heavy Chain 6 |
| NAFL | nonalcoholic fatty liver |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NFS | NAFLD Fibrosis Score |
| NF-κB | nuclear factor κB |
| NLRP3 | NOD-like receptor protein 3 |
| NO | nitric oxide |
| PAI-1 | plasminogen inhibitor activator 1 |
| PKA | protein kinase A |
| ROS | reactive oxygen species |
| TGF-β1 | transforming growth factor-β1 |
| TTC | Takotsubo cardiomyopathy |
| VLDL-C | low-density lipoprotein cholesterol |
References
- Khadem, E.; Toosi, M.N.; Ilkhani, R. Liver- Heart Inter- Relationship in Fatty Liver Disease Based on the Avicenna’s Point of View. Iran. J. Public Health 2013, 42, 648–649. [Google Scholar]
- Asrani, N.S.; Freese, D.K.; Phillips, S.D.; Heimbach, J.; Asrani, S.K.; Warnes, C.A.; Kamath, P.S. Congenital heart disease and the liver. Hepatology 2012, 56, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Giallourakis, C.C.; Rosenberg, P.M.; Friedman, L.S. The liver in heart failure. Clin. Liver Dis. 2002, 6, 947–967. [Google Scholar] [CrossRef]
- De Gonzalez, A.K.K.; Lefkowitch, J.H. Heart Disease and the Liver. Gastroenterol. Clin. N. Am. 2017, 46, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.P.; Cerini, R.; Sayegh, R.; Moreau, R.; Degott, C.; Lebrec, D.; Lee, S.S. Cardiac hepatopathy: Clinical, hemodynamic, and histologic characteristics and correlations. Hepatology 2003, 37, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Téllez, L.; Rodriguez-Santiago, E.; Albillos, A. Fontan-Associated Liver Disease: A Review. Ann. Hepatol. 2018, 17, 192–204. [Google Scholar] [CrossRef]
- Fauci, A.S.; Braunwald, E.; Hauser, S.L.; Longo, D.L.; Jameson, J.; Loscalzo, J. Harrison’s Principles of Internal Medicine; McGraw-Hill Medical: New York, NY, USA, 2008; Volume 2. [Google Scholar]
- Kiesewetter, C.H.; Sheron, N.; Vettukattill, J.J.; Hacking, N.; Stedman, B.; Millward-Sadler, H.; Haw, M.; Cope, R.; Salmon, A.P.; Sivaprakasam, M.C.; et al. Hepatic changes in the failing Fontan circulation. Heart 2006, 93, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, I.S.; Jacobson, I.M. Cardiovascular Diseases and the Liver. Clin. Liver Dis. 2011, 15, 1–20. [Google Scholar] [CrossRef]
- Vasconcelos, L.A.B.A.; De Almeida, E.A.; Bachur, L.F. Clinical evaluation and hepatic laboratory assessment in individuals with congestive heart failure. Arq. Bras. Cardiol. 2007, 88, 590–595. [Google Scholar] [CrossRef] [Green Version]
- Poelzl, G.; Eberl, C.; Achrainer, H.; Doerler, J.; Pachinger, O.; Frick, M.; Ulmer, H. Prevalence and Prognostic Significance of Elevated γ-Glutamyltransferase in Chronic Heart Failure. Circ. Heart Fail. 2009, 2, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, V.; Jäger, B.; Zubkova, A.; Drolz, A. Hypoxic hepatitis – epidemiology, pathophysiology and clinical management. Wien. Klin. Wochenschr. 2010, 122, 129–139. [Google Scholar] [CrossRef]
- Dunn, G.D.; Hayes, P.; Breen, K.J.; Schenker, S. The liver in congestive heart failure: A review. Am. J. Med. Sci. 1973, 265, 174–189. [Google Scholar] [CrossRef]
- Shah, H.; Kuehl, K.; Sherker, A.H. Liver Disease After the Fontan Procedure. J. Clin. Gastroenterol. 2010, 44, 1. [Google Scholar] [CrossRef]
- Wells, M.L.; Fenstad, E.R.; Poterucha, J.T.; Hough, D.M.; Young, P.M.; Araoz, P.A.; Ehman, R.L.; Venkatesh, S.K. Imaging Findings of Congestive Hepatopathy. Radiographics 2016, 36, 1024–1037. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.-F.; Swanson, P.; Krieger, E.; Liou, I.W.; Carithers, R.L.; Yeh, M.M. Congestive hepatic fibrosis score: A novel histologic assessment of clinical severity. Mod. Pathol. 2014, 27, 1552–1558. [Google Scholar] [CrossRef] [Green Version]
- Sherlock, S. The Liver in Heart Failure Relation of Anatomical, Functional, and Circulatory Changes. Heart 1951, 13, 273–293. [Google Scholar] [CrossRef] [Green Version]
- Maleki, M.; Vakilian, F.; Amin, A. Liver diseases in heart failure. Heart Asia 2011, 3, 143–149. [Google Scholar]
- Russell, S.D.; Rogers, J.; Milano, C.A.; Dyke, D.B.; Pagani, F.D.; Aranda, J.M.; Klodell, C.T.; Boyle, A.J.; John, R.; Chen, L.; et al. Renal and Hepatic Function Improve in Advanced Heart Failure Patients During Continuous-Flow Support With the HeartMate II Left Ventricular Assist Device. Circulation 2009, 120, 2352–2357. [Google Scholar] [CrossRef] [Green Version]
- Dichtl, W.; Vogel, W.; Dunst, K.M.; Grander, W.; Alber, H.F.; Frick, M.; Antretter, H.; Laufer, G.; Pachinger, O.; Pölzl, G. Cardiac hepatopathy before and after heart. transplantation. Transpl. Int. 2005, 18, 697–702. [Google Scholar] [CrossRef]
- Seeto, R.K.; Fenn, B.; Rockey, D.C. Ischemic hepatitis: Clinical presentation and pathogenesis. Am. J. Med. 2000, 109, 109–113. [Google Scholar] [CrossRef]
- Harjola, V.-P.; Mullens, W.; Banaszewski, M.; Bauersachs, J.; Rocca, H.-P.B.-L.; Chioncel, O.; Collins, S.P.; Doehner, W.; Filippatos, G.S.; Flammer, A.; et al. Organ dysfunction, injury and failure in acute heart failure: From pathophysiology to diagnosis and management. A review on behalf of the Acute Heart Failure Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2017, 19, 821–836. [Google Scholar] [CrossRef]
- Eipel, C.; Abshagen, K.; Vollmar, B. Regulation of hepatic blood flow: The hepatic arterial buffer response revisited. World J. Gastroenterol. 2010, 16, 6046–6057. [Google Scholar] [CrossRef]
- Henrion, J.; Descamps, O.; Luwaert, R.; Schapira, M.; Parfonry, A.; Heller, F. Hypoxic hepatitis in patients with cardiac failure: Incidence in a coronary care unit and measurement of hepatic blood flow. J. Hepatol. 1994, 21, 696–703. [Google Scholar] [CrossRef]
- Naschitz, J.E.; Yeshurun, D.; Shahar, J. Cardiogenic Hepatorenal Syndrome. Angiology 1990, 41, 893–900. [Google Scholar] [CrossRef]
- Birrer, R.; Takuda, Y.; Takara, T. Hypoxic hepatopathy: Pathophysiology and prognosis. Intern. Med. 2007, 46, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Henrion, J.; Schapira, M.; Luwaert, R.; Colin, L.; Delannoy, A.; Heller, F.R. Hypoxic hepatitis: Clinical and hemodynamic study in 142 consecutive cases. Medicine 2003, 82, 392–406. [Google Scholar] [CrossRef]
- Denis, C.; De Kerguennec, C.; Bernuau, J.; Beauvais, F.; Cohen-Solal, A. Acute hypoxic hepatitis (‘liver shock’): Still a frequently overlooked cardiological diagnosis. Eur. J. Heart Fail. 2004, 6, 561–565. [Google Scholar] [CrossRef]
- Giannini, E.G.; Testa, R.; Savarino, V. Liver enzyme alteration: A guide for clinicians. Can. Med. Assoc. J. 2005, 172, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, W.M.; Reynolds, T.B. Serum Lactic Dehydrogenase in the Differential Diagnosis of Acute Hepatocellular Injury. J. Clin. Gastroenterol. 1994, 19, 118–121. [Google Scholar] [CrossRef]
- Alvarez, A.M.; Mukherjee, D. Liver Abnormalities in Cardiac Diseases and Heart Failure. Int. J. Angiol. 2011, 20, 135–142. [Google Scholar] [CrossRef] [Green Version]
- De La Monte, S.M.; Arcidi, J.M.; Moore, G.W.; Hutchins, G.M. Midzonal Necrosis as a Pattern of Hepatocellular Injury After Shock. Gastroenterology 1984, 86, 627–631. [Google Scholar] [CrossRef]
- Waseem, N.; Chen, P.-H. Hypoxic Hepatitis: A Review and Clinical Update. J. Clin. Transl. Hepatol. 2016, 4, 263–268. [Google Scholar]
- Limas, C.J.; Guiha, N.H.; Lekagul, O.; Cohn, J.N. Impaired Left Ventricular Function in Alcoholic Cirrhosis with Ascites. Circulation 1974, 49, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Møller, S.; Henriksen, J.H. Cirrhotic cardiomyopathy. J. Hepatol. 2010, 53, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Alqahtani, S.A.; Fouad, T.R.; Lee, S.S. Cirrhotic Cardiomyopathy. Semin. Liver Dis. 2008, 28, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Wiese, S.; Hove, J.; Bendtsen, F.; Møller, S. Cirrhotic cardiomyopathy: Pathogenesis and clinical relevance. Nat. Rev. Gastroenterol. Hepatol. 2013, 11, 177–186. [Google Scholar] [CrossRef]
- Goldberg, D.; Fallon, M.B. The Art and Science of Diagnosing and Treating Lung and Heart Disease Secondary to Liver Disease. Clin. Gastroenterol. Hepatol. 2015, 13, 2118–2127. [Google Scholar] [CrossRef] [Green Version]
- Bolognesi, M.; Di Pascoli, M.; Verardo, A.; Gatta, A. Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis. World J. Gastroenterol. 2014, 20, 2555–2563. [Google Scholar] [CrossRef]
- Møller, S. Cirrhotic cardiomyopathy: A pathophysiological review of circulatory dysfunction in liver disease. Heart 2002, 87, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, J.H.; Fuglsang, S.; Bendtsen, F.; Christensen, E.; Møller, S. Arterial compliance in patients with cirrhosis: Stroke volume-pulse pressure ratio as simplified index. Am. J. Physiol. Liver Physiol. 2001, 280, G584–G594. [Google Scholar] [CrossRef] [Green Version]
- Dümcke, C.W.; Møller, S. Autonomic dysfunction in cirrhosis and portal hypertension. Scand. J. Clin. Lab. Investig. 2008, 68, 437–447. [Google Scholar] [CrossRef]
- Kelbæk, H.; Rabøl, A.; Brynjolf, I.; Eriksen, J.; Bonnevie, O.; Godtfredsen, J.; Munck, O.; Lund, J.O. Haemodynamic response to exercise in patients with alcoholic liver cirrhosis. Clin. Physiol. 1987, 7, 35–41. [Google Scholar] [CrossRef]
- Møller, S.; Dümcke, C.W.; Krag, A. The heart and the liver. Expert. Rev. Gastroenterol. Hepatol. 2009, 3, 51–64. [Google Scholar] [CrossRef]
- Lee, S.S.; Marty, J.; Mantz, J.; Samain, E.; Braillon, A.; Lebrec, D. Desensitization of myocardial beta-adrenergic receptors in cirrhotic rats. Hepatology 1990, 12, 481–485. [Google Scholar] [CrossRef]
- Hausdorff, W.P.; Caron, M.G.; Lefkowitz, R.J. Turning off the signal: Desensitization of beta-adrenergic receptor function. FASEB J. 1990, 4, 2881–2889. [Google Scholar] [CrossRef]
- Van Obbergh, L.; Vallieres, Y.; Blaise, G. Cardiac modifications occurring in the ascitic rat with biliary cirrhosis are nitric oxide related. J. Hepatol. 1996, 24, 747–752. [Google Scholar] [CrossRef]
- Liu, H.; Ma, Z.; Lee, S.S. Contribution of nitric oxide to the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Gastroenterology 2000, 118, 937–944. [Google Scholar] [CrossRef]
- Gaskari, S.A.; Liu, H.; Moezi, L.; Li, Y.; Baik, S.K.; Lee, S.S. Role of endocannabinoids in the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Br. J. Pharmacol. 2005, 146, 315–323. [Google Scholar] [CrossRef]
- Basu, P.P.; Aloysius, M.M.; Shah, N.J.; Brown, R.S., Jr. Review article: The endocannabinoid system in liver disease, a potential therapeutic target. Aliment. Pharmacol. Ther. 2014, 39, 790–801. [Google Scholar] [CrossRef]
- Bátkai, S.; Mukhopadhyay, P.; Harvey-White, J.; Kechrid, R.; Pacher, P.; Kunos, G. Endocannabinoids acting at CB1 receptors mediate the cardiac contractile dysfunction in vivo in cirrhotic rats. Am. J. Physiol. Circ. Physiol. 2007, 293, H1689–H1695. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.Y.; Liu, H.; Nam, S.W.; Kunos, G.; Lee, S.S. Mechanisms of TNF α-induced cardiac dysfunction in cholestatic bile ductligated mice: Interaction between TNF α and endocannabinoids. J. Hepatol. 2010, 53, 298–306. [Google Scholar] [CrossRef]
- Nayor, M.; Cooper, L.L.; Enserro, D.M.; Xanthakis, V.; Larson, M.G.; Benjamin, E.J.; Aragam, J.; Mitchell, G.F.; Vasan, R.S. Left Ventricular Diastolic Dysfunction in the Community: Impact of Diagnostic Criteria on the Burden, Correlates, and Prognosis. J. Am. Heart Assoc. 2018, 7, e008291. [Google Scholar] [CrossRef] [Green Version]
- Baik, S.K.; Fouad, T.R.; Lee, S.S. Cirrhotic cardiomyopathy. Orphanet J. Rare Dis. 2007, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Glenn, T.K.; Honar, H.; Liu, H.; Ter Keurs, H.E.; Lee, S.S. Role of cardiac myofilament proteins titin and collagen in the pathogenesis of diastolic dysfunction in cirrhotic rats. J. Hepatol. 2011, 55, 1249–1255. [Google Scholar] [CrossRef]
- Li, M.X.; Hwang, P.M. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; George, B.; Alcivar-Franco, D.; Campbell, C.L.; Charnigo, R.; Delisle, B.; Hundley, J.; Darrat, Y.; Morales, G.; Elayi, S.-C.; et al. QT prolongation is associated with increased mortality in end stage liver disease. World J. Cardiol. 2017, 9, 347–354. [Google Scholar] [CrossRef]
- Țieranu, E.; Donoiu, I.; Istrătoaie, O.; Găman, A.; Țieranu, L.; Gheonea, D.; Ciurea, T. Q-T Interval Prolongation in Patients with Liver Cirrhosis. Curr. Health Sci. J. 2018, 44, 274–279. [Google Scholar]
- Genovesi, S.; Pizzala, D.M.P.; Pozzi, M.; Ratti, L.; Milanese, M.; Pieruzzi, F.U.E.G.; Vincenti, A.; Stella, A.; Mancia, G.; Stramba-Badiale, M. QT interval prolongation and decreased heart rate variability in cirrhotic patients: Relevance of hepatic venous pressure gradient and serum calcium. Clin. Sci. 2009, 116, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Hendrickse, M.; Triger, D. Peripheral and cardiovascular autonomic impairment in chronic liver disease: Prevalence and relation to hepatic function. J. Hepatol. 1992, 16, 177–183. [Google Scholar] [CrossRef]
- Licata, A.; Novo, G.; Colomba, D.; Tuttolomondo, A.; Galia, M.; Cammà, C. Cardiac involvement in patients with cirrhosis. J. Cardiovasc. Med. 2016, 17, 26–36. [Google Scholar] [CrossRef]
- Wiese, S.; Mortensen, C.; Gøtze, J.P.; Christensen, E.; Andersen, O.; Bendtsen, F.; Møller, S. Cardiac and proinflammatory markers predict prognosis in cirrhosis. Liver Int. 2014, 34, e19–e30. [Google Scholar] [CrossRef]
- Møller, S.; Danielsen, K.V.; Wiese, S.; Hove, J.; Bendtsen, F. An update on cirrhotic cardiomyopathy. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 497–505. [Google Scholar] [CrossRef]
- Liu, H.; Jayakumar, S.; Traboulsi, M.; Lee, S.S. Cirrhotic cardiomyopathy: Implications for liver transplantation. Liver Transplant. 2017, 23, 826–835. [Google Scholar] [CrossRef] [Green Version]
- Schnell, F.; Donal, E.; Lorho, R.; Lavoué, S.; Gacouin, A.; Compagnon, P.; Boudjema, K.; Mabo, P.; Le Tulzo, Y.; Camus, C. Severe left-sided heart failure early after liver transplantation. Liver Transplant. 2009, 15, 1296–1305. [Google Scholar] [CrossRef]
- Dote, K.; Sato, H.; Tateishi, H.; Uchida, T.; Ishihara, M. [Myocardial stunning due to simultaneous multivessel coronary spasms: A review of 5 cases]. J. Cardiol. 1991, 21, 203–214. [Google Scholar]
- Saner, F.H.; Plicht, B.; Treckmann, J.; Máthé, Z.; Sotiropoulos, G.C.; Radtke, A.; Beckebaum, S.; Cicinnati, V.; Paul, A. Tako-Tsubo syndrome as a rare cause of cardiac failure in liver transplantation. Liver Int. 2010, 30, 159–160. [Google Scholar] [CrossRef]
- Bainbridge, D.; Cheng, D. Stress-induced cardiomyopathy in the perioperative setting. Can. J. Anesth. 2009, 56, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Pilgrim, T.; Wyss, T. Takotsubo cardiomyopathy or transient left ventricular apical ballooning syndrome: A systematic review. Int. J. Cardiol. 2008, 124, 283–292. [Google Scholar] [CrossRef]
- Tomescu, D.R.; Tulbure, D.; Dima, S.; Ungureanu, D.; Popescu, M.; Popescu, I. First Report of Cytokine Removal using CytoSorb® in Severe Noninfectious Inflammatory Syndrome after Liver Transplantation. Int. J. Artif. Organs 2016, 39, 136–140. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Taiteishi, H.; Uchida, T. Takotsubo-type cardiomyopathy due to multivessel spasm. In Clinical Aspect of Myocardial Injury: From Ischemia to Heart Failure; Kodama, K., Haze, K., Hon, M., Eds.; Kagakuhyouronsha: Tokyo, Japan, 1990; pp. 56–64. [Google Scholar]
- Nakano, T.; Onoue, K.; Nakada, Y.; Nakagawa, H.; Kumazawa, T.; Ueda, T.; Nishida, T.; Soeda, T.; Okayama, S.; Watanabe, M.; et al. Alteration of β-Adrenoceptor Signaling in Left Ventricle of Acute Phase Takotsubo Syndrome: A Human Study. Sci. Rep. 2018, 8, 12731. [Google Scholar] [CrossRef]
- Sandhu, G.; Servetnyk, Z.; Croitor, S.; Herzog, E. Atropine aggravates signs and symptoms of Takotsubo cardiomyopathy. Am. J. Emerg. Med. 2010, 28, 258.e5–258.e7. [Google Scholar] [CrossRef]
- Fouad, T.R.; Abdel-Razek, W.; Burak, K.W.; Bain, V.G.; Lee, S.S. Prediction of Cardiac Complications After Liver Transplantation. Transplantation 2009, 87, 763–770. [Google Scholar] [CrossRef]
- Josefsson, A.; Fu, M.; Allayhari, P.; Björnsson, E.S.; Castedal, M.; Olausson, M.; Kalaitzakis, E. Impact of peri-transplant heart failure & left-ventricular diastolic dysfunction on outcomes following liver transplantation. Liver Int. 2012, 32, 1262–1269. [Google Scholar]
- Sonny, A.; Ibrahim, A.; Schuster, A.; Jaber, W.A.; Cywinski, J.B. Impact and persistence of cirrhotic cardiomyopathy after liver transplantation. Clin. Transplant. 2016, 30, 986–993. [Google Scholar] [CrossRef]
- Qureshi, W.; Mittal, C.; Ahmad, U.; Alirhayim, Z.; Hassan, S.; Qureshi, S.; Khalid, F. Clinical predictors of post-liver transplant new-onset heart failure. Liver Transplant. 2013, 19, 701–710. [Google Scholar] [CrossRef]
- Albeldawi, M.; Aggarwal, A.; Madhwal, S.; Cywinski, J.; Lopez, R.; Eghtesad, B.; Zein, N.N. Cumulative risk of cardiovascular events after orthotopic liver transplantation. Liver Transplant. 2012, 18, 370–375. [Google Scholar] [CrossRef]
- Ikegami, T.; Shirabe, K.; Soejima, Y.; Taketomi, A.; Yoshizumi, T.; Uchiyama, H.; Harada, N.; Maehara, Y. The impact of renal replacement therapy before or after living donor liver transplantation. Clin. Transplant. 2011, 26, 143–148. [Google Scholar] [CrossRef]
- Sehgal, L.; Srivastava, P.; Pandey, C.K.; Jha, A. Preoperative cardiovascular investigations in liver transplant candidate: An update. Indian J. Anaesth. 2016, 60, 12–18. [Google Scholar] [CrossRef]
- Sattar, Y.; Siew, K.S.W.; Connerney, M.; Ullah, W.; Alraies, M.C. Management of Takotsubo Syndrome: A Comprehensive Review. Cureus 2020, 12, e6556. [Google Scholar] [CrossRef] [Green Version]
- Petäjä, E.M.; Yki-Järvinen, H. Definitions of Normal Liver Fat and the Association of Insulin Sensitivity with Acquired and Genetic NAFLD—A Systematic Review. Int. J. Mol. Sci. 2016, 17, 633. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, P.; Kowdley, K.V. The Metabolic Syndrome and Its Influence on Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2016, 20, 225–243. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Björnsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motamed, N.; Rabiee, B.; Poustchi, H.; Dehestani, B.; Hemmasi, G.R.; Khansari, M.; Maadi, M.; Saeedian, F.S.; Zamani, F. Non-alcoholic fatty liver disease (NAFLD) and 10-year risk of cardiovascular diseases. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Haddad, T.M.; Hamdeh, S.; Kanmanthareddy, A.; Alla, V.M. Nonalcoholic fatty liver disease and the risk of clinical cardiovascular events: A systematic review and meta-analysis. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, S209–S216. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.N.; Djibre, A.; Farah, R.; Grosovski, M.; Marmor, A. Presence of Coronary Plaques in Patients with Nonalcoholic Fatty Liver Disease 1. Radiology 2010, 254, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Pinarbasi, B.; Demir, K.; Oflaz, H.; Ahishali, E.; Akyuz, F.; Elitok, A.; Cimen, A.O.; Golcuk, E.; Gulluoglu, M.; Issever, H.; et al. Measurement of the coronary flow velocity reserve in patients with non-alcoholic fatty liver disease. Turk. J. Gastroenterol. 2012, 23, 720–726. [Google Scholar] [CrossRef]
- Wan, S.-H.; Vogel, M.W.; Chen, H.H. Pre-clinical diastolic dysfunction. J. Am. Coll. Cardiol. 2013, 63, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Zoppini, G.; Targher, G.; Golia, G.; Bonora, E. Non-alcoholic fatty liver disease is independently associated with left ventricular hypertrophy in hypertensive Type 2 diabetic individuals. J. Endocrinol. Investig. 2012, 35, 215–218. [Google Scholar] [CrossRef]
- Markus, M.R.P.; Baumeister, S.E.; Stritzke, J.; Dörr, M.; Wallaschofski, H.; Völzke, H.; Lieb, W. Hepatic Steatosis Is Associated With Aortic Valve Sclerosis in the General Population. Arter. Thromb. Vasc. Boil. 2013, 33, 1690–1695. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Pernigo, M.; Bergamini, C.; Bonapace, S.; Lipari, P.; Valbusa, F.; Bertolini, L.; Zenari, L.; Pichiri, I.; Dauriz, M.; et al. Heart valve calcification in patients with type 2 diabetes and nonalcoholic fatty liver disease. Metabolism 2015, 64, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A. Nonalcoholic Fatty Liver Disease (NAFLD) and Risk of Cardiac Arrhythmias: A New Aspect of the Liver-heart Axis. J. Clin. Transl. Hepatol. 2017, 5, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Tana, C.; Ballestri, S.; Ricci, F.; Di Vincenzo, A.; Ticinesi, A.; Gallina, S.; Giamberardino, M.A.; Cipollone, F.; Sutton, R.; Vettor, R.; et al. Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease: Mechanisms and Therapeutic Implications. Int. J. Environ. Res. Public Health 2019, 16, 3104. [Google Scholar] [CrossRef] [Green Version]
- Fotbolcu, H.; Zorlu, E. Nonalcoholic fatty liver disease as a multi-systemic disease. World J. Gastroenterol. 2016, 22, 4079–4090. [Google Scholar] [CrossRef]
- Gaziano, J.; Hennekens, C.H.; O’Donnell, C.J.; Breslow, J.L.; Buring, J.E. Fasting Triglycerides, High-Density Lipoprotein, and Risk of Myocardial Infarction. Circulation 1997, 96, 2520–2525. [Google Scholar] [CrossRef]
- Niederreiter, L.; Tilg, H. Cytokines and fatty liver diseases. Liver Res. 2018, 2, 14–20. [Google Scholar] [CrossRef]
- Francque, S.; Van Der Graaff, D.; Kwanten, W. Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J. Hepatol. 2016, 65, 425–443. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lu, H.-Y. Nonalcoholic fatty liver disease and cardiovascular disease. World J. Gastroenterol. 2014, 20, 8407–8415. [Google Scholar] [CrossRef]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef] [Green Version]
- Tariq, Z.; Green, C.J.; Hodson, L. Are oxidative stress mechanisms the common denominator in the progression from hepatic steatosis towards non-alcoholic steatohepatitis (NASH)? Liver Int. 2014, 34, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Polimeni, L.; Del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Semenkovich, C.F. Insulin resistance and atherosclerosis. J. Clin. Investig. 2006, 116, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Joutsi-Korhonen, L.; Sevastianova, K.; Bergholm, R.; Hakkarainen, A.; Pietiläinen, K.H.; Lundbom, N.; Rissanen, A.; Lassila, R.; Yki-Järvinen, H. Increased coagulation factor VIII, IX, XI and XII activities in non-alcoholic fatty liver disease. Liver Int. 2010, 31, 176–183. [Google Scholar] [CrossRef]
- Loeffen, R.; Spronk, H.; Cate, H.T. The impact of blood coagulability on atherosclerosis and cardiovascular disease. J. Thromb. Haemost. 2012, 10, 1207–1216. [Google Scholar] [CrossRef]
- Verrijken, A.; Francque, S.; Mertens, I.; Prawitt, J.; Caron, S.; Hubens, G.; Van Marck, E.; Staels, B.; Michielsen, P.; Van Gaal, L. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2013, 59, 121–129. [Google Scholar] [CrossRef]
- Tofler, G.H.; Massaro, J.; O’Donnell, C.; Wilson, P.; Vasan, R.; Sutherland, P.; Meigs, J.; Levy, D.; D’Agostino, R. Plasminogen activator inhibitor and the risk of cardiovascular disease: The Framingham Heart Study. Thromb. Res. 2016, 140, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Barb, D.; Bril, F.; Kalavalapalli, S.; Cusi, K. Plasma Fibroblast Growth Factor 21 Is Associated with Severity of Nonalcoholic Steatohepatitis in Patients with Obesity and Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 3327–3336. [Google Scholar] [CrossRef]
- Chow, W.; Xu, A.; Woo, Y.C.; Tso, A.W.; Cheung, S.C.; Fong, C.H.; Tse, H.-F.; Chau, M.T.; Cheung, B.M.Y.; Lam, K.S.L. Serum Fibroblast Growth Factor-21 Levels Are Associated With Carotid Atherosclerosis Independent of Established Cardiovascular Risk Factors. Arter. Thromb. Vasc. Boil. 2013, 33, 2454–2459. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Ma, X.; Zhou, J.; Pan, X.; Hao, Y.; Zhou, M.; Lu, Z.; Gao, M.; Bao, Y.; Jia, W. Additive relationship between serum fibroblast growth factor 21 level and coronary artery disease. Cardiovasc. Diabetol. 2013, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Haukeland, J.W.; Dahl, T.B.; Yndestad, A.; Gladhaug, I.P.; Løberg, E.M.; Haaland, T.; Konopski, Z.; Wium, C.; Aasheim, E.T.; Johansen, O.E.; et al. Fetuin A in nonalcoholic fatty liver disease: In vivo and in vitro studies. Eur. J. Endocrinol. 2012, 166, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Weikert, C.; Stefan, N.; Schulze, M.B.; Pischon, T.; Berger, K.; Joost, H.-G.; Häring, H.-U.; Boeing, H.; Fritsche, A. Plasma Fetuin-A Levels and the Risk of Myocardial Infarction and Ischemic Stroke. Circulation 2008, 118, 2555–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.-S.; Adams, L.A.; De Lédinghen, V.; Wong, G.L.-H.; Sookoian, S. Noninvasive biomarkers in NAFLD and NASH—Current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Corey, K.E.; Cannon, C.P.; Blazing, M.; Park, J.-G.; O’Donoghue, M.L.; Chung, R.T.; Giugliano, R.P. The nonalcoholic fatty liver disease (NAFLD) fibrosis score, cardiovascular risk stratification and a strategy for secondary prevention with ezetimibe. Int. J. Cardiol. 2018, 270, 245–252. [Google Scholar] [CrossRef] [PubMed]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Cardio-Metabolic Disorders in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 2215. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; De Bold, A.J. The heart as an endocrine organ. Endocr. Connect. 2014, 3, R31–R44. [Google Scholar] [CrossRef]
- Wu, Y.-S.; Zhu, B.; Luo, A.-L.; Yang, L.; Yang, C. The Role of Cardiokines in Heart Diseases: Beneficial or Detrimental? BioMed Res. Int. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Shimano, M.; Ouchi, N.; Walsh, K. Cardiokines: Recent progress in elucidating the cardiac secretome. Circulation 2012, 126, e327–e332. [Google Scholar] [CrossRef]
- Rashed, H.M.; Nair, B.G.; Patel, T.B. Regulation of hepatic glycolysis and gluconeogenesis by atrial natriuretic peptide. Arch. Biochem. Biophys. 1992, 298, 640–645. [Google Scholar] [CrossRef]
- Jahng, J.W.S.; Song, E.; Sweeney, G. Crosstalk between the heart and peripheral organs in heart failure. Exp. Mol. Med. 2016, 48, e217. [Google Scholar] [CrossRef]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Kawamoto, A.; Tamaki, Y.; Iwanaga, Y.; Soga, T.; Kita, T.; et al. Analysis of liver metabolism in a rat model of heart failure. Int. J. Cardiol. 2012, 161, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Grueter, C.E.; Van Rooij, E.; Johnson, B.A.; DeLeon, S.; Sutherland, L.B.; Qi, X.; Gautron, L.; Elmquist, J.K.; Bassel-Duby, R.; Olson, E.N. A Cardiac MicroRNA Governs Systemic Energy Homeostasis by Regulation of MED13. Cell 2012, 149, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskin, K.K.; E Grueter, C.; Kusminski, C.M.; Holland, W.L.; Bookout, A.L.; Satapati, S.; Kong, Y.M.; Burgess, S.C.; Malloy, C.R.; E Scherer, P.; et al. MED 13-dependent signaling from the heart confers leanness by enhancing metabolism in adipose tissue and liver. EMBO Mol. Med. 2014, 6, 1610–1621. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Heart over mind: Metabolic control of white adipose tissue and liver. EMBO Mol. Med. 2014, 6, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Alter, P.; Glück, T.; Figiel, J.H.; Koczulla, A.R.; Vogelmeier, C.F.; Rupp, H.; Information, P.E.K.F.C. From Heart Failure to Highly Unsaturated Fatty Acid Deficiency and Vice Versa: Bidirectional Heart and Liver Interactions. Can. J. Cardiol. 2016, 32, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Jobe, L.J.; Meléndez, G.C.; Levick, S.P.; Du, Y.; Brower, G.L.; Janicki, J.S. TNF-alpha inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am. J. Physiol. Circ. Physiol. 2009, 297, H1462–H1468. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Relationship between Heart Disease and Liver Disease: A Two-Way Street. Cells 2020, 9, 567. https://doi.org/10.3390/cells9030567
El Hadi H, Di Vincenzo A, Vettor R, Rossato M. Relationship between Heart Disease and Liver Disease: A Two-Way Street. Cells. 2020; 9(3):567. https://doi.org/10.3390/cells9030567
Chicago/Turabian StyleEl Hadi, Hamza, Angelo Di Vincenzo, Roberto Vettor, and Marco Rossato. 2020. "Relationship between Heart Disease and Liver Disease: A Two-Way Street" Cells 9, no. 3: 567. https://doi.org/10.3390/cells9030567