Cellular Metabolic Regulation in the Differentiation and Function of Regulatory T Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
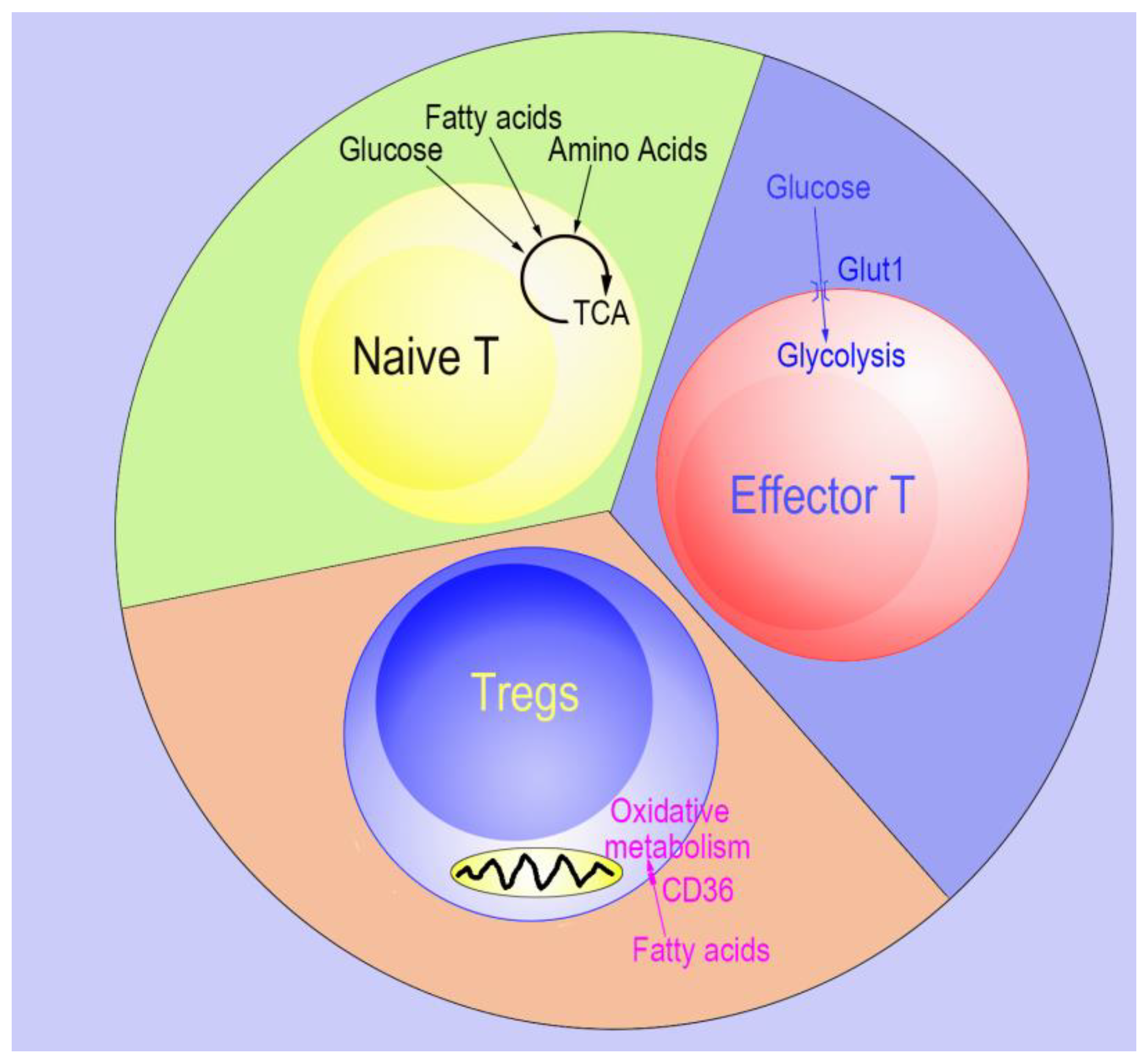
:1. Overview of the Effects of Cellular Metabolism on Tregs
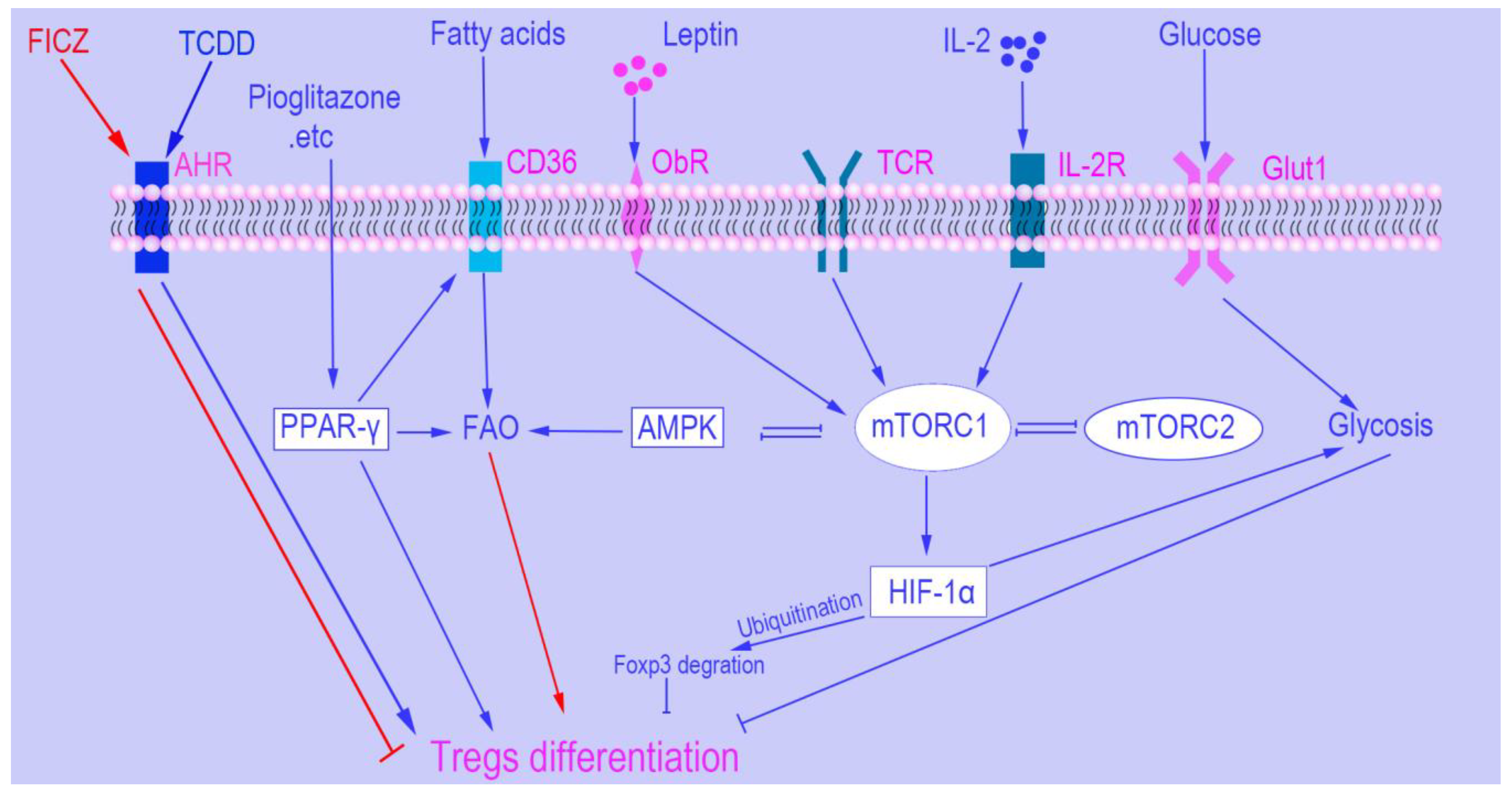
2. mTOR
2.1. mTOR and Tregs Differentiation
2.2. mTOR and Tregs Function
2.3. mTOR and Tregs Expansion
2.4. mTOR and Tregs Migration
3. Promising Metabolic Targets to Manipulate Tregs Frequency and Function
3.1. Hypoxia-Inducible Factor 1α (HIF1α)
3.2. AMP-Activated Protein Kinase (AMPK)
3.3. Leptin
3.4. Peroxisome Proliferator-Activated Receptors (PPARs)
3.5. The Aryl Hydrocarbon Receptor (AHR)
3.6. Interleukin 2 (IL-2)
4. Concluding Remarks and Perspectives
- The different roles of mTORC1 and mTORC2 in Treg induction, migration, expansion, and function;
- The different metabolic profiles of Tregs during steady states and inflammatory conditions;
- Identification of metabolic factors that correlate Tregs development, function, and expansion with environment cues;
- The basis of the requirement of high doses of IL-2 (mTOR activator) along with rapamycin (inhibit mTOR) to expand Tregs in vitro.
Funding
Conflicts of Interest
References
- Qian, X.; Wang, K.; Wang, X.; Zheng, S.G.; Lu, L. Generation of human regulatory t cells de novo with suppressive function prevent xenogeneic graft versus host disease. Int. Immunopharmacol. 2011, 11, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Lan, Q.; Chen, M.; Wang, J.; Shi, W.; Horwitz, D.A.; Quesniaux, V.; Ryffel, B.; Liu, Z.; Brand, D.; et al. Antigen-specific transforming growth factor beta-induced treg cells, but not natural treg cells, ameliorate autoimmune arthritis in mice by shifting the th17/treg cell balance from th17 predominance to treg cell predominance. Arthritis Rheum. 2012, 64, 2548–2558. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Zhou, X.; Fan, H.; Chen, M.; Wang, J.; Ryffel, B.; Brand, D.; Ramalingam, R.; Kiela, P.R.; Horwitz, D.A.; et al. Polyclonal cd4+foxp3+ treg cells induce tgfbeta-dependent tolerogenic dendritic cells that suppress the murine lupus-like syndrome. J. Mol. Cell Biol. 2012, 4, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zheng, S.G. How regulatory t cells sense and adapt to inflammation. Cell. Mol. Immunol. 2015, 12, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; Thornton, A.M. Ttregs, ptregs, and itregs: Similarities and differences. Immunol. Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Bour-Jordan, H. Current and future immunomodulation strategies to restore tolerance in autoimmune diseases. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Juvet, S.C.; Whatcott, A.G.; Bushell, A.R.; Wood, K.J. Harnessing regulatory t cells for clinical use in transplantation: The end of the beginning. Am. J. Transplant. 2014, 14, 750–763. [Google Scholar] [CrossRef]
- Horwitz, D.A.; Zheng, S.G.; Gray, J.D.; Wang, J.H.; Ohtsuka, K.; Yamagiwa, S. Regulatory t cells generated ex vivo as an approach for the therapy of autoimmune disease. Semin. Immunol. 2004, 16, 135–143. [Google Scholar] [CrossRef]
- Horwitz, D.A.; Gray, J.D.; Zheng, S.G. The potential of human regulatory t cells generated ex vivo as a treatment for lupus and other chronic inflammatory diseases. Arthritis Res. 2002, 4, 241–246. [Google Scholar] [CrossRef]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. Il-2 is essential for tgf-beta to convert naive cd4+cd25- cells to cd25+foxp3+ regulatory t cells and for expansion of these cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef]
- Zheng, S.G.; Wang, J.; Horwitz, D.A. Cutting edge: Foxp3+cd4+cd25+ regulatory t cells induced by il-2 and tgf-beta are resistant to th17 conversion by il-6. J. Immunol. 2008, 180, 7112–7116. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Kong, N.; Wang, J.; Fan, H.; Zou, H.; Horwitz, D.; Brand, D.; Liu, Z.; Zheng, S.G. Cutting edge: All-trans retinoic acid sustains the stability and function of natural regulatory t cells in an inflammatory milieu. J. Immunol. 2010, 185, 2675–2679. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Lu, L.; Chen, M.; Xu, L.; Lan, Q.; Li, Q.; Liu, Z.; Chen, G.; Wang, P.; Wang, X.; et al. Tgf-beta-induced cd4+foxp3+ t cells attenuate acute graft-versus-host disease by suppressing expansion and killing of effector cd8+ cells. J. Immunol. 2014, 193, 3388–3397. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; Carbone, F.; Di Silvestre, D.; Brambilla, F.; De Rosa, V.; Galgani, M.; Faicchia, D.; Marone, G.; Tramontano, D.; Corona, M.; et al. The proteomic landscape of human ex vivo regulatory and conventional t cells reveals specific metabolic requirements. Immunity 2016, 44, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory cd4+ t cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L. Metabolism in t cell activation and differentiation. Curr. Opin. Immunol. 2010, 22, 314–320. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 reprograms t cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef]
- Kabat, A.M.; Harrison, O.J.; Riffelmacher, T.; Moghaddam, A.E.; Pearson, C.F.; Laing, A.; Abeler-Dorner, L.; Forman, S.P.; Grencis, R.K.; Sattentau, Q.; et al. The autophagy gene atg16l1 differentially regulates treg and th2 cells to control intestinal inflammation. Elife 2016, 5, e12444. [Google Scholar] [CrossRef]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory t cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef]
- Haghikia, A.; Jorg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef]
- Chen, X.; Su, W.; Wan, T.; Yu, J.; Zhu, W.; Tang, F.; Liu, G.; Olsen, N.; Liang, D.; Zheng, S.G. Sodium butyrate regulates th17/treg cell balance to ameliorate uveitis via the nrf2/ho-1 pathway. Biochem. Pharmacol. 2017, 142, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Raud, B.; Roy, D.G.; Divakaruni, A.S.; Tarasenko, T.N.; Franke, R.; Ma, E.H.; Samborska, B.; Hsieh, W.Y.; Wong, A.H.; Stuve, P.; et al. Etomoxir actions on regulatory and memory t cells are independent of cpt1a-mediated fatty acid oxidation. Cell Metab. 2018, 28, 504–515.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Guan, K.L. Expanding mtor signaling. Cell Res. 2007, 17, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Blouet, C.; Ono, H.; Schwartz, G.J. Mediobasal hypothalamic p70 s6 kinase 1 modulates the control of energy homeostasis. Cell Metab. 2008, 8, 459–467. [Google Scholar] [CrossRef]
- Verbist, K.C.; Guy, C.S.; Milasta, S.; Liedmann, S.; Kaminski, M.M.; Wang, R.; Green, D.R. Metabolic maintenance of cell asymmetry following division in activated t lymphocytes. Nature 2016, 532, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mtor regulates the differentiation of helper t cells through the selective activation of signaling by mtorc1 and mtorc2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mtor. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mtor network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Chi, H. Regulation and function of mtor signalling in t cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Zheng, Y.; Collins, S.L.; Lutz, M.A.; Allen, A.N.; Kole, T.P.; Zarek, P.E.; Powell, J.D. A role for mammalian target of rapamycin in regulating t cell activation versus anergy. J. Immunol. 2007, 178, 2163–2170. [Google Scholar] [CrossRef]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of immune responses by mtor. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. Mtorc1 couples immune signals and metabolic programming to establish t(reg)-cell function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef]
- Chapman, N.M.; Zeng, H.; Nguyen, T.M.; Wang, Y.; Vogel, P.; Dhungana, Y.; Liu, X.; Neale, G.; Locasale, J.W.; Chi, H. Mtor coordinates transcriptional programs and mitochondrial metabolism of activated treg subsets to protect tissue homeostasis. Nat. Commun. 2018, 9, 2095. [Google Scholar] [CrossRef]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and toll-like receptor signaling balance treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands cd4+cd25+foxp3+ regulatory t cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Wang, G.Y.; Zhang, Q.; Yang, Y.; Chen, W.J.; Liu, W.; Jiang, N.; Chen, G.H. Rapamycin combined with allogenic immature dendritic cells selectively expands cd4+cd25+foxp3+ regulatory t cells in rats. Hepatobiliary Pancreat. Dis. Int. 2012, 11, 203–208. [Google Scholar] [CrossRef]
- Ikeda, K.; Kinoshita, M.; Kayama, H.; Nagamori, S.; Kongpracha, P.; Umemoto, E.; Okumura, R.; Kurakawa, T.; Murakami, M.; Mikami, N.; et al. Slc3a2 mediates branched-chain amino-acid-dependent maintenance of regulatory t cells. Cell Rep. 2017, 21, 1824–1838. [Google Scholar] [CrossRef]
- Asanuma, S.; Tanaka, J.; Sugita, J.; Kosugi, M.; Shiratori, S.; Wakasa, K.; Shono, Y.; Shigematsu, A.; Kondo, T.; Kobayashi, T.; et al. Expansion of cd4(+)cd25 (+) regulatory t cells from cord blood cd4(+) cells using the common gamma-chain cytokines (il-2 and il-15) and rapamycin. Ann. Hematol. 2011, 90, 617–624. [Google Scholar] [CrossRef]
- Cybulski, N.; Hall, M.N. Tor complex 2: A signaling pathway of its own. Trends Biochem. Sci. 2009, 34, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Jacinto, E. Mtor complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tang, J.; Chen, W.; Li, Q.; Nie, J.; Lin, F.; Wu, Q.; Chen, Z.; Gao, Z.; Fan, H.; et al. Inflammation negatively regulates foxp3 and regulatory t-cell function via dbc1. Proc. Natl. Acad. Sci. USA 2015, 112, E3246–E3254. [Google Scholar] [CrossRef] [PubMed]
- Kishore, M.; Cheung, K.C.P.; Fu, H.; Bonacina, F.; Wang, G.; Coe, D.; Ward, E.J.; Colamatteo, A.; Jangani, M.; Baragetti, A.; et al. Regulatory t cell migration is dependent on glucokinase-mediated glycolysis. Immunity 2017, 47, 875–889.e10. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. Hif1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of th17 and treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.Y.; Leng, R.X.; Tao, J.H.; Li, X.P.; Ye, D.Q.; Olsen, N.; Zheng, S.G.; Pan, H.F. Hypoxia-inducible factor-1alpha: A promising therapeutic target for autoimmune diseases. Expert Opin. Ther. Targets 2017, 21, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of t(h)17/t(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef]
- Kong, N.; Lan, Q.; Chen, M.; Zheng, T.; Su, W.; Wang, J.; Yang, Z.; Park, R.; Dagliyan, G.; Conti, P.S.; et al. Induced t regulatory cells suppress osteoclastogenesis and bone erosion in collagen-induced arthritis better than natural t regulatory cells. Ann. Rheum. Dis. 2012, 71, 1567–1572. [Google Scholar] [CrossRef]
- Xu, L.; Kitani, A.; Fuss, I.; Strober, W. Cutting edge: Regulatory t cells induce cd4+cd25-foxp3- t cells or are self-induced to become th17 cells in the absence of exogenous tgf-beta. J. Immunol. 2007, 178, 6725–6729. [Google Scholar] [CrossRef]
- Deng, W.; Ren, Y.; Feng, X.; Yao, G.; Chen, W.; Sun, Y.; Wang, H.; Gao, X.; Sun, L. Hypoxia inducible factor-1 alpha promotes mesangial cell proliferation in lupus nephritis. Am. J. Nephrol. 2014, 40, 507–515. [Google Scholar] [CrossRef]
- Hu, F.; Shi, L.; Mu, R.; Zhu, J.; Li, Y.; Ma, X.; Li, C.; Jia, R.; Yang, D.; Li, Y.; et al. Hypoxia-inducible factor-1alpha and interleukin 33 form a regulatory circuit to perpetuate the inflammation in rheumatoid arthritis. PLoS ONE 2013, 8, e72650. [Google Scholar]
- Giatromanolaki, A.; Sivridis, E.; Maltezos, E.; Athanassou, N.; Papazoglou, D.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Upregulated hypoxia inducible factor-1alpha and -2alpha pathway in rheumatoid arthritis and osteoarthritis. Arthritis Res. Ther. 2003, 5, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Loukovaara, S.; Koivunen, P.; Ingles, M.; Escobar, J.; Vento, M.; Andersson, S. Elevated protein carbonyl and hif-1alpha levels in eyes with proliferative diabetic retinopathy. Acta Ophthalmol. 2014, 92, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Graumann, U.; Reynolds, R.; Steck, A.J.; Schaeren-Wiemers, N. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol. 2003, 13, 554–573. [Google Scholar] [CrossRef] [PubMed]
- Vasilopoulos, Y.; Sourli, F.; Zafiriou, E.; Klimi, E.; Ioannou, M.; Mamuris, Z.; Simos, G.; Koukoulis, G.; Roussaki-Schulze, A. High serum levels of hif-1alpha in psoriatic patients correlate with an over-expression of il-6. Cytokine 2013, 62, 38–39. [Google Scholar] [CrossRef]
- Mimouna, S.; Goncalves, D.; Barnich, N.; Darfeuille-Michaud, A.; Hofman, P.; Vouret-Craviari, V. Crohn disease-associated escherichia coli promote gastrointestinal inflammatory disorders by activation of hif-dependent responses. Gut Microbes 2011, 2, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Bai, A.; Ma, A.G.; Yong, M.; Weiss, C.R.; Ma, Y.; Guan, Q.; Bernstein, C.N.; Peng, Z. Ampk agonist downregulates innate and adaptive immune responses in tnbs-induced murine acute and relapsing colitis. Biochem. Pharmacol. 2010, 80, 1708–1717. [Google Scholar] [CrossRef]
- Gualdoni, G.A.; Mayer, K.A.; Goschl, L.; Boucheron, N.; Ellmeier, W.; Zlabinger, G.J. The amp analog aicar modulates the treg/th17 axis through enhancement of fatty acid oxidation. FASEB J. 2016, 30, 3800–3809. [Google Scholar] [CrossRef]
- Park, M.J.; Lee, S.Y.; Moon, S.J.; Son, H.J.; Lee, S.H.; Kim, E.K.; Byun, J.K.; Shin, D.Y.; Park, S.H.; Yang, C.W.; et al. Metformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between t helper 17 and tregs. Transl. Res. 2016, 173, 115–130. [Google Scholar]
- Tamas, P.; Hawley, S.A.; Clarke, R.G.; Mustard, K.J.; Green, K.; Hardie, D.G.; Cantrell, D.A. Regulation of the energy sensor amp-activated protein kinase by antigen receptor and ca2+ in t lymphocytes. J. Exp. Med. 2006, 203, 1665–1670. [Google Scholar] [CrossRef]
- Barnes, K.; Ingram, J.C.; Porras, O.H.; Barros, L.F.; Hudson, E.R.; Fryer, L.G.; Foufelle, F.; Carling, D.; Hardie, D.G.; Baldwin, S.A. Activation of glut1 by metabolic and osmotic stress: Potential involvement of amp-activated protein kinase (ampk). J. Cell Sci. 2002, 115, 2433–2442. [Google Scholar] [PubMed]
- Sun, Y.; Tian, T.; Gao, J.; Liu, X.; Hou, H.; Cao, R.; Li, B.; Quan, M.; Guo, L. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating t helper 17 and regulatory t cells in mice. J. Neuroimmunol. 2016, 292, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, S.H.; Yang, E.J.; Kim, E.K.; Kim, J.K.; Shin, D.Y.; Cho, M.L. Metformin ameliorates inflammatory bowel disease by suppression of the stat3 signaling pathway and regulation of the between th17/treg balance. PLoS ONE 2015, 10, e0135858. [Google Scholar] [CrossRef] [PubMed]
- Waickman, A.T.; Powell, J.D. Mtor, metabolism, and the regulation of t-cell differentiation and function. Immunol. Rev. 2012, 249, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Blanco, D.B.; Neale, G.; Vogel, P.; Avila, J.; Clish, C.B.; Wu, C.; Shrestha, S.; Rankin, S.; Long, L.; et al. Homeostatic control of metabolic and functional fitness of treg cells by lkb1 signalling. Nature 2017, 548, 602–606. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Fan, W.; Henriquez, B.; Yu, R.T.; Atkins, A.R.; Liddle, C.; Zheng, Y.; Downes, M.; Evans, R.M. Metabolic control of regulatory t cell (treg) survival and function by lkb1. Proc. Natl. Acad. Sci. USA 2017, 114, 12542–12547. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Matarese, G.; Lord, G.M.; Keogh, J.M.; Lawrence, E.; Agwu, C.; Sanna, V.; Jebb, S.A.; Perna, F.; Fontana, S.; et al. Beneficial effects of leptin on obesity, t cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Investig. 2002, 110, 1093–1103. [Google Scholar] [CrossRef]
- Matarese, G.; Procaccini, C.; De Rosa, V.; Horvath, T.L.; La Cava, A. Regulatory t cells in obesity: The leptin connection. Trends Mol. Med. 2010, 16, 247–256. [Google Scholar] [CrossRef]
- De Rosa, V.; Procaccini, C.; La Cava, A.; Chieffi, P.; Nicoletti, G.F.; Fontana, S.; Zappacosta, S.; Matarese, G. Leptin neutralization interferes with pathogenic t cell autoreactivity in autoimmune encephalomyelitis. J. Clin. Investig. 2006, 116, 447–455. [Google Scholar] [CrossRef]
- Matarese, G.; Di Giacomo, A.; Sanna, V.; Lord, G.M.; Howard, J.K.; Di Tuoro, A.; Bloom, S.R.; Lechler, R.I.; Zappacosta, S.; Fontana, S. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J. Immunol. 2001, 166, 5909–5916. [Google Scholar] [CrossRef]
- Wang, X.; Qiao, Y.; Yang, L.; Song, S.; Han, Y.; Tian, Y.; Ding, M.; Jin, H.; Shao, F.; Liu, A. Leptin levels in patients with systemic lupus erythematosus inversely correlate with regulatory t cell frequency. Lupus 2017, 26, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Procaccini, C.; Cali, G.; Pirozzi, G.; Fontana, S.; Zappacosta, S.; La Cava, A.; Matarese, G. A key role of leptin in the control of regulatory t cell proliferation. Immunity 2007, 26, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Umesono, K.; Noonan, D.J.; Heyman, R.A.; Evans, R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992, 358, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Bothwell, A.L. The nuclear receptor ppars as important regulators of t-cell functions and autoimmune diseases. Mol. Cells 2012, 33, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of pparalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Zhu, Y.; Ni, Y.; Liu, R.; Hou, M.; Yang, B.; Song, J.; Sun, H.; Xu, Z.; Ji, M. Ppar-gamma agonist alleviates liver and spleen pathology via inducing treg cells during schistosoma japonicum infection. J. Immunol. Res. 2018, 2018, 6398078. [Google Scholar] [CrossRef]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. Ppar-gamma is a major driver of the accumulation and phenotype of adipose tissue treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef]
- Hontecillas, R.; Bassaganya-Riera, J. Peroxisome proliferator-activated receptor gamma is required for regulatory cd4+ t cell-mediated protection against colitis. J. Immunol. 2007, 178, 2940–2949. [Google Scholar] [CrossRef]
- Cipolletta, D. Adipose tissue-resident regulatory t cells: Phenotypic specialization, functions and therapeutic potential. Immunology 2014, 142, 517–525. [Google Scholar] [CrossRef]
- Bapat, S.P.; Myoung Suh, J.; Fang, S.; Liu, S.; Zhang, Y.; Cheng, A.; Zhou, C.; Liang, Y.; LeBlanc, M.; Liddle, C.; et al. Depletion of fat-resident treg cells prevents age-associated insulin resistance. Nature 2015, 528, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of t(reg) and t(h)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A. Interleukin-2: Inception, impact, and implications. Science 1988, 240, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising foxp3-expressing cd25+cd4+ regulatory t cells in immunological tolerance to self and non-self. Nat. Immunol. 2005, 6, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Mizui, M.; Tsokos, G.C. Low-dose il-2 in the treatment of lupus. Curr. Rheumatol. Rep. 2016, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Von Spee-Mayer, C.; Siegert, E.; Abdirama, D.; Rose, A.; Klaus, A.; Alexander, T.; Enghard, P.; Sawitzki, B.; Hiepe, F.; Radbruch, A.; et al. Low-dose interleukin-2 selectively corrects regulatory t cell defects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2016, 75, 1407–1415. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Churlaud, G.; Mallone, R.; Six, A.; Derian, N.; Chaara, W.; Lorenzon, R.; Long, S.A.; Buckner, J.H.; Afonso, G.; et al. Low-dose interleukin-2 fosters a dose-dependent regulatory t cell tuned milieu in t1d patients. J. Autoimmun. 2015, 58, 48–58. [Google Scholar] [CrossRef]
- Koreth, J.; Kim, H.T.; Jones, K.T.; Lange, P.B.; Reynolds, C.G.; Chammas, M.J.; Dusenbury, K.; Whangbo, J.; Nikiforow, S.; Alyea, E.P., 3rd; et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood 2016, 128, 130–137. [Google Scholar] [CrossRef]
- Matsuoka, K.; Koreth, J.; Kim, H.T.; Bascug, G.; McDonough, S.; Kawano, Y.; Murase, K.; Cutler, C.; Ho, V.T.; Alyea, E.P.; et al. Low-dose interleukin-2 therapy restores regulatory t cell homeostasis in patients with chronic graft-versus-host disease. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Colello, J.; Jarjour, W.; Zheng, S.G. Cellular Metabolic Regulation in the Differentiation and Function of Regulatory T Cells. Cells 2019, 8, 188. https://doi.org/10.3390/cells8020188
Chen Y, Colello J, Jarjour W, Zheng SG. Cellular Metabolic Regulation in the Differentiation and Function of Regulatory T Cells. Cells. 2019; 8(2):188. https://doi.org/10.3390/cells8020188
Chicago/Turabian StyleChen, Ye, Jacob Colello, Wael Jarjour, and Song Guo Zheng. 2019. "Cellular Metabolic Regulation in the Differentiation and Function of Regulatory T Cells" Cells 8, no. 2: 188. https://doi.org/10.3390/cells8020188