Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives

Abstract
:1. Introduction
2. Mechanistic Concepts of Liver Fibrosis
2.1. Hepatocyte Cell Death and Apoptosis
2.2. HSC Activation and Myofibroblast Progenitor Cells
2.3. Liver Macrophages
2.4. Lymphocytes
2.5. Gut Dysbiosis
2.6. Molecular Signaling Pathways Involved in Liver Fibrogenesis
2.6.1. PDGF Signaling
2.6.2. TGF-β Signaling
2.6.3. Oxidative Stress
2.6.4. The Inflammasome (NLRP3)-Caspase1 Pathway
2.6.5. Wnt/β-Catenin Signaling
3. Disease-Related Pro-fibrogenic Mechanisms in Chronic Liver Diseases
3.1. Chronic Hepatitis C
3.2. Chronic Hepatitis B
3.3. Alcoholic Liver Disease
3.4. Non-Alcoholic Liver Disease
4. Resolution of Liver Fibrosis
4.1. Molecular Mechanisms of Fibrosis Regression
4.2. Candidate Targets and Pathways for Therapeutic Intervention
4.2.1. Hepatic Protection via Inhibition of Apoptosis
4.2.2. Hepatic Protection via Reduction of Oxidative Stress
4.2.3. Hepatic Protection via Restoration of Gut Microbiome
4.2.4. Hepatic Protection via Lipid-Lowering Agents
4.2.5. Inhibition of HSC Activation
4.2.6. Reduction of Fibrotic Scar evolution and Contractility
4.2.7. Immune Modulation
4.2.8. Phytodrugs with Multi-Dimensional Effects on Liver Fibrosis
4.3. From Mouse to Men: Challenges in the Clinical Development of Anti-Fibrotic Compounds
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. New concepts on the clinical course and stratification of compensated and decompensated cirrhosis. Hepatol. Int. 2018, 12, 34–43. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. Clinical states of cirrhosis and competing risks. J. Hepatol. 2018, 68, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. Off. J. Int. Assoc. Study Liver 2018, 38, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Investig. 2007, 117, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Elpek, G.O. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef]
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar] [CrossRef]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, V.; Harris, E.N.; Kidambi, S. SECs (Sinusoidal Endothelial Cells), Liver Microenvironment, and Fibrosis. Biomed. Res. Int 2017, 2017, 4097205. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Barron, L.; Wynn, T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G723–G728. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Fallowfield, J.A.; Mizuno, M.; Kendall, T.J.; Constandinou, C.M.; Benyon, R.C.; Duffield, J.S.; Iredale, J.P. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J. Immunol. 2007, 178, 5288–5295. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Nishikawa, K.; Osawa, Y.; Kimura, K. Wnt/beta-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. Int. J. Mol. Sci. 2018, 19, 3103. [Google Scholar] [CrossRef] [Green Version]
- van Grunsven, L.A. 3D in vitro models of liver fibrosis. Drug Deliv. Rev. 2017, 121, 133–146. [Google Scholar] [CrossRef]
- Van de Bovenkamp, M.; Groothuis, G.M.; Meijer, D.K.; Olinga, P. Liver fibrosis in vitro: Cell culture models and precision-cut liver slices. Toxicol. Vitr. 2007, 21, 545–557. [Google Scholar] [CrossRef]
- Kim, Y.O.; Popov, Y.; Schuppan, D. Optimized Mouse Models for Liver Fibrosis. Methods Mol. Biol 2017, 1559, 279–296. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68-69, 435–451. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef]
- Li, J.; Wang, F.-P.; She, W.-M.; Yang, C.-Q.; Li, L.; Tu, C.-T.; Wang, J.-Y.; Jiang, W. Enhanced high-mobility group box 1 (HMGB1) modulates regulatory T cells (Treg)/T helper 17 (Th17) balance via toll-like receptor (TLR)-4-interleukin (IL)-6 pathway in patients with chronic hepatitis B. J. Viral Hepat. 2014, 21, 129–140. [Google Scholar] [CrossRef]
- Li, J.; Zeng, C.; Zheng, B.; Liu, C.; Tang, M.; Jiang, Y.; Chang, Y.; Song, W.; Wang, Y.; Yang, C. HMGB1-induced autophagy facilitates hepatic stellate cells activation: A new pathway in liver fibrosis. Clin. Sci. 2018, 132, 1645–1667. [Google Scholar] [CrossRef]
- Huebener, P.; Pradere, J.-P.; Hernandez, C.; Gwak, G.-Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Jenkins, R.E.; Antoine, D.J.; Schwabe, R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2015, 125, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Canbay, A.; Higuchi, H.; Bronk, S.F.; Taniai, M.; Sebo, T.J.; Gores, G.J. Fas enhances fibrogenesis in the bile duct ligated mouse: A link between apoptosis and fibrosis. Gastroenterology 2002, 123, 1323–1330. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Zhan, S.-S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive Lipid Species and Metabolic Pathways in Progression and Resolution of Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 282–302 e288. [Google Scholar] [CrossRef]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallee, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, B.; Summers, S.A. Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.C.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Fingas, C.D.; Meng, X.W.; Finnberg, N.; El-Deiry, W.S.; Kaufmann, S.H.; Gores, G.J. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J. Boil. Chem. 2011, 286, 39336–39348. [Google Scholar] [CrossRef] [Green Version]
- Cazanave, S.C.; Wang, X.; Zhou, H.; Rahmani, M.; Grant, S.; Durrant, D.E.; Klaassen, C.D.; Yamamoto, M.; Sanyal, A.J. Degradation of Keap1 activates BH3-only proteins Bim and PUMA during hepatocyte lipoapoptosis. Cell Death Differ. 2014, 21, 1303–1312. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Waldrop, S.L.; Bronk, S.F.; Gores, G.J.; Davis, L.S.; Kilic, G. Lipoapoptosis induced by saturated free fatty acids stimulates monocyte migration: A novel role for Pannexin1 in liver cells. Purinergic Signal. 2015, 11, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Haigh, W.G.; Thorning, D.; Savard, C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J. Lipid Res. 2013, 54, 1326–1334. [Google Scholar] [CrossRef] [Green Version]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164 e110. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testerink, N.; Ajat, M.; Houweling, M.; Brouwers, J.F.; Pully, V.V.; van Manen, H.-J.; Otto, C.; Helms, J.B.; Vaandrager, A.B. Replacement of Retinyl Esters by Polyunsaturated Triacylglycerol Species in Lipid Droplets of Hepatic Stellate Cells during Activation. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 153–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Hazra, S.; Xiong, S.; Wang, J.; Rippe, R.A.; Krishna, V.; Chatterjee, K.; Tsukamoto, H. Peroxisome Proliferator-activated Receptor γ Induces a Phenotypic Switch from Activated to Quiescent Hepatic Stellate Cells. J. Biol. Chem. 2004, 279, 11392–11401. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Dijke, P.t. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [Green Version]
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular Matrix Molecular Remodeling in Human Liver Fibrosis Evolution. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisseleva, T.; Uchinami, H.; Feirt, N.; Quintana-Bustamante, O.; Segovia, J.C.; Schwabe, R.F.; Brenner, D.A. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol. 2006, 45, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G.; Kruglov, E.; Dranoff, J.A. Autocrine release of TGF-β by portal fibroblasts regulates cell growth. FEBS Lett. 2004, 559, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.J.; Russo, F.P.; Rey, V.; Burra, P.; Rugge, M.; Wright, N.A.; Alison, M.R. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology 2004, 126, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts Derive from Hepatocytes in Liver Fibrosis via Epithelial to Mesenchymal Transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [Green Version]
- Beaussier, M.; Wendum, D.; Schiffer, E.; Dumont, S.; Rey, C.; Lienhart, A.; Housset, C. Prominent contribution of portal mesenchymal cells to liver fibrosis in ischemic and obstructive cholestatic injuries. Lab. Invest. 2007, 87, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–3305. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, J.; Asahina, K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial–mesenchymal transition in liver injury. Proc. Natl. Acad. Sci. USA 2013, 110, 2324–2329. [Google Scholar] [CrossRef] [Green Version]
- Baertschiger, R.M.; Serre-Beinier, V.; Morel, P.; Bosco, D.; Peyrou, M.; Clément, S.; Sgroi, A.; Kaelin, A.; Buhler, L.H.; Gonelle-Gispert, C. Fibrogenic Potential of Human Multipotent Mesenchymal Stromal Cells in Injured Liver. PLoS ONE 2009, 4, e6657. [Google Scholar] [CrossRef]
- Watanabe, Y.; Tsuchiya, A.; Seino, S.; Kawata, Y.; Kojima, Y.; Ikarashi, S.; Starkey Lewis, P.J.; Lu, W.Y.; Kikuta, J.; Kawai, H.; et al. Mesenchymal Stem Cells and Induced Bone Marrow-Derived Macrophages Synergistically Improve Liver Fibrosis in Mice. Stem Cells Transl. Med. 2018, 8, 271–284. [Google Scholar] [CrossRef] [Green Version]
- An, S.Y.; Jang, Y.J.; Lim, H.-J.; Han, J.; Lee, J.; Lee, G.; Park, J.Y.; Park, S.-Y.; Kim, J.H.; Do, B.-R.; et al. Milk Fat Globule-EGF Factor 8, Secreted by Mesenchymal Stem Cells, Protects Against Liver Fibrosis in Mice. Gastroenterology 2017, 152, 1174–1186. [Google Scholar] [CrossRef] [Green Version]
- ZHAO, Y.-L.; ZHU, R.-T.; SUN, Y.-L. Epithelial-mesenchymal transition in liver fibrosis. Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taura, K.; Miura, K.; Iwaisako, K.; Österreicher, C.H.; Kodama, Y.; Penz-Österreicher, M.; Brenner, D.A. Hepatocytes Do Not Undergo Epithelial-Mesenchymal Transition in Liver Fibrosis in Mice. Hepatol. (Baltimore, Md.) 2010, 51, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, A.S.; Diaz, R.; Hui, J.-J.; Yanger, K.; Zong, Y.; Alpini, G.; Stanger, B.Z.; Wells, R.G. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatol. (Baltimore, Md.) 2011, 53, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishitsuji, H.; Funami, K.; Shimizu, Y.; Ujino, S.; Sugiyama, K.; Seya, T.; Takaku, H.; Shimotohno, K. Hepatitis C Virus Infection Induces Inflammatory Cytokines and Chemokines Mediated by the Cross Talk between Hepatocytes and Stellate Cells. J. Virol. 2013, 87, 8169–8178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell Death and Cell Death Responses in Liver Disease: Mechanisms and Clinical Relevance. Gastroenterology 2014, 147, 765–783.e764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Bieghs, V.; Hendrikx, T.; Gorp, P.J.V.; Verheyen, F.; Guichot, Y.D.; Walenbergh, S.M.A.; Jeurissen, M.L.J.; Gijbels, M.; Rensen, S.S.; Bast, A.; et al. The Cholesterol Derivative 27-Hydroxycholesterol Reduces Steatohepatitis in Mice. Gastroenterology 2013, 144, 167–178.e161. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Minicis, S.d.; Inokuchi, S.; Taura, K.; Miyai, K.; Rooijen, N.v.; Schwabe, R.F.; Brenner, D.A. CCR2 promotes hepatic fibrosis in mice. Hepatology 2009, 50, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014, 147, 577–594.e571. [Google Scholar] [CrossRef]
- Sahin, H.; Trautwein, C.; Wasmuth, H.E. Functional role of chemokines in liver disease models. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Lee, U.E.; Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Best Pr. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tao, Q.; Sun, M.; Wu, J.Z.; Yang, W.; Jian, P.; Peng, J.; Hu, Y.; Liu, C.; Liu, P. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab. Invest. 2010, 90, 1805–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, A.P.; Salmon, M.; Buckley, C.D.; Adams, D.H. Immune interactions in hepatic fibrosis. or “Leucocyte-stromal interactions in hepatic fibrosis”. Clin. Liver Dis. 2008, 12, 861. [Google Scholar] [CrossRef] [Green Version]
- Muhanna, N.; Horani, A.; Doron, S.; Safadi, R. Lymphocyte–hepatic stellate cell proximity suggests a direct interaction. Clin. Exp. Immunol. 2007, 148, 338–347. [Google Scholar] [CrossRef]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11–mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Li, Z.; Zhang, Q.; Qu, Y.; Xu, M.; Wan, X.; Lu, L. CXCL6-EGFR-induced Kupffer cells secrete TGF-β1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2 pathway in liver fibrosis. J. Cell. Mol. Med. 2018, 22, 5050–5061. [Google Scholar] [CrossRef]
- Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Resolution of liver fibrosis: Basic mechanisms and clinical relevance. Semin. Liver Dis. 2015, 35, 119–131. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [Green Version]
- Barenwaldt, A.; Laubli, H. The sialoglycan-Siglec glyco-immune checkpoint - a target for improving innate and adaptive anti-cancer immunity. Expert Opin. Ther. Targets 2019, 23, 839–853. [Google Scholar] [CrossRef]
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Adham, Z.A.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity 2014, 40, 706–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.-S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship With clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Holt, A.P.; Stamataki, Z.; Adams, D.H. Attenuated liver fibrosis in the absence of B cells. Hepatology 2006, 43, 868–871. [Google Scholar] [CrossRef]
- Safadi, R.; Ohta, M.; Alvarez, C.E.; Fiel, M.I.; Bansal, M.; Mehal, W.Z.; Friedman, S.L. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology 2004, 127, 870–882. [Google Scholar] [CrossRef]
- Novobrantseva, T.I.; Majeau, G.R.; Amatucci, A.; Kogan, S.; Brenner, I.; Casola, S.; Shlomchik, M.J.; Koteliansky, V.; Hochman, P.S.; Ibraghimov, A. Attenuated liver fibrosis in the absence of B cells. J. Clin. Invest. 2005, 115, 3072–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, C. The Roles of Liver-Resident Lymphocytes in Liver Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-Y.; Kubes, P. Leukocyte adhesion in the liver: Distinct adhesion paradigm from other organs. J. Hepatol. 2008, 48, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Patsenker, E.; Stickel, F. Role of integrins in fibrosing liver diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G425–434. [Google Scholar] [CrossRef] [PubMed]
- Curbishley, S.M.; Eksteen, B.; Gladue, R.P.; Lalor, P.; Adams, D.H. CXCR3 Activation Promotes Lymphocyte Transendothelial Migration across Human Hepatic Endothelium under Fluid Flow. Am. J. Pathol. 2005, 167, 887–899. [Google Scholar] [CrossRef] [Green Version]
- Weng, H.-L.; Liu, Y.; Chen, J.-L.; Huang, T.; Xu, L.-J.; Godoy, P.; Hu, J.-H.; Zhou, C.; Stickel, F.; Marx, A.; et al. The etiology of liver damage imparts cytokines transforming growth factor β1 or interleukin-13 as driving forces in fibrogenesis. Hepatology 2009, 50, 230–243. [Google Scholar] [CrossRef]
- Tan, Z.; Qian, X.; Jiang, R.; Liu, Q.; Wang, Y.; Chen, C.; Wang, X.; Ryffel, B.; Sun, B. IL-17A Plays a Critical Role in the Pathogenesis of Liver Fibrosis through Hepatic Stellate Cell Activation. J. Immunol. 2013, 191, 1835–1844. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Su, Y.; Hua, X.; Xie, C.; Liu, J.; Huang, Y.; Zhou, L.; Zhang, M.; Li, X.; Gao, Z. Levels of hepatic Th17 cells and regulatory T cells upregulated by hepatic stellate cells in advanced HBV-related liver fibrosis. J. Transl. Med. 2017, 15, s12967–s13017. [Google Scholar] [CrossRef] [Green Version]
- Tu, J.-F.; Ding, Y.-H.; Ying, X.-H.; Wu, F.-Z.; Zhou, X.-M.; Zhang, D.-K.; Zou, H.; Ji, J.-S. Regulatory T cells, especially ICOS + FOXP3 + regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased Regulatory T Cells Correlate With CD8 T-Cell Impairment and Poor Survival in Hepatocellular Carcinoma Patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef]
- Faggioli, F.; Palagano, E.; Tommaso, L.D.; Donadon, M.; Marrella, V.; Recordati, C.; Mantero, S.; Villa, A.; Vezzoni, P.; Cassani, B. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 2018, 67, 1970–1985. [Google Scholar] [CrossRef] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Licona-Limon, P.; Flavell, R.A. Role of the intestinal microbiome in liver disease. J. Autoimmun 2013, 46, 66–73. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef] [PubMed]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Claudel, T.; Staels, B.; Kuipers, F. The Farnesoid X receptor: A molecular link between bile acid and lipid and glucose metabolism. Arter. Thromb. Vasc. Biol. 2005, 25, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Gunnarsdottir, S.A.; Sadik, R.; Shev, S.; Simren, M.; Sjovall, H.; Stotzer, P.O.; Abrahamsson, H.; Olsson, R.; Bjornsson, E.S. Small intestinal motility disturbances and bacterial overgrowth in patients with liver cirrhosis and portal hypertension. Am. J. Gastroenterol. 2003, 98, 1362–1370. [Google Scholar] [CrossRef]
- Kakiyama, G.; Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Heuman, D.M.; Kang, D.J.; Takei, H.; Nittono, H.; Ridlon, J.M.; Fuchs, M.; et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am. J. Physiol. Liver Physiol. 2014, 306, G929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teltschik, Z.; Wiest, R.; Beisner, J.; Nuding, S.; Hofmann, C.; Schoelmerich, J.; Bevins, C.L.; Stange, E.F.; Wehkamp, J. Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology 2012, 55, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; Lario, M.; Alvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Idilman, R.; Mabudian, L.; Hood, M.; Fagan, A.; Turan, D.; White, M.B.; Karakaya, F.; Wang, J.; Atalay, R.; et al. Diet affects gut microbiota and modulates hospitalization risk differentially in an international cirrhosis cohort. Hepatology 2018, 68, 234–247. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Hylemon, P.B.; Ridlon, J.M.; Heuman, D.M.; Daita, K.; White, M.B.; Monteith, P.; Noble, N.A.; Sikaroodi, M.; Gillevet, P.M. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Liver Physiol. 2012, 303, G675–685. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef]
- Breitkopf, K.; Roeyen, C.V.; Sawitza, I.; Wickert, L.; Floege, J.; Gressner, A.M. Expression patterns of PDGF-A, -B, -C and -D and the PDGF-receptors α and β in activated rat hepatic stellate cells (HSC). Cytokine 2005, 31, 349–357. [Google Scholar] [CrossRef]
- Czochra, P.; Klopcic, B.; Meyer, E.; Herkel, J.; Garcia-Lazaro, J.F.; Thieringer, F.; Schirmacher, P.; Biesterfeld, S.; Galle, P.R.; Lohse, A.W.; et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J. Hepatol. 2006, 45, 419–428. [Google Scholar] [CrossRef]
- Campbell, J.S.; Hughes, S.D.; Gilbertson, D.G.; Palmer, T.E.; Holdren, M.S.; Haran, A.C.; Odell, M.M.; Bauer, R.L.; Ren, H.-P.; Haugen, H.S.; et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 3389–3394. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.J.; Riehle, K.J.; Shimizu-Albergine, M.; Bauer, R.L.; Hudkins, K.L.; Johansson, F.; Yeh, M.M.; Jr, W.M.M.; Yeung, R.S.; Campbell, J.S. Activation of Platelet-Derived Growth Factor Receptor Alpha Contributes to Liver Fibrosis. PLoS ONE 2014, 9, e92925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.-C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dropmann, A.; Dediulia, T.; Breitkopf-Heinlein, K.; Korhonen, H.; Janicot, M.; Weber, S.N.; Thomas, M.; Piiper, A.; Bertran, E.; Fabregat, I.; et al. TGF-β1 and TGF-β2 abundance in liver diseases of mice and men. Oncotarget 2016, 7, 19499–19518. [Google Scholar] [CrossRef] [Green Version]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; Xia, L.; Ahamed, J. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014, 59, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Zhou, D.; Meng, X.; Wang, X.; Liu, C.; Huang, C.; Li, J.; Zhang, L. Smad2 increases the apoptosis of activated human hepatic stellate cells induced by TRAIL. Int. Immunopharmacol. 2016, 32, 76–86. [Google Scholar] [CrossRef]
- Inagaki, Y.; Okazaki, I. Emerging insights into Transforming growth factor β Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-H. Fine Tuning and Cross-talking of TGF-β Signal by Inhibitory Smads. BMB Rep. 2005, 38, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Dooley, S.; Hamzavi, J.; Ciuclan, L.; Godoy, P.; Ilkavets, I.; Ehnert, S.; Ueberham, E.; Gebhardt, R.; Kanzler, S.; Geier, A.; et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008, 135, 642–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, W.; Hu, H.; Tang, N.; Zhang, C.; Liang, W.; Wang, M. Smad3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-beta1 in cultured rat hepatic stellate cells. Pharm Res. 2006, 23, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Aquino, E.; Zarco, N.; Casas-Grajales, S.; Ramos-Tovar, E.; Flores-Beltran, R.E.; Arauz, J.; Shibayama, M.; Favari, L.; Tsutsumi, V.; Segovia, J.; et al. Naringenin prevents experimental liver fibrosis by blocking TGFbeta-Smad3 and JNK-Smad3 pathways. World J. Gastroenterol. 2017, 23, 4354–4368. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Murata, M.; Yamaguchi, T.; Matsuzaki, K.; Okazaki, K. Reversible Human TGF-beta Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions. J. Clin. Med. 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Valle, V.; Chávez-Tapia, N.C.; Uribe, M.; Méndez-Sánchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; James Ou, J.-H. Mechanisms of Liver Injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Liver Physiol. 2006, 290, G847–G851. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2016, 8, 3895–3932. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Sancho-bru, P.; Ginès, P.; Lora, J.M.; Al-garawi, A.; Solé, M.; Colmenero, J.; Nicolás, J.M.; Jiménez, W.; Weich, N.; et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 2003, 125, 117–125. [Google Scholar] [CrossRef]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernández-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH Oxidase NOX4 Mediates Stellate Cell Activation and Hepatocyte Cell Death during Liver Fibrosis Development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.-I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte NADPH Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schroder, K.; Brandes, R.P.; Devaraj, S.; Torok, N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Muriel, P. NF-κB in liver diseases: A target for drug therapy. J. Appl. Toxicol. 2009, 29, 91–100. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Mann, J. Epigenetics and Liver Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Götze, S.; Schumacher, E.C.; Kordes, C.; Häussinger, D. Epigenetic Changes during Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0128745. [Google Scholar] [CrossRef] [Green Version]
- El Taghdouini, A.; Sørensen, A.L.; Reiner, A.H.; Coll, M.; Verhulst, S.; Mannaerts, I.; Øie, C.I.; Smedsrød, B.; Najimi, M.; Sokal, E.; et al. Genome-wide analysis of DNA methylation and gene expression patterns in purified, uncultured human liver cells and activated hepatic stellate cells. Oncotarget 2015, 6, 26729–26745. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, Y.; Waku, T.; Iwasaki, N.; Ono, W.; Yamaguchi, C.; Yanagisawa, J. Global analysis of DNA methylation in early-stage liver fibrosis. BMC Med. Genom. 2012, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. (Berl) 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Yang, L.; van Rooijen, N.; Brenner, D.A.; Ohnishi, H.; Seki, E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013, 57, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; DeLanghe, S.; Al Alam, D.; Utley, S.; Estrada, J.; Wang, K.S. beta-catenin regulates mesenchymal progenitor cell differentiation during hepatogenesis. J. Surg. Res. 2010, 164, 276–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavanchy, D. The global burden of hepatitis C. L Liver Int. 2009, 29 (Suppl. 1), 74–81. [Google Scholar] [CrossRef]
- Lingala, S.; Ghany, M.G. Natural History of Hepatitis, C. Gastroenterol. Clin. North. Am. 2015, 44, 717–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection and Remaining Challenges. Gastroenterology 2019, 156, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Jacobson Brown, P.M.; Neuman, M.G. Immunopathogenesis of hepatitis C viral infection: Th1/Th2 responses and the role of cytokines. Clin. Biochem. 2001, 34, 167–171. [Google Scholar] [CrossRef]
- Boltjes, A.; Movita, D.; Boonstra, A.; Woltman, A.M. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J. Hepatol. 2014, 61, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Baskic, D.; Vukovic, V.; Popovic, S.; Jovanovic, D.; Mitrovic, S.; Djurdjevic, P.; Avramovic, D.; Arsovic, A.; Bankovic, D.; Cukic, J.; et al. Chronic Hepatitis C: Conspectus of immunological events in the course of fibrosis evolution. PLoS ONE 2019, 14, e0219508. [Google Scholar] [CrossRef] [Green Version]
- Schulze-Krebs, A.; Preimel, D.; Popov, Y.; Bartenschlager, R.; Lohmann, V.; Pinzani, M.; Schuppan, D. Hepatitis C virus-replicating hepatocytes induce fibrogenic activation of hepatic stellate cells. Gastroenterology 2005, 129, 246–258. [Google Scholar] [CrossRef]
- Bataller, R.; Paik, Y.H.; Lindquist, J.N.; Lemasters, J.J.; Brenner, D.A. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology 2004, 126, 529–540. [Google Scholar] [CrossRef]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C Virus Core Protein Inhibits Mitochondrial Electron Transport and Increases Reactive Oxygen Species (ROS) Production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [Green Version]
- Okuda, M.; Li, K.; Beard, M.R.; Showalter, L.A.; Scholle, F.; Lemon, S.M.; Weinman, S.A. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002, 122, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Thorén, F.; Romero, A.; Lindh, M.; Dahlgren, C.; Hellstrand, K. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: Role of NADPH oxidase-derived oxygen radicals. J. Leukoc. Biol. 2004, 76, 1180–1186. [Google Scholar] [CrossRef]
- Mazzocca, A.; Sciammetta, S.C.; Carloni, V.; Cosmi, L.; Annunziato, F.; Harada, T.; Abrignani, S.; Pinzani, M. Binding of hepatitis C virus envelope protein E2 to CD81 up-regulates matrix metalloproteinase-2 in human hepatic stellate cells. J. Boil. Chem. 2005, 280, 11329–11339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoudjehane, L.; Bisch, G.; Scatton, O.; Granier, C.; Gaston, J.; Housset, C.; Roingeard, P.; Cosset, F.L.; Perdigao, F.; Balladur, P.; et al. Infection of Human Liver Myofibroblasts by Hepatitis C Virus: A Direct Mechanism of Liver Fibrosis in Hepatitis C. PLoS ONE 2015, 10, e0134141. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005 e1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwal, F.; Kramer, J.R.; Asch, S.M.; Cao, Y.; Li, L.; El-Serag, H.B. Long-Term Risk of Hepatocellular Carcinoma in HCV Patients Treated With Direct Acting Antiviral Agents. Hepatology 2020, 71, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Hamdane, N.; Juhling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Roca Suarez, A.A.; et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019, 156, 2313–2329.e2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, S.; Kaspi, A.; Domovitz, T.; Davidovich, A.; Lavi-Itzkovitz, A.; Meirson, T.; Alison Holmes, J.; Dai, C.Y.; Huang, C.F.; Chung, R.T.; et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019, 15, e1008181. [Google Scholar] [CrossRef]
- Lohmann, V.; Bartenschlager, R. Indelibly Stamped by Hepatitis C Virus Infection: Persistent Epigenetic Signatures Increasing Liver Cancer Risk. Gastroenterology 2019, 156, 2130–2133. [Google Scholar] [CrossRef] [Green Version]
- Childs, L.; Roesel, S.; Tohme, R.A. Status and progress of hepatitis B control through vaccination in the South-East Asia Region, 1992-2015. Vaccine 2018, 36, 6–14. [Google Scholar] [CrossRef]
- Milich, D.; Liang, T.J. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 2003, 38, 1075–1086. [Google Scholar] [CrossRef]
- Shin, E.C.; Sung, P.S.; Park, S.H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef]
- Tang, C.M.; Yau, T.O.; Yu, J. Management of chronic hepatitis B infection: Current treatment guidelines, challenges, and new developments. World J. Gastroenterol. 2014, 20, 6262–6278. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Tu, W.; Han, J.; He, J.; Liu, J.; Han, P.; Wang, Y.; Li, M.; Liu, M.; Liao, J.; et al. Hepatic SATB1 induces paracrine activation of hepatic stellate cells and is upregulated by HBx. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. Hepatitis B virus infects hepatic stellate cells and affects their proliferation and expression of collagen type I. Chin. Med J. 2009, 122, 1455–1461. [Google Scholar] [PubMed]
- Martinez, M.G.; Villeret, F.; Testoni, B.; Zoulim, F. Can we cure hepatitis B virus with novel direct-acting antivirals? Liver Int. 2020, 40 (Suppl. 1), 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Cytochrome P450 2E1-derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. J. Boil. Chem. 2002, 277, 9853–9864. [Google Scholar] [CrossRef] [Green Version]
- Svegliati-Baroni, G.; Inagaki, Y.; Rincon-Sanchez, A.R.; Else, C.; Saccomanno, S.; Benedetti, A.; Ramirez, F.; Rojkind, M. Early response of alpha2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology 2005, 42, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Greenwel, P.; Dominguez-Rosales, J.A.; Mavi, G.; Rivas-Estilla, A.M.; Rojkind, M. Hydrogen peroxide: A link between acetaldehyde-elicited alpha1(I) collagen gene up-regulation and oxidative stress in mouse hepatic stellate cells. Hepatology 2000, 31, 109–116. [Google Scholar] [CrossRef]
- Natori, S.; Rust, C.; Stadheim, L.M.; Srinivasan, A.; Burgart, L.J.; Gores, G.J. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J. Hepatol. 2001, 34, 248–253. [Google Scholar] [CrossRef]
- Faouzi, S.; Burckhardt, B.E.; Hanson, J.C.; Campe, C.B.; Schrum, L.W.; Rippe, R.A.; Maher, J.J. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J. Biol. Chem. 2001, 276, 49077–49082. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, M.D.; Kono, H.; Yin, M.; Nakagami, M.; Uesugi, T.; Arteel, G.E.; Gabele, E.; Rusyn, I.; Yamashina, S.; Froh, M.; et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic. Biol. Med. 2001, 31, 1544–1549. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. TNF-alpha-induced cell death in ethanol-exposed cells depends on p38 MAPK signaling but is independent of Bid and caspase-8. Am. J. Physiol. Liver Physiol. 2003, 285, G503–516. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, M.; Tsukamoto, H. Stimulation of hepatic lipocyte collagen production by Kupffer cell-derived transforming growth factor beta: Implication for a pathogenetic role in alcoholic liver fibrogenesis. Hepatology 1990, 11, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.T. Alcohol abuse, alcoholism, and damage to the immune system—A review. Alcohol. Clin. Exp. Res. 1998, 22, 1927–1942. [Google Scholar]
- Guo, T.L.; Zhang, L.X.; Chen, J.P.; Nguyen, V.A.; White, K.L., Jr.; Gao, B. Differential STAT5 activation and phenotypic marker expression by immune cells following low levels of ethanol consumption in mice. Immunopharmacol. Immunotoxicol. 2002, 24, 121–138. [Google Scholar] [CrossRef]
- Collier, S.D.; Pruett, S.B. Mechanisms of suppression of poly I:C-induced activation of NK cells by ethanol. Alcohology 2000, 21, 87–95. [Google Scholar] [CrossRef]
- Inagaki, Y.; Nemoto, T.; Kushida, M.; Sheng, Y.; Higashi, K.; Ikeda, K.; Kawada, N.; Shirasaki, F.; Takehara, K.; Sugiyama, K.; et al. Interferon alfa down-regulates collagen gene transcription and suppresses experimental hepatic fibrosis in mice. Hepatology 2003, 38, 890–899. [Google Scholar] [CrossRef]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef]
- Friedman, S.L. Mac the knife? Macrophages- the double-edged sword of hepatic fibrosis. J. Clin. Investig. 2005, 115, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; an international consensus panel. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef]
- Wong, V.W.; Chitturi, S.; Wong, G.L.; Yu, J.; Chan, H.L.; Farrell, G.C. Pathogenesis and novel treatment options for non-alcoholic steatohepatitis. Lancet Gastroenterol. Hepatol. 2016, 1, 56–67. [Google Scholar] [CrossRef]
- Sell, S. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatology 2001, 33, 738–750. [Google Scholar] [CrossRef]
- Roskams, T.A.; Theise, N.D.; Balabaud, C.; Bhagat, G.; Bhathal, P.S.; Bioulac-Sage, P.; Brunt, E.M.; Crawford, J.M.; Crosby, H.A.; Desmet, V.; et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology 2004, 39, 1739–1745. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; De Minicis, S.; Marzioni, M. Hepatic fibrogenesis in response to chronic liver injury: Novel insights on the role of cell-to-cell interaction and transition. Liver Int. 2008, 28, 1052–1064. [Google Scholar] [CrossRef]
- Xia, J.L.; Dai, C.; Michalopoulos, G.K.; Liu, Y. Hepatocyte growth factor attenuates liver fibrosis induced by bile duct ligation. Am. J. Pathol. 2006, 168, 1500–1512. [Google Scholar] [CrossRef] [Green Version]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Van Hul, N.K.; Abarca-Quinones, J.; Sempoux, C.; Horsmans, Y.; Leclercq, I.A. Relation between liver progenitor cell expansion and extracellular matrix deposition in a CDE-induced murine model of chronic liver injury. Hepatology 2009, 49, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M. Challenges and opportunities in drug development for nonalcoholic steatohepatitis. Eur. J. Pharmacol. 2020, 870, 172913. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e310. [Google Scholar] [CrossRef] [Green Version]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Stepanova, M.; Rafiq, N.; Makhlouf, H.; Younoszai, Z.; Agrawal, R.; Goodman, Z. Pathologic criteria for nonalcoholic steatohepatitis: Interprotocol agreement and ability to predict liver-related mortality. Hepatology 2011, 53, 1874–1882. [Google Scholar] [CrossRef]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Aguilar Schall, R.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- D’Ambrosio, R.; Aghemo, A.; Rumi, M.G.; Ronchi, G.; Donato, M.F.; Paradis, V.; Colombo, M.; Bedossa, P. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology 2012, 56, 532–543. [Google Scholar] [CrossRef]
- Levrero, M.; Subic, M.; Villeret, F.; Zoulim, F. Perspectives and limitations for nucleo(t)side analogs in future HBV therapies. Curr. Opin. Virol. 2018, 30, 80–89. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Troeger, J.S.; Mederacke, I.; Gwak, G.Y.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.P.; Friedman, R.A.; Schwabe, R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012, 143, 1073–1083 e1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, H.D.; Otano, I.; Rombouts, K.; Singh, K.P.; Peppa, D.; Gill, U.S.; Böttcher, K.; Kennedy, P.T.F.; Oben, J.; Pinzani, M.; et al. TRAIL regulatory receptors constrain human hepatic stellate cell apoptosis. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Arabpour, M.; Cool, R.H.; Faber, K.N.; Quax, W.J.; Haisma, H.J. Receptor-specific TRAIL as a means to achieve targeted elimination of activated hepatic stellate cells. J. Drug Target. 2017, 25, 360–369. [Google Scholar] [CrossRef]
- Park, S.-J.; Sohn, H.-Y.; Yoon, J.; Park, S.I. Down-regulation of FoxO-dependent c-FLIP expression mediates TRAIL-induced apoptosis in activated hepatic stellate cells. Cell. Signal. 2009, 21, 1495–1503. [Google Scholar] [CrossRef]
- Fukushima, J.; Kamada, Y.; Matsumoto, H.; Yoshida, Y.; Ezaki, H.; Takemura, T.; Saji, Y.; Igura, T.; Tsutsui, S.; Kihara, S.; et al. Adiponectin prevents progression of steatohepatitis in mice by regulating oxidative stress and Kupffer cell phenotype polarization. Hepatol. Res. 2009, 39, 724–738. [Google Scholar] [CrossRef]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like Receptors in Microbial Recognition and Host Defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Glässner, A.; Eisenhardt, M.; Kokordelis, P.; Krämer, B.; Wolter, F.; Nischalke, H.D.; Boesecke, C.; Sauerbruch, T.; Rockstroh, J.K.; Spengler, U.; et al. Impaired CD4+ T cell stimulation of NK cell anti-fibrotic activity may contribute to accelerated liver fibrosis progression in HIV/HCV patients. J. Hepatol. 2013, 59, 427–433. [Google Scholar] [CrossRef]
- Muhanna, N.; Tair, L.A.; Doron, S.; Amer, J.; Azzeh, M.; Mahamid, M.; Friedman, S.; Safadi, R. Amelioration of hepatic fibrosis by NK cell activation. Gut 2011, 60, 90–98. [Google Scholar] [CrossRef]
- Melhem, A.; Muhanna, N.; Bishara, A.; Alvarez, C.E.; Ilan, Y.; Bishara, T.; Horani, A.; Nassar, M.; Friedman, S.L.; Safadi, R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J. Hepatol. 2006, 45, 60–71. [Google Scholar] [CrossRef] [PubMed]
- JEONG, W.I.; PARK, O.; GAO, B. Abrogation of the Antifibrotic Effects of Natural Killer Cells/Interferon-γ Contributes to Alcohol Acceleration of Liver Fibrosis. Gastroenterology 2008, 134, 248–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gur, C.; Doron, S.; Kfir-Erenfeld, S.; Horwitz, E.; Abu-tair, L.; Safadi, R.; Mandelboim, O. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut 2012, 61, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Gao, B. Natural Killer Cells in Liver Disease. Hepatol. (Baltimore, Md.) 2013, 57, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.B.; Chamroonkul, N. Antifibrotics in liver disease: Are we getting closer to clinical use? Hepatol. Int. 2019, 13, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Latief, U.; Ahmad, R. Herbal remedies for liver fibrosis: A review on the mode of action of fifty herbs. J. Tradit Complement. Med. 2018, 8, 352–360. [Google Scholar] [CrossRef]
- Duval, F.; Moreno-Cuevas, J.E.; Gonzalez-Garza, M.T.; Maldonado-Bernal, C.; Cruz-Vega, D.E. Liver fibrosis and mechanisms of the protective action of medicinal plants targeting inflammation and the immune response. Int. J. Inflam. 2015, 2015, 943497. [Google Scholar] [CrossRef] [Green Version]
- Neong, S.F.; Adebayo, D.; Wong, F. An update on the pathogenesis and clinical management of cirrhosis with refractory ascites. Expert. Rev. Gastroenterol. Hepatol. 2019, 13, 293–305. [Google Scholar] [CrossRef]
- Wree, A.; Mehal, W.Z.; Feldstein, A.E. Targeting Cell Death and Sterile Inflammation Loop for the Treatment of Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2016, 36, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Thapaliya, S.; Wree, A.; Povero, D.; Inzaugarat, M.E.; Berk, M.; Dixon, L.; Papouchado, B.G.; Feldstein, A.E. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Dig. Dis. Sci. 2014, 59, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witek, R.P.; Stone, W.C.; Karaca, F.G.; Syn, W.K.; Pereira, T.A.; Agboola, K.M.; Omenetti, A.; Jung, Y.; Teaberry, V.; Choi, S.S.; et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Manicardi, N.; Ortega-Ribera, M.; Maeso-Diaz, R.; Guixe-Muntet, S.; Fernandez-Iglesias, A.; Hide, D.; Garcia-Caldero, H.; Boyer-Diaz, Z.; Contreras, P.C.; et al. Emricasan Ameliorates Portal Hypertension and Liver Fibrosis in Cirrhotic Rats Through a Hepatocyte-Mediated Paracrine Mechanism. Hepatol. Commun. 2019, 3, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Goodman, Z.; Jabbar, A.; Vemulapalli, R.; Younes, Z.H.; Freilich, B.; Sheikh, M.Y.; Schattenberg, J.M.; Kayali, Z.; Zivony, A.; et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol. 2019. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Bosch, J.; Kayali, Z.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.; Satapathy, S.K.; Ghabril, M.; Shiffman, M.L.; Younes, Z.H.; et al. Randomized Placebo-Controlled Trial of Emricasan in Non-alcoholic Steatohepatitis (NASH) Cirrhosis with Severe Portal Hypertension. J. Hepatol. 2019. [Google Scholar] [CrossRef]
- Mehta, G.; Rousell, S.; Burgess, G.; Morris, M.; Wright, G.; McPherson, S.; Frenette, C.; Cave, M.; Hagerty, D.T.; Spada, A.; et al. A Placebo-Controlled, Multicenter, Double-Blind, Phase 2 Randomized Trial of the Pan-Caspase Inhibitor Emricasan in Patients with Acutely Decompensated Cirrhosis. J. Clin. Exp. Hepatol. 2018, 8, 224–234. [Google Scholar] [CrossRef]
- Budas, G.; Karnik, S.; Jonnson, T.; Watkins, S.; Breckenridge, D. Reduction of liver steatosis and fibrosis with an ASK1 inhibitor in a murine model of NASH is accomplished by improvements in cholesterol, bile acid and lipid metabolism. J. Hepatol. 2016, 64, S170. [Google Scholar] [CrossRef]
- Yamamoto, E.; Dong, Y.F.; Kataoka, K.; Yamashita, T.; Tokutomi, Y.; Matsuba, S.; Ichijo, H.; Ogawa, H.; Kim-Mitsuyama, S. Olmesartan prevents cardiovascular injury and hepatic steatosis in obesity and diabetes, accompanied by apoptosis signal regulating kinase-1 inhibition. Hypertension 2008, 52, 573–580. [Google Scholar] [CrossRef]
- Wang, P.X.; Ji, Y.X.; Zhang, X.J.; Zhao, L.P.; Yan, Z.Z.; Zhang, P.; Shen, L.J.; Yang, X.; Fang, J.; Tian, S.; et al. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat. Med. 2017, 23, 439–449. [Google Scholar] [CrossRef]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, Q.; Mo, W.; Feng, J.; Li, S.; Li, J.; Liu, T.; Xu, S.; Wang, W.; Lu, X.; et al. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-beta1/Smads and PI3K/Akt pathways. Sci. Rep. 2017, 7, 9289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Lou, Q.; Wang, F.; Li, E.; Sun, J.; Fang, H.; Xi, J.; Ju, L. N-acetylcysteine protects against liver injure induced by carbon tetrachloride via activation of the Nrf2/HO-1 pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 8655–8662. [Google Scholar] [PubMed]
- Kessoku, T.; Imajo, K.; Honda, Y.; Kato, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; Mawatari, H.; Fujita, K.; Yoneda, M.; et al. Resveratrol ameliorates fibrosis and inflammation in a mouse model of nonalcoholic steatohepatitis. Sci. Rep. 2016, 6, 22251. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Osterreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, T.; Paik, Y.H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef] [Green Version]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Radovanovic Spurnic, A.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N.; et al. Gut-Liver Axis, Gut Microbiota, and Its Modulation in the Management of Liver Diseases: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef] [Green Version]
- Ferrere, G.; Wrzosek, L.; Cailleux, F.; Turpin, W.; Puchois, V.; Spatz, M.; Ciocan, D.; Rainteau, D.; Humbert, L.; Hugot, C.; et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 2017, 66, 806–815. [Google Scholar] [CrossRef]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatol. 2003, 37, 343–350. [Google Scholar] [CrossRef]
- Chen, L.; Pan, D.D.; Zhou, J.; Jiang, Y.Z. Protective effect of selenium-enriched Lactobacillus on CCl4-induced liver injury in mice and its possible mechanisms. World J. Gastroenterol. 2005, 11, 5795–5800. [Google Scholar] [CrossRef]
- Velayudham, A.; Dolganiuc, A.; Ellis, M.; Petrasek, J.; Kodys, K.; Mandrekar, P.; Szabo, G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 2009, 49, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.Y.; Li, L.; Yu, C.H.; Shen, Z.; Chen, L.H.; Li, Y.M. Effects of probiotics on nonalcoholic fatty liver disease: A meta-analysis. World J. Gastroenterol. 2013, 19, 6911–6918. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Zhang, Y.; Huang, X.B.; You, N.; Zheng, L.; Li, J. Fecal microbiota transplantation prevents hepatic encephalopathy in rats with carbon tetrachloride-induced acute hepatic dysfunction. World J. Gastroenterol. 2017, 23, 6983–6994. [Google Scholar] [CrossRef] [PubMed]
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A.; et al. Healthy Donor Fecal Microbiota Transplantation in Steroid-Ineligible Severe Alcoholic Hepatitis: A Pilot Study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Kassam, Z.; Fagan, A.; Gavis, E.A.; Liu, E.; Cox, I.J.; Kheradman, R.; Heuman, D.; Wang, J.; Gurry, T.; et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology 2017, 66, 1727–1738. [Google Scholar] [CrossRef]
- DeFilipp, Z.; Bloom, P.P.; Torres Soto, M.; Mansour, M.K.; Sater, M.R.A.; Huntley, M.H.; Turbett, S.; Chung, R.T.; Chen, Y.B.; Hohmann, E.L.; et al. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N. Engl. J. Med. 2019, 381, 2043–2050. [Google Scholar] [CrossRef]
- Chou, R.; Dana, T.; Blazina, I.; Daeges, M.; Jeanne, T.L. Statins for Prevention of Cardiovascular Disease in Adults: Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2016, 316, 2008–2024. [Google Scholar] [CrossRef]
- Schierwagen, R.; Maybuchen, L.; Zimmer, S.; Hittatiya, K.; Back, C.; Klein, S.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5, 12931. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Garcia-Caldero, H.; Hide, D.; Marrone, G.; Guixe-Muntet, S.; Peralta, C.; Garcia-Pagan, J.C.; Abraldes, J.G.; Bosch, J. Simvastatin maintains function and viability of steatotic rat livers procured for transplantation. J. Hepatol. 2013, 58, 1140–1146. [Google Scholar] [CrossRef]
- Schierwagen, R.; Maybuchen, L.; Hittatiya, K.; Klein, S.; Uschner, F.E.; Braga, T.T.; Franklin, B.S.; Nickenig, G.; Strassburg, C.P.; Plat, J.; et al. Statins improve NASH via inhibition of RhoA and Ras. Am. J. Physiol. Liver Physiol. 2016, 311, G724–G733. [Google Scholar] [CrossRef] [PubMed]
- Pose, E.; Trebicka, J.; Mookerjee, R.P.; Angeli, P.; Gines, P. Statins: Old drugs as new therapy for liver diseases? J. Hepatol. 2019, 70, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, L.W.; Hsu, Y.C.; Lee, T.F.; Lin, Y.; Chiu, Y.T.; Yang, K.C.; Wu, J.C.; Huang, Y.T. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015, 15, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arab, J.P.; Shah, V.H. Statins and portal hypertension: A tale of two models. Hepatology 2016, 63, 2044–2047. [Google Scholar] [CrossRef]
- Moreno, M.; Ramalho, L.N.; Sancho-Bru, P.; Ruiz-Ortega, M.; Ramalho, F.; Abraldes, J.G.; Colmenero, J.; Dominguez, M.; Egido, J.; Arroyo, V.; et al. Atorvastatin attenuates angiotensin II-induced inflammatory actions in the liver. Am. J. Physiol. Liver Physiol. 2009, 296, G147–156. [Google Scholar] [CrossRef] [PubMed]
- Kamal, S.; Khan, M.A.; Seth, A.; Cholankeril, G.; Gupta, D.; Singh, U.; Kamal, F.; Howden, C.W.; Stave, C.; Nair, S.; et al. Beneficial Effects of Statins on the Rates of Hepatic Fibrosis, Hepatic Decompensation, and Mortality in Chronic Liver Disease: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2017, 112, 1495–1505. [Google Scholar] [CrossRef]
- Pollo-Flores, P.; Soldan, M.; Santos, U.C.; Kunz, D.G.; Mattos, D.E.; da Silva, A.C.; Marchiori, R.C.; Rezende, G.F. Three months of simvastatin therapy vs. placebo for severe portal hypertension in cirrhosis: A randomized controlled trial. Dig. Liver Dis. 2015, 47, 957–963. [Google Scholar] [CrossRef]
- Chang, F.M.; Wang, Y.P.; Lang, H.C.; Tsai, C.F.; Hou, M.C.; Lee, F.Y.; Lu, C.L. Statins decrease the risk of decompensation in hepatitis B virus- and hepatitis C virus-related cirrhosis: A population-based study. Hepatology 2017, 66, 896–907. [Google Scholar] [CrossRef] [Green Version]
- Abraldes, J.G.; Albillos, A.; Banares, R.; Turnes, J.; Gonzalez, R.; Garcia-Pagan, J.C.; Bosch, J. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: A randomized controlled trial. Gastroenterology 2009, 136, 1651–1658. [Google Scholar] [CrossRef]
- Naci, H.; Brugts, J.J.; Fleurence, R.; Tsoi, B.; Toor, H.; Ades, A.E. Comparative benefits of statins in the primary and secondary prevention of major coronary events and all-cause mortality: A network meta-analysis of placebo-controlled and active-comparator trials. Eur. J. Prev Cardiol. 2013, 20, 641–657. [Google Scholar] [CrossRef]
- Pose, E.; Napoleone, L.; Amin, A.; Campion, D.; Jimenez, C.; Piano, S.; Roux, O.; Uschner, F.E.; de Wit, K.; Zaccherini, G.; et al. Safety of two different doses of simvastatin plus rifaximin in decompensated cirrhosis (LIVERHOPE-SAFETY): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 31–41. [Google Scholar] [CrossRef]
- Cheng, J.H.; She, H.; Han, Y.P.; Wang, J.; Xiong, S.; Asahina, K.; Tsukamoto, H. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am. J. Physiol. Liver Physiol. 2008, 294, G39–49. [Google Scholar] [CrossRef] [PubMed]
- Kordes, C.; Sawitza, I.; Haussinger, D. Canonical Wnt signaling maintains the quiescent stage of hepatic stellate cells. Biochem. Biophys. Res. Commun. 2008, 367, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Kim, H.Y.; Moon, S.H.; et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcora, B.O.; Storm, G.; Bansal, R. Inhibition of canonical WNT signaling pathway by beta-catenin/CBP inhibitor ICG-001 ameliorates liver fibrosis in vivo through suppression of stromal CXCL12. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R., Jr.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; He, W.; Li, Y.; Ding, H.; Hou, Y.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. J. Am. Soc. Nephrol 2011, 22, 1642–1653. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Osawa, Y.; Ohtsuki, T.; Hayashi, Y.; Yamaji, K.; Yamane, D.; Hara, M.; Munekata, K.; Tsukiyama-Kohara, K.; Hishima, T.; et al. Selective inhibitor of Wnt/beta-catenin/CBP signaling ameliorates hepatitis C virus-induced liver fibrosis in mouse model. Sci. Rep. 2017, 7, 325. [Google Scholar] [CrossRef] [Green Version]
- Osawa, Y.; Oboki, K.; Imamura, J.; Kojika, E.; Hayashi, Y.; Hishima, T.; Saibara, T.; Shibasaki, F.; Kohara, M.; Kimura, K. Inhibition of Cyclic Adenosine Monophosphate (cAMP)-response Element-binding Protein (CREB)-binding Protein (CBP)/beta-Catenin Reduces Liver Fibrosis in Mice. EBioMedicine 2015, 2, 1751–1758. [Google Scholar] [CrossRef] [Green Version]
- Osawa, Y.; Kojika, E.; Hayashi, Y.; Kimura, M.; Nishikawa, K.; Yoshio, S.; Doi, H.; Kanto, T.; Kimura, K. Tumor necrosis factor-alpha-mediated hepatocyte apoptosis stimulates fibrosis in the steatotic liver in mice. Hepatol. Commun. 2018, 2, 407–420. [Google Scholar] [CrossRef]
- Ubeda, M.; Lario, M.; Munoz, L.; Borrero, M.J.; Rodriguez-Serrano, M.; Sanchez-Diaz, A.M.; Del Campo, R.; Lledo, L.; Pastor, O.; Garcia-Bermejo, L.; et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057. [Google Scholar] [CrossRef]
- Schwabl, P.; Hambruch, E.; Seeland, B.A.; Hayden, H.; Wagner, M.; Garnys, L.; Strobel, B.; Schubert, T.L.; Riedl, F.; Mitteregger, D.; et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J. Hepatol. 2017, 66, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Bowlus, C.L.; Pockros, P.J.; Kremer, A.E.; Pares, A.; Forman, L.M.; Drenth, J.P.H.; Ryder, S.D.; Terracciano, L.; Jin, Y.; Liberman, A.; et al. Long-Term Obeticholic Acid Therapy Improves Histological Endpoints in Patients With Primary Biliary Cholangitis. Clin. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Rizzo, G.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pellicciari, R.; Morelli, A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004, 127, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Modica, S.; Gadaleta, R.M.; Moschetta, A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl. Recept. Signal. 2010, 8, e005. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D. Structure of the extracellular matrix in normal and fibrotic liver: Collagens and glycoproteins. Semin. Liver Dis. 1990, 10, 1–10. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis-Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Jimenez Calvente, C.; Sehgal, A.; Popov, Y.; Kim, Y.O.; Zevallos, V.; Sahin, U.; Diken, M.; Schuppan, D. Specific hepatic delivery of procollagen alpha1(I) small interfering RNA in lipid-like nanoparticles resolves liver fibrosis. Hepatology 2015, 62, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molokanova, O.; Schonig, K.; Weng, S.Y.; Wang, X.; Bros, M.; Diken, M.; Ohngemach, S.; Karsdal, M.; Strand, D.; Nikolaev, A.; et al. Inducible knockdown of procollagen I protects mice from liver fibrosis and leads to dysregulated matrix genes and attenuated inflammation. Matrix Biol. 2018, 66, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Murase, K.; Kato, J.; Kobune, M.; Sato, T.; Kawano, Y.; Takimoto, R.; Takada, K.; Miyanishi, K.; Matsunaga, T.; et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 2008, 26, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Soule, B.; Tirucherai, G.; Kavita, U.; Kundu, S.R.C. Safety, tolerability, and pharmacokinetics of BMS-986263/ND-L02-s0201, a novel targeted lipid nanoparticle delivering HSP47siRNA, in healthy participants: A randomised, placebo-controlled, double-blind, phase 1 study. J. Hepatol. 2018, 68, S112. [Google Scholar] [CrossRef]
- Perepelyuk, M.; Terajima, M.; Wang, A.Y.; Georges, P.C.; Janmey, P.A.; Yamauchi, M.; Wells, R.G. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am. J. Physiol. Liver Physiol. 2013, 304, G605–G614. [Google Scholar] [CrossRef] [PubMed]
- Smith-Mungo, L.I.; Kagan, H.M. Lysyl oxidase: Properties, regulation and multiple functions in biology. Matrix Biol. 1998, 16, 387–398. [Google Scholar] [CrossRef]
- Elbjeirami, W.M.; Yonter, E.O.; Starcher, B.C.; West, J.L. Enhancing mechanical properties of tissue-engineered constructs via lysyl oxidase crosslinking activity. J. Biomed. Mater. Res. A 2003, 66, 513–521. [Google Scholar] [CrossRef]
- van der Slot-Verhoeven, A.J.; van Dura, E.A.; Attema, J.; Blauw, B.; Degroot, J.; Huizinga, T.W.; Zuurmond, A.M.; Bank, R.A. The type of collagen cross-link determines the reversibility of experimental skin fibrosis. Biochim Biophys Acta 2005, 1740, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Chang, T.R.; Aggarwal, A.; Lee, R.C.; Ehrlich, H.P. Mechanisms and dynamics of mechanical strengthening in ligament-equivalent fibroblast-populated collagen matrices. Ann. Biomed. Eng. 1993, 21, 289–305. [Google Scholar] [CrossRef]
- Giampuzzi, M.; Botti, G.; Di Duca, M.; Arata, L.; Ghiggeri, G.; Gusmano, R.; Ravazzolo, R.; Di Donato, A. Lysyl oxidase activates the transcription activity of human collagene III promoter. Possible involvement of Ku antigen. J. Boil. Chem. 2000, 275, 36341–36349. [Google Scholar] [CrossRef] [Green Version]
- Atsawasuwan, P.; Mochida, Y.; Katafuchi, M.; Kaku, M.; Fong, K.S.; Csiszar, K.; Yamauchi, M. Lysyl oxidase binds transforming growth factor-beta and regulates its signaling via amine oxidase activity. J. Boil. Chem. 2008, 283, 34229–34240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucero, H.A.; Ravid, K.; Grimsby, J.L.; Rich, C.B.; DiCamillo, S.J.; Maki, J.M.; Myllyharju, J.; Kagan, H.M. Lysyl oxidase oxidizes cell membrane proteins and enhances the chemotactic response of vascular smooth muscle cells. J. Boil. Chem. 2008, 283, 24103–24117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiume, L.; Favilli, G. Inhibition of experimental cirrhosis by carbon tetrachloride following treatment with aminoacetonitrile. Nature 1961, 189, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B.; Ikenaga, N.; Peng, Z.W.; Sverdlov, D.Y.; Greenstein, A.; Smith, V.; Schuppan, D.; Popov, Y. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J. 2016, 30, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Ikenaga, N.; Peng, Z.W.; Vaid, K.A.; Liu, S.B.; Yoshida, S.; Sverdlov, D.Y.; Mikels-Vigdal, A.; Smith, V.; Schuppan, D.; Popov, Y.V. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 2017, 66, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients With Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef]
- Muir, A.J.; Levy, C.; Janssen, H.L.A.; Montano-Loza, A.J.; Shiffman, M.L.; Caldwell, S.; Luketic, V.; Ding, D.; Jia, C.; McColgan, B.J.; et al. Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results With Insights on the Natural History of the Disease. Hepatology 2019, 69, 684–698. [Google Scholar] [CrossRef] [Green Version]
- Meissner, E.G.; McLaughlin, M.; Matthews, L.; Gharib, A.M.; Wood, B.J.; Levy, E.; Sinkus, R.; Virtaneva, K.; Sturdevant, D.; Martens, C.; et al. Simtuzumab treatment of advanced liver fibrosis in HIV and HCV-infected adults: Results of a 6-month open-label safety trial. Liver Int. 2016, 36, 1783–1792. [Google Scholar] [CrossRef] [Green Version]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS ONE 2016, 11, e0158156. [Google Scholar] [CrossRef] [PubMed]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef]
- Puengel, T.; Krenkel, O.; Mossanen, J.; Longerich, E.; Trautwein, C. The dual CCR/CCR5 antagonist Cenicriviroc ameliorates steatohepatitis and fibrosis in vivo by inhibiting the infiltration of inflammatory monocytes into injured liver. J. Hepatol. 2016, s159–s182. [Google Scholar]
- Friedman, S.; Sanyal, A.; Goodman, Z.; Lefebvre, E.; Gottwald, M.; Fischer, L.; Ratziu, V. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp Clin. Trials 2016, 47, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, E.; Gottwald, M.; Lasseter, K.; Chang, W.; Willett, M.; Smith, P.F.; Somasunderam, A.; Utay, N.S. Pharmacokinetics, Safety, and CCR2/CCR5 Antagonist Activity of Cenicriviroc in Participants With Mild or Moderate Hepatic Impairment. Clin. Transl. Sci. 2016, 9, 139–148. [Google Scholar] [CrossRef]
- Barondes, S.H.; Castronovo, V.; Cooper, D.N.; Cummings, R.D.; Drickamer, K.; Feizi, T.; Gitt, M.A.; Hirabayashi, J.; Hughes, C.; Kasai, K.; et al. Galectins: A family of animal beta-galactoside-binding lectins. Cell 1994, 76, 597–598. [Google Scholar] [CrossRef]
- Davidson, P.J.; Li, S.Y.; Lohse, A.G.; Vandergaast, R.; Verde, E.; Pearson, A.; Patterson, R.J.; Wang, J.L.; Arnoys, E.J. Transport of galectin-3 between the nucleus and cytoplasm. I. Conditions and signals for nuclear import. Glycobiology 2006, 16, 602–611. [Google Scholar] [CrossRef]
- Ochieng, J.; Fridman, R.; Nangia-Makker, P.; Kleiner, D.E.; Liotta, L.A.; Stetler-Stevenson, W.G.; Raz, A. Galectin-3 is a novel substrate for human matrix metalloproteinases-2 and -9. Biochemistry 1994, 33, 14109–14114. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Niki, T.; Nomura, T.; Oura, K.; Tadokoro, T.; Sakamoto, T.; Tani, J.; Yoneyama, H.; Morishita, A.; Kuroda, N.; et al. Correlation between serum galectin-9 levels and liver fibrosis. J. Gastroenterol. Hepatol. 2018, 33, 492–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matter, M.S.; Marquardt, J.U.; Andersen, J.B.; Quintavalle, C.; Korokhov, N.; Stauffer, J.K.; Kaji, K.; Decaens, T.; Quagliata, L.; Elloumi, F.; et al. Oncogenic driver genes and the inflammatory microenvironment dictate liver tumor phenotype. Hepatology 2016, 63, 1888–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacigalupo, M.L.; Manzi, M.; Rabinovich, G.A.; Troncoso, M.F. Hierarchical and selective roles of galectins in hepatocarcinogenesis, liver fibrosis and inflammation of hepatocellular carcinoma. World, J. Gastroenterol. 2013, 19, 8831–8849. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Y.; Hsu, D.K.; Liu, F.T. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 6737–6742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, K.C.; Frigeri, L.G.; Liu, F.T. An endogenous lectin, galectin-3 (epsilon BP/Mac-2), potentiates IL-1 production by human monocytes. Immunol. Lett. 1994, 42, 113–116. [Google Scholar] [CrossRef]
- Sano, H.; Hsu, D.K.; Yu, L.; Apgar, J.R.; Kuwabara, I.; Yamanaka, T.; Hirashima, M.; Liu, F.T. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J. Immunol. 2000, 165, 2156–2164. [Google Scholar] [CrossRef] [Green Version]
- Traber, P.G.; Zomer, E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE 2013, 8, e83481. [Google Scholar] [CrossRef] [Green Version]
- Traber, P.G.; Chou, H.; Zomer, E.; Hong, F.; Klyosov, A.; Fiel, M.I.; Friedman, S.L. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE 2013, 8, e75361. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Marri, S.R.; Chalasani, N.; Kohli, R.; Aronstein, W.; Thompson, G.A.; Irish, W.; Miles, M.V.; Xanthakos, S.A.; Lawitz, E.; et al. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment. Pharmacol.. 2016, 44, 1183–1198. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients with Nonalcoholic Steatohepatitis With Cirrhosis And Portal Hypertension. Gastroenterology 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muriel, P.; Rivera-Espinoza, Y. Beneficial drugs for liver diseases. J. Appl. Toxicol. 2008, 28, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Faghihzadeh, F.; Adibi, P.; Rafiei, R.; Hekmatdoost, A. Resveratrol supplementation improves inflammatory biomarkers in patients with nonalcoholic fatty liver disease. Nutr. Res. 2014, 34, 837–843. [Google Scholar] [CrossRef]
- Ferenci, P. Silymarin in the treatment of liver diseases: What is the clinical evidence? Clin. Liver Dis. (Hoboken) 2016, 7, 8–10. [Google Scholar] [CrossRef]
- Dehmlow, C.; Erhard, J.; de Groot, H. Inhibition of Kupffer cell functions as an explanation for the hepatoprotective properties of silibinin. Hepatology 1996, 23, 749–754. [Google Scholar] [CrossRef]
- Boigk, G.; Stroedter, L.; Herbst, H.; Waldschmidt, J.; Riecken, E.O.; Schuppan, D. Silymarin retards collagen accumulation in early and advanced biliary fibrosis secondary to complete bile duct obliteration in rats. Hepatology 1997, 26, 643–649. [Google Scholar] [CrossRef]
- Enjalbert, F.; Rapior, S.; Nouguier-Soule, J.; Guillon, S.; Amouroux, N.; Cabot, C. Treatment of amatoxin poisoning: 20-year retrospective analysis. J. Toxicol. Clin. Toxicol. 2002, 40, 715–757. [Google Scholar] [CrossRef]
- Sonnenbichler, J.; Zetl, I. Biochemical effects of the flavonolignane silibinin on RNA, protein and DNA synthesis in rat livers. Prog Clin. Biol Res. 1986, 213, 319–331. [Google Scholar] [PubMed]
- Loguercio, C.; Federico, A.; Trappoliere, M.; Tuccillo, C.; de Sio, I.; Di Leva, A.; Niosi, M.; D’Auria, M.V.; Capasso, R.; Del Vecchio Blanco, C.; et al. The effect of a silybin-vitamin e-phospholipid complex on nonalcoholic fatty liver disease: A pilot study. Dig. Dis. Sci. 2007, 52, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Scherzer, T.M.; Kerschner, H.; Rutter, K.; Beinhardt, S.; Hofer, H.; Schoniger-Hekele, M.; Holzmann, H.; Steindl-Munda, P. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology 2008, 135, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Andrade, R.J.; de la Cruz, J.P.; Rodriguez-Mendizabal, M.; Blanco, E.; Sanchez de la Cuesta, F. Effects of silymarin MZ-80 on oxidative stress in patients with alcoholic cirrhosis. Results of a randomized, double-blind, placebo-controlled clinical study. Int J. Clin. Pharmacol. Ther. 2002, 40, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Velussi, M.; Cernigoi, A.M.; De Monte, A.; Dapas, F.; Caffau, C.; Zilli, M. Long-term (12 months) treatment with an anti-oxidant drug (silymarin) is effective on hyperinsulinemia, exogenous insulin need and malondialdehyde levels in cirrhotic diabetic patients. J. Hepatol. 1997, 26, 871–879. [Google Scholar] [CrossRef]
- Clichici, S.; Olteanu, D.; Nagy, A.L.; Oros, A.; Filip, A.; Mircea, P.A. Silymarin inhibits the progression of fibrosis in the early stages of liver injury in CCl(4)-treated rats. J. Med. Food 2015, 18, 290–298. [Google Scholar] [CrossRef]
- Tsai, J.H.; Liu, J.Y.; Wu, T.T.; Ho, P.C.; Huang, C.Y.; Shyu, J.C.; Hsieh, Y.S.; Tsai, C.C.; Liu, Y.C. Effects of silymarin on the resolution of liver fibrosis induced by carbon tetrachloride in rats. J. Viral. Hepat. 2008, 15, 508–514. [Google Scholar] [CrossRef]
- Wu, J.W.; Lin, L.C.; Hung, S.C.; Chi, C.W.; Tsai, T.H. Analysis of silibinin in rat plasma and bile for hepatobiliary excretion and oral bioavailability application. J. Pharm. Biomed. Anal. 2007, 45, 635–641. [Google Scholar] [CrossRef]
- Zarrelli, A.; Romanucci, V.; Tuccillo, C.; Federico, A.; Loguercio, C.; Gravante, R.; Di Fabio, G. New silibinin glyco-conjugates: Synthesis and evaluation of antioxidant properties. Bioorg. Med. Chem. Lett. 2014, 24, 5147–5149. [Google Scholar] [CrossRef]
- Loguercio, C.; Andreone, P.; Brisc, C.; Brisc, M.C.; Bugianesi, E.; Chiaramonte, M.; Cursaro, C.; Danila, M.; de Sio, I.; Floreani, A.; et al. Silybin combined with phosphatidylcholine and vitamin E in patients with nonalcoholic fatty liver disease: A randomized controlled trial. Free Radic. Biol. Med. 2012, 52, 1658–1665. [Google Scholar] [CrossRef] [Green Version]
- Tu, C.T.; Han, B.; Liu, H.C.; Zhang, S.C. Curcumin protects mice against concanavalin A-induced hepatitis by inhibiting intrahepatic intercellular adhesion molecule-1 (ICAM-1) and CXCL10 expression. Mol. Cell Biochem 2011, 358, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Espinoza, Y.; Muriel, P. Pharmacological actions of curcumin in liver diseases or damage. Liver Int. 2009, 29, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Vizzutti, F.; Provenzano, A.; Galastri, S.; Milani, S.; Delogu, W.; Novo, E.; Caligiuri, A.; Zamara, E.; Arena, U.; Laffi, G.; et al. Curcumin limits the fibrogenic evolution of experimental steatohepatitis. Lab. Invest. 2010, 90, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Wang, L.; Lu, Q.; Da, W. Liver injury attenuation by curcumin in a rat NASH model: An Nrf2 activation-mediated effect? Ir. J. Med. Sci. 2016, 185, 93–100. [Google Scholar] [CrossRef]
- Ireson, C.; Orr, S.; Jones, D.J.; Verschoyle, R.; Lim, C.K.; Luo, J.L.; Howells, L.; Plummer, S.; Jukes, R.; Williams, M.; et al. Characterization of metabolites of the chemopreventive agent curcumin in human and rat hepatocytes and in the rat in vivo, and evaluation of their ability to inhibit phorbol ester-induced prostaglandin E2 production. Cancer Res. 2001, 61, 1058–1064. [Google Scholar]
- Huang, Y.; Cao, S.; Zhang, Q.; Zhang, H.; Fan, Y.; Qiu, F.; Kang, N. Biological and pharmacological effects of hexahydrocurcumin, a metabolite of curcumin. Arch. Biochem. Biophys. 2018, 646, 31–37. [Google Scholar] [CrossRef]
- Wang, J.; Yu, X.; Zhang, L.; Wang, L.; Peng, Z.; Chen, Y. The pharmacokinetics and tissue distribution of curcumin and its metabolites in mice. Biomed. Chromatogr. 2018, e4267. [Google Scholar] [CrossRef]
- Panahi, Y.; Kianpour, P.; Mohtashami, R.; Jafari, R.; Simental-Mendia, L.E.; Sahebkar, A. Efficacy and Safety of Phytosomal Curcumin in Non-Alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Drug Res. (Stuttg) 2017, 67, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, S.; Asgary, S.; Askari, G.; Keshvari, M.; Hatamipour, M.; Feizi, A.; Sahebkar, A. Treatment of Non-alcoholic Fatty Liver Disease with Curcumin: A Randomized Placebo-controlled Trial. Phytother Res. 2016, 30, 1540–1548. [Google Scholar] [CrossRef]
- Saadati, S.; Sadeghi, A.; Mansour, A.; Yari, Z.; Poustchi, H.; Hedayati, M.; Hatami, B.; Hekmatdoost, A. Curcumin and inflammation in non-alcoholic fatty liver disease: A randomized, placebo controlled clinical trial. BMC Gastroenterol. 2019, 19, 133. [Google Scholar] [CrossRef]
- Torok, N.J.; Dranoff, J.A.; Schuppan, D.; Friedman, S.L. Strategies and endpoints of antifibrotic drug trials: Summary and recommendations from the AASLD Emerging Trends Conference, Chicago, June 2014. Hepatology 2015, 62, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Popov, Y.; Schuppan, D. Targeting liver fibrosis: Strategies for development and validation of antifibrotic therapies. Hepatology 2009, 50, 1294–1306. [Google Scholar] [CrossRef] [PubMed]
- Reimer, K.C.; Wree, A.; Roderburg, C.; Tacke, F. New drugs for NAFLD: Lessons from basic models to the clinic. Hepatol. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Igal, G.; Evelyn, A.; Jennifer, C.X.; Natalia, K.; Alexandra, N.; Eddie, G.; John, D. Fibrotic Human Lung Extracellular Matrix as a Disease-Specific Substrate for Models of pulmonary Fibrosis. J. Respir Med. Lund Dis. 2019, 4, 1043. [Google Scholar]
- Loomba, R.L.E.; Ghalib, R.; Elkhashab, M.; Caldwell, S.; Abdelmalek, M. Longitudinal changes in liver stiffness by magnetic resonance elastography (MRE), liver fibrosis, and serum markers of fibrosis in a multi-center clinical trial in nonalcoholic steatohepatitis (NASH). J. Hepatol. 2017, 66, S671. [Google Scholar] [CrossRef]
- Nishikawa, K.; Iwaya, K.; Kinoshita, M.; Fujiwara, Y.; Akao, M.; Sonoda, M.; Thiruppathi, S.; Suzuki, T.; Hiroi, S.; Seki, S.; et al. Resveratrol increases CD68(+) Kupffer cells colocalized with adipose differentiation-related protein and ameliorates high-fat-diet-induced fatty liver in mice. Mol. Nutr. Food Res. 2015, 59, 1155–1170. [Google Scholar] [CrossRef]
- Kimura, K.; Ikoma, A.; Shibakawa, M.; Shimoda, S.; Harada, K.; Saio, M.; Imamura, J.; Osawa, Y.; Kimura, M.; Nishikawa, K.; et al. Safety, Tolerability, and Preliminary Efficacy of the Anti-Fibrotic Small Molecule PRI-724, a CBP/beta-Catenin Inhibitor, in Patients with Hepatitis C Virus-related Cirrhosis: A Single-Center, Open-Label, Dose Escalation Phase 1 Trial. EBioMedicine 2017, 23, 79–87. [Google Scholar] [CrossRef]



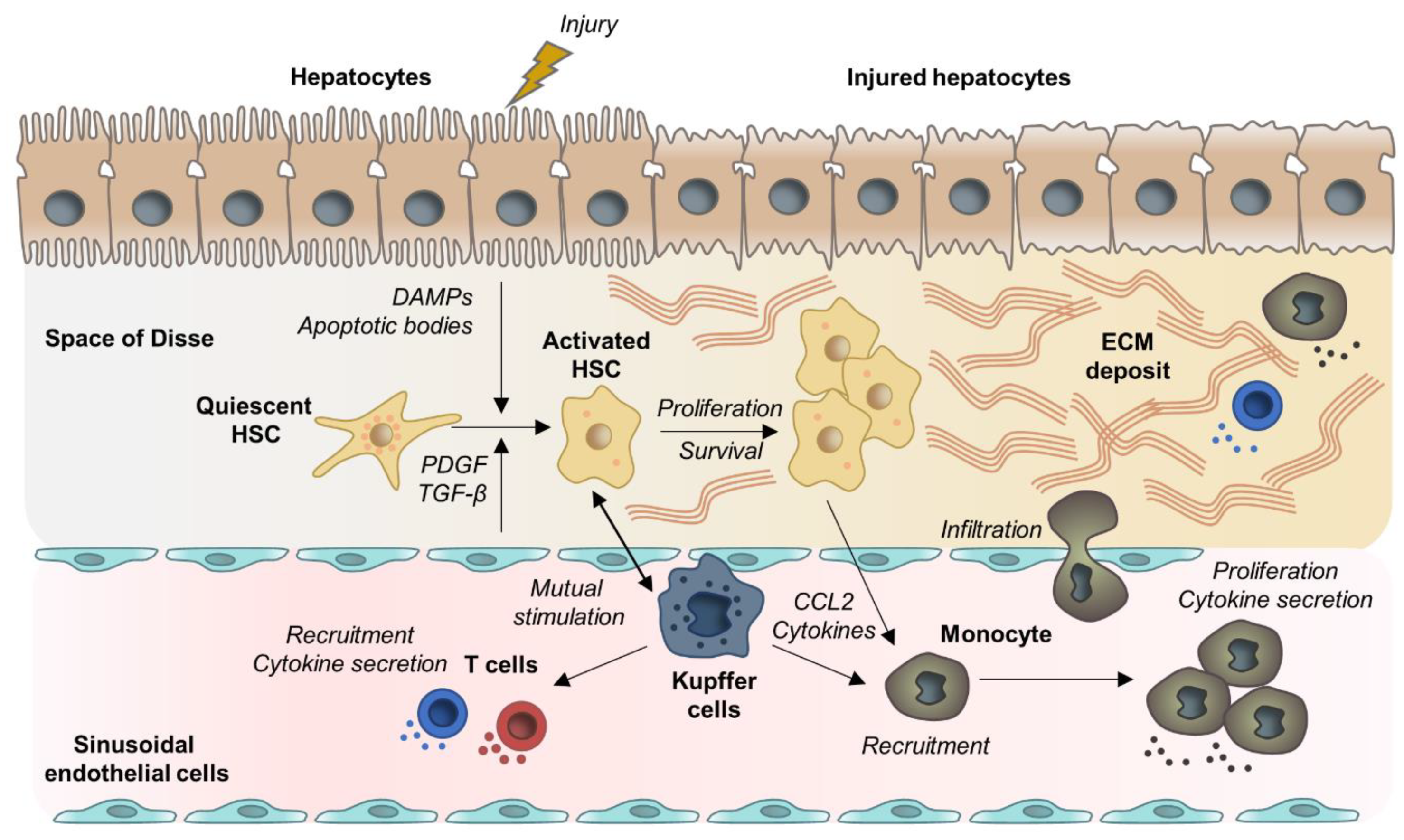{kind=link}
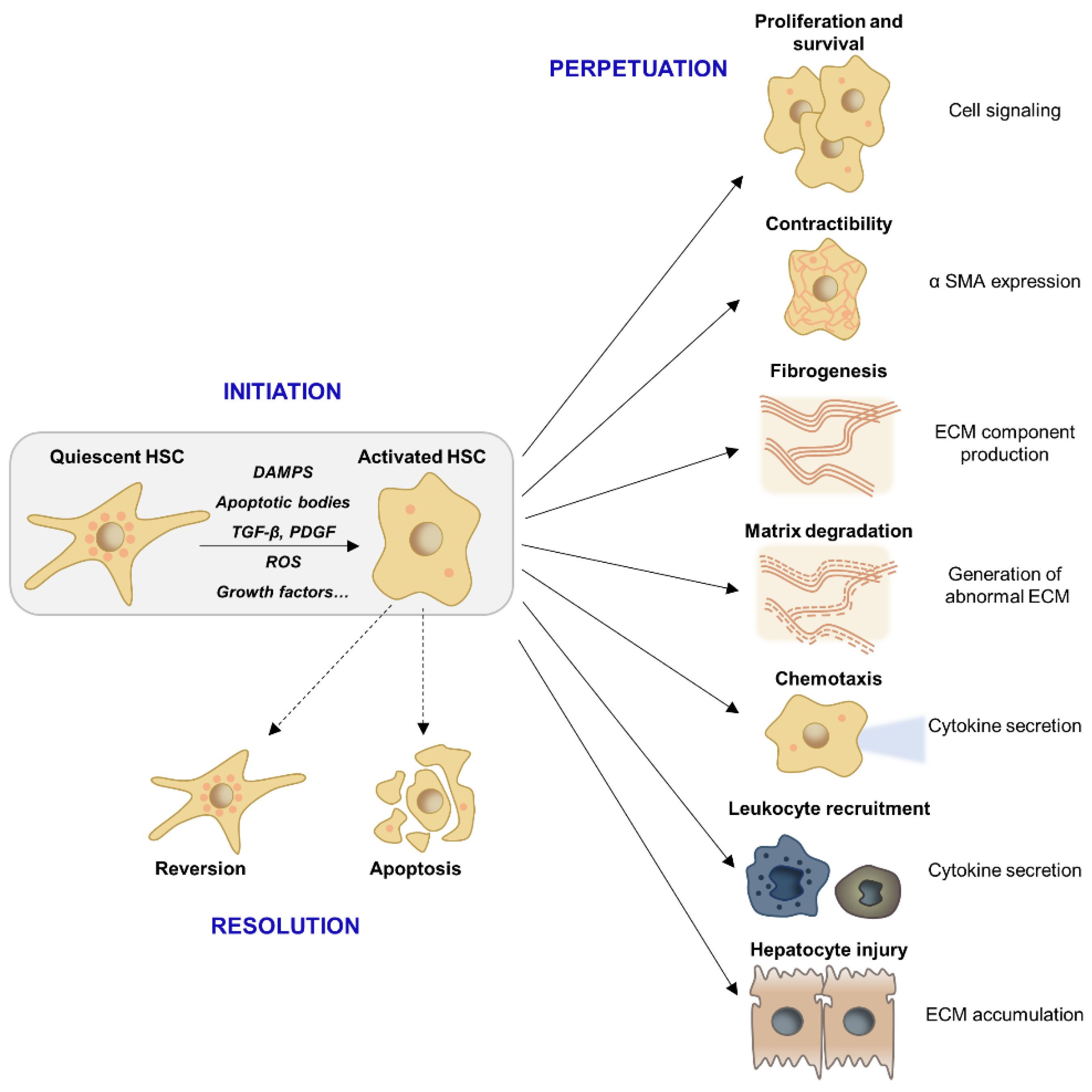{kind=link}
| Anti-fibrotic Mechanism | Agent | Rationale | Molecular Mode of Action in Preclinical Studies | Key Findings in Clinical Trials |
|---|---|---|---|---|
| Inhibition of hepatocyte apoptosis | Pan-caspase inhibitor Emricasan | Hepatocyte apoptosis is a major trigger of inflammation and HSC activation [249,250]. | Decreased HSCs activation and improvement of liver function in rat CCl4 model [253]. | Phase 2: Improvement of liver inflammation or fibrosis and tendency towards worsening of hepatocyte ballooning in NASH patients with F1-F3 fibrosis [254]. Small reductive effect on HVPG in cirrhotic NASH patients [255]. No effects in patients with acutely decompensated cirrhosis [256]. NCT02138253: clinical trial of Emricasan in the setting of post-transplant HCV-induced fibrosis after SVR: awaited 2020. |
| ASK1 inhibitor, selonsertib | Mediation of hepatocyte apoptosis via activation of JNK and p38 MAP kinases [257]. | Improvement of steatosis and fibrosis in NASH mouse model [257]. | Phase 2: Improvement of histological degree of fibrosis in patients with NASH F2-3 [260] Decrease of liver stiffness by MRE and improvements of non-invasive markers of fibrosis and inflammation [386]. Phase 3: STELLAR-3 and 4: Selonsertib in NASH patients and bridging fibrosis or cirrhosis: ongoing (NCT03053050; NCT03053063) | |
| Reduction of oxidative stress | Natural antioxidant with several targets, Resveratrol | Anti-inflammatory and antioxidant activity | Resveratrol reduces inflammation, fibrosis [264] as well as steatosis [387] in a mice models of NASH. | Phase 2: significant protective effects of resveratrol on markers of liver inflammation and hepatic steatosis grade within 12 weeks of treatment, no effect on fibrosis [357]. |
| Dual NOX1/4 inhibitor, GKT137831 | Activation of HSCs (NOX1) and induction of apoptosis in hepatocytes (NOX4) by production of superoxide radicals [155,266]. | Anti-fibrotic effect in CCl4 and bile duct ligation based mouse models of liver fibrosis via suppression of ROS production in HSCs in-vitro and in-vivo [267]. | Phase 2: significant effects on serological cholestasis parameters after 6 weeks of treatment in PBC. Ongoing study (NCT03226067). |
| Anti-fibrotic Mechanism | Agent | Rationale | Molecular Mode of Action in Preclinical Studies | Key Findings in Clinical Trials |
|---|---|---|---|---|
| Inhibition of HSC activation | FXR agonist, Obeticholic acid | Transcriptional regulation of fibrogenic genes in HSCs [306]. Improvement of intestinal mucosal barrier and homeostasis of gut-liver axis [301,302]. | Downregulation of collagen 1 synthesis in HSCs, potent anti-fibrotic effect in animal models of liver fibrosis [306]. | Phase 2: Improvement of fibrosis after 72 weeks treatment with OCA [309] Phase 3 (CENTAUR): dose-dependent improvement of fibrosis in 23% of OCA 25 mg treated compared to 12% placebo treated participants. Reduction of hepatocellular inflammation and ballooning [310]. |
| CBP/β-catenin small molecule inhibitor PRI-724 | Implication of Wnt/β-catenin signaling in HSC activation and liver fibrosis [169,292,293]. | Inhibition of HSC activation in HCV transgenic mice as well as CCl4 based murine liver fibrosis [298]. Beneficial effects on fibrosis resolution by activating anti-fibrotic macrophage subpopulations [299]. Decrease of hepatocyte apoptosis as well as fibrosis degree in NASH mouse model [300]. | Phase 1: dose dependent histological improvement (>2 point decrease in histologic activity index score) in 3/12 patients, but deterioration by 2 points in 2/12 patients with HCV associated cirrhosis [388]. Phase 2: PRI-724 in patients with hepatitis B or C related liver cirrhosis: expected to be completed in July 2020 (NCT03620474). | |
| Reduction of fibrotic scar evolution and contractility | Hsp47 siRNA delivering lipid nanoparticle, BMS 986263 | Function of Hsp47 as a collagen 1 chaperone [315]. | Significant anti-fibrotic effects in 3 and in-vivo models of liver fibrosis [315]. | Phase 1b/2: open label dose escalation study of BMS 986,263 in patients with moderate to severe fibrosis: completed, not yet published (NCT02227459). |
| LOXL2 specific monoclonal antibody, AB0023 (Simtuzumab) | Contributing of LOXL2 to ECM stiffness and hampered degradation of deposited collagen fibrils [317,318,319,320] Implication in Collagen 3 expression [322] and PDGFR sensitivity [324]. | Potent anti-fibrotic activity in bleomycin based mouse model of liver fibrosis via inhibition of collagen-crosslinking and its downstream activating effect on TGF-β1 signaling that contributes to myofibroblast simulation [328]. | Phase 2: No effect on fibrosis in NASH, PSC, or patients with HIV and/or HCV-infected patients with liver fibrosis [329,330,331]. |
| Anti-fibrotic Mechanism | Agent | Rationale | Molecular Mechanism of Action in Preclinical Studies | Key Findings in Clinical Trials |
|---|---|---|---|---|
| Immune modulation | CCR2/CCR5 inhibitor, Cenicriviroc | Involvement of CCR2/CCR5 mediated monocyte and macrophage recruitment during early pro-fibrogenic response [15,332,333]. | Dose-dependent decrease in monocyte/macrophage recruitment [334,335,336].Significant decrease in lobular inflammation, hepatocellular ballooning as well as collagen 1 and α-SMA protein expression in NASH mouse model [334]. | Phase 2: Improvement of fibrosis stage (> 1 stage) without worsening of steatohepatitis especially in patients with high disease activity (NAS > 5, prominent hepatocyte ballooning, F2-F3 fibrosis) [338]. No effect on lobular inflammation, but decrease in serological markers of systemic inflammation (hsCRP, IL6, fibrinogen) [338]. Phase 3: AURORA, NASH patients with advanced fibrosis and cirrhosis (NCT03028740): ongoing |
| Inhibitor of galectin-3, Belapectin | Function of galectin-3 as a chemoattractant for macrophages and monocytes, hereby accelerating further pro-inflammatory and pro-fibrogenic immune responses [347,348]. Activator of MMP2 and MMP9 [342]. | Dose-dependent reduction of NAS, fibrosis and portal pressure in rat and murine models of NASH potentially due to an impact on macrophage polarization and reduced activation of HSCs [349,350]. | Phase 2: No effect on fibrosis following within 52 weeks of treatment in NASH patients. Significant protective effects on hepatocyte ballooning as well as significant lower HPVG and varices development in a subgroup of patients with NASH cirrhosis [352]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. https://doi.org/10.3390/cells9040875
Roehlen N, Crouchet E, Baumert TF. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells. 2020; 9(4):875. https://doi.org/10.3390/cells9040875
Chicago/Turabian StyleRoehlen, Natascha, Emilie Crouchet, and Thomas F. Baumert. 2020. "Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives" Cells 9, no. 4: 875. https://doi.org/10.3390/cells9040875