
Mesenchymal-Stem-Cell-Based Therapy against Gliomas


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical and Therapeutic Use of MSCs
2.1. Therapeutic Gene Delivery
2.2. Oncolytic Virus Delivery
2.3. miRNA Delivery
3. Methods to Improve MSCs’ Tropism
3.1. CAR-MSC Cells
3.2. Conjugation of MSCs with Nanoparticles
4. Exosomes Derived from MSCs
5. MSCs Associated with Gliomas
6. Do MSCs Support or Suppress Tumor Progression of Gliomas?
6.1. BM-MSC
6.2. UC-MSC
6.3. AT-MSC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grossman, S.A.; Batara, J.F. Current management of glioblastoma multiforme. Semin. Oncol. 2004, 31, 635–644. [Google Scholar] [CrossRef] [PubMed]
- McGirt, M.J.; Chaichana, K.L.; Attenello, F.J.; Weingart, J.D.; Than, K.; Burger, P.C.; Olivi, A.; Brem, H.; Quinoñes-Hinojosa, A. Extent of surgical resection is independently associated with survival in patients with hemispheric infiltrating low-grade gliomas. Neurosurgery 2008, 63, 700–707. [Google Scholar] [CrossRef] [PubMed]
- de Melo, S.M.; Bittencourt, S.; Ferrazoli, E.G.; da Silva, C.S.; da Cunha, F.F.; da Silva, F.H.; Stilhano, R.S.; Denapoli, P.M.; Zanetti, B.F.; Martin, P.K.; et al. The Anti-Tumor Effects of Adipose Tissue Mesenchymal Stem Cell Transduced with HSV-Tk Gene on U-87-Driven Brain Tumor. PLoS ONE 2015, 10, e0128922. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Yoo, K.H.; Sym, S.J.; Khang, D. Mesenchymal stem cell therapy assisted by nanotechnology: A possible combinational treatment for brain tumor and central nerve regeneration. Int. J. Nanomed. 2019, 14, 5925–5942. [Google Scholar] [CrossRef] [PubMed]
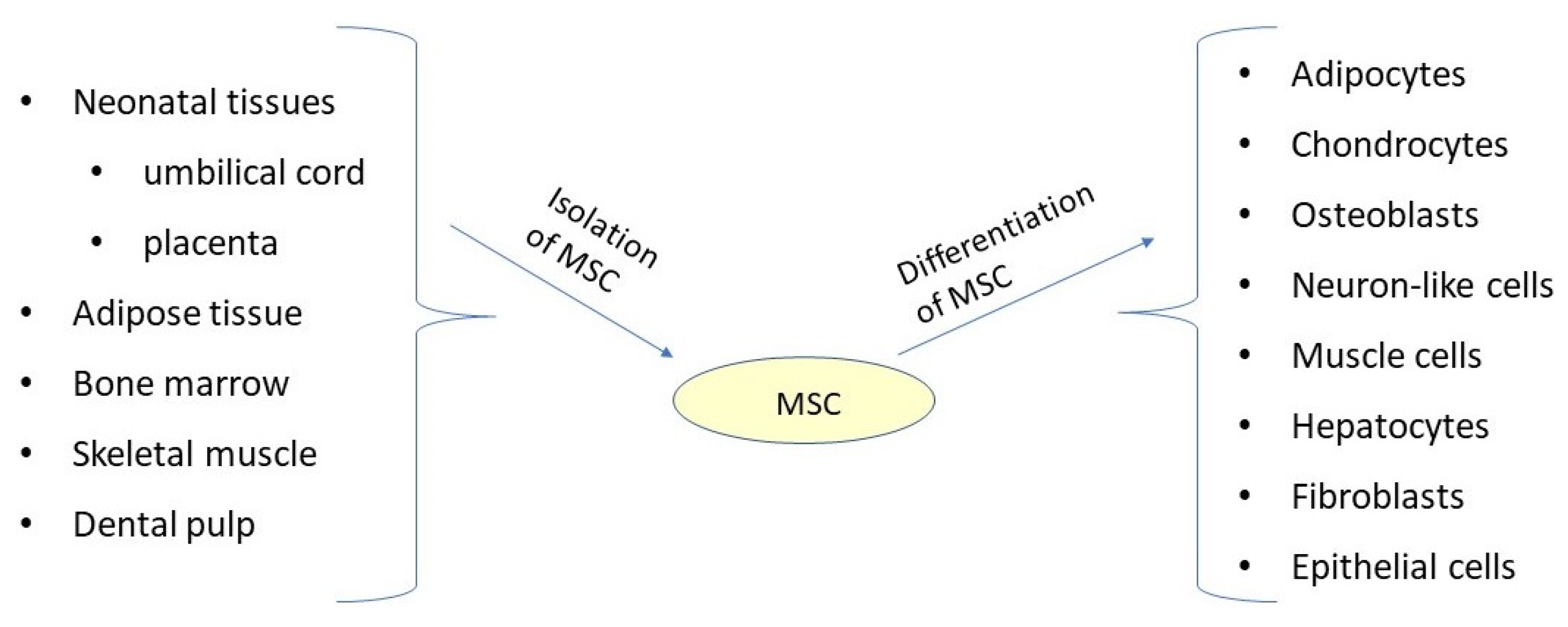
- Friedenstein, A.J.; Piatetzky, S., II; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, C.; Li, Q.; Chesler, D.A.; Yuan, K.; Guerrero-Cazares, H.; Quinones-Hinojosa, A. Mesenchymal stem cells derived from adipose tissue vs bone marrow: In vitro comparison of their tropism towards gliomas. PLoS ONE 2013, 8, e58198. [Google Scholar] [CrossRef] [PubMed]
- Safford, K.M.; Rice, H.E. Stem cell therapy for neurologic disorders: Therapeutic potential of adipose-derived stem cells. Curr. Drug Targets 2005, 6, 57–62. [Google Scholar] [CrossRef]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef]
- Erices, A.; Conget, P.; Minguell, J.J. Mesenchymal progenitor cells in human umbilical cord blood. Br. J. Haematol. 2000, 109, 235–242. [Google Scholar] [CrossRef]
- Covas, D.T.; Siufi, J.L.; Silva, A.R.; Orellana, M.D. Isolation and culture of umbilical vein mesenchymal stem cells. Braz. J. Med. Biol. Res. 2003, 36, 1179–1183. [Google Scholar] [CrossRef]
- Pierdomenico, L.; Bonsi, L.; Calvitti, M.; Rondelli, D.; Arpinati, M.; Chirumbolo, G.; Becchetti, E.; Marchionni, C.; Alviano, F.; Fossati, V.; et al. Multipotent mesenchymal stem cells with immunosuppressive activity can be easily isolated from dental pulp. Transplantation 2005, 80, 836–842. [Google Scholar] [CrossRef]
- Rodríguez-Lozano, F.J.; Bueno, C.; Insausti, C.L.; Meseguer, L.; Ramírez, M.C.; Blanquer, M.; Marín, N.; Martínez, S.; Moraleda, J.M. Mesenchymal stem cells derived from dental tissues. Int. Endod. J. 2011, 44, 800–806. [Google Scholar] [CrossRef]
- In’t Anker, P.S.; Scherjon, S.A.; Kleijburg-van der Keur, C.; de Groot-Swings, G.M.; Claas, F.H.; Fibbe, W.E.; Kanhai, H.H. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells 2004, 22, 1338–1345. [Google Scholar] [CrossRef]
- Miao, Z.; Jin, J.; Chen, L.; Zhu, J.; Huang, W.; Zhao, J.; Qian, H.; Zhang, X. Isolation of mesenchymal stem cells from human placenta: Comparison with human bone marrow mesenchymal stem cells. Cell Biol. Int. 2006, 30, 681–687. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Mushahary, D.; Spittler, A.; Kasper, C.; Weber, V.; Charwat, V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytometry A 2018, 93, 19–31. [Google Scholar] [CrossRef]
- Baghaei, K.; Hashemi, S.M.; Tokhanbigli, S.; Asadi Rad, A.; Assadzadeh-Aghdaei, H.; Sharifian, A.; Zali, M.R. Isolation, differentiation, and characterization of mesenchymal stem cells from human bone marrow. Gastroenterol. Hepatol. Bed Bench 2017, 10, 208–213. [Google Scholar]
- Feng, X.; Qi, F.; Wang, H.; Li, W.; Gan, Y.; Qi, C.; Lin, Z.; Chen, L.; Wang, P.; Hu, Z.; et al. Sorting Technology for Mesenchymal Stem Cells from a Single Tissue Source. Stem Cell Rev. Rep. 2024, 20, 524–537. [Google Scholar] [CrossRef]
- Prakash, N.; Kim, J.; Jeon, J.; Kim, S.; Arai, Y.; Bello, A.B.; Park, H.; Lee, S.H. Progress and emerging techniques for biomaterial-based derivation of mesenchymal stem cells (MSCs) from pluripotent stem cells (PSCs). Biomater. Res. 2023, 27, 31. [Google Scholar] [CrossRef]
- Yang, H.; Chen, J.; Li, J. Isolation, culture, and delivery considerations for the use of mesenchymal stem cells in potential therapies for acute liver failure. Front. Immunol. 2023, 14, 1243220. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M.; Nadri, S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat. Protoc. 2009, 4, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Chen, L.; Wang, X.; Xiang, C. Mesenchymal stem cells as therapeutic agents and in gene delivery for the treatment of glioma. J. Zhejiang Univ. Sci. B 2017, 18, 737–746. [Google Scholar] [CrossRef]
- Bajetto, A.; Thellung, S.; Dellacasagrande, I.; Pagano, A.; Barbieri, F.; Florio, T. Cross talk between mesenchymal and glioblastoma stem cells: Communication beyond controversies. Stem Cells Transl. Med. 2020, 9, 1310–1330. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Jiao, X.; Jiang, W.; Yang, L.; Gong, Q.; Wang, X.; Wei, M.; Gong, S. Mesenchymal stem cells influence monocyte/macrophage phenotype: Regulatory mode and potential clinical applications. Biomed. Pharmacother. 2023, 165, 115042. [Google Scholar] [CrossRef] [PubMed]
- Luque-Campos, N.; Bustamante-Barrientos, F.A.; Pradenas, C.; García, C.; Araya, M.J.; Bohaud, C.; Contreras-López, R.; Elizondo-Vega, R.; Djouad, F.; Luz-Crawford, P.; et al. The Macrophage Response Is Driven by Mesenchymal Stem Cell-Mediated Metabolic Reprogramming. Front. Immunol. 2021, 12, 624746. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Xu, Y.; Liu, Q.; Zhang, Q. Mesenchymal Stem Cell-Macrophage Crosstalk and Maintenance of Inflammatory Microenvironment Homeostasis. Front. Cell Dev. Biol. 2021, 9, 681171. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.I.; Kim, M.R.; Jeong, H.Y.; Jeong, H.C.; Jeong, M.H.; Yoon, S.H.; Kim, Y.S.; Ahn, Y. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Exp. Mol. Med. 2014, 46, e70. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Deng, Z.; Zhang, J.; Yang, C.; Liu, J.; Han, W.; Ye, P.; Si, Y.; Chen, G. Mesenchymal stem cells promote type 2 macrophage polarization to ameliorate the myocardial injury caused by diabetic cardiomyopathy. J. Transl. Med. 2019, 17, 251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiang, W.; Yi, D.Y.; Xue, B.Z.; Wen, W.W.; Abdelmaksoud, A.; Xiong, N.X.; Jiang, X.B.; Zhao, H.Y.; Fu, P. Current status and potential challenges of mesenchymal stem cell-based therapy for malignant gliomas. Stem Cell Res. Ther. 2018, 9, 228. [Google Scholar] [CrossRef]
- Pavon, L.F.; Sibov, T.T.; de Souza, A.V.; da Cruz, E.F.; Malheiros, S.M.F.; Cabral, F.R.; de Souza, J.G.; Boufleur, P.; de Oliveira, D.M.; de Toledo, S.R.C.; et al. Tropism of mesenchymal stem cell toward CD133(+) stem cell of glioblastoma in vitro and promote tumor proliferation in vivo. Stem Cell Res. Ther. 2018, 9, 310. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Doucette, T.; Rao, G.; Yang, Y.; Gumin, J.; Shinojima, N.; Bekele, B.N.; Qiao, W.; Zhang, W.; Lang, F.F. Mesenchymal stem cells display tumor-specific tropism in an RCAS/Ntv-a glioma model. Neoplasia 2011, 13, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Do, P.T.; Wu, C.C.; Chiang, Y.H.; Hu, C.J.; Chen, K.Y. Mesenchymal Stem/Stromal Cell Therapy in Blood-Brain Barrier Preservation Following Ischemia: Molecular Mechanisms and Prospects. Int. J. Mol. Sci. 2021, 22, 10045. [Google Scholar] [CrossRef]
- Karsy, M.; Guan, J.; Jensen, R.; Huang, L.E.; Colman, H. The Impact of Hypoxia and Mesenchymal Transition on Glioblastoma Pathogenesis and Cancer Stem Cells Regulation. World Neurosurg. 2016, 88, 222–236. [Google Scholar] [CrossRef]
- Conaty, P.; Sherman, L.S.; Naaldijk, Y.; Ulrich, H.; Stolzing, A.; Rameshwar, P. Methods of Mesenchymal Stem Cell Homing to the Blood-Brain Barrier. Methods Mol. Biol. 2018, 1842, 81–91. [Google Scholar] [CrossRef]
- Chen, M.; Li, X.; Zhang, X.; He, X.; Lai, L.; Liu, Y.; Zhu, G.; Li, W.; Li, H.; Fang, Q.; et al. The inhibitory effect of mesenchymal stem cell on blood-brain barrier disruption following intracerebral hemorrhage in rats: Contribution of TSG-6. J. Neuroinflamm. 2015, 12, 61. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.; Li, J.; Zhong, L.; Chen, X.; Chen, S. SDF-1 mediates mesenchymal stem cell recruitment and migration via the SDF-1/CXCR4 axis in bone defect. J. Bone Miner. Metab. 2021, 39, 126–138. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, H.; Guo, L.; Wang, S.; Cheng, W.; Wan, L.; Zhang, Z.; Xing, L.; Zhou, Q.; Yang, X.; et al. SDF-1 secreted by mesenchymal stem cells promotes the migration of endothelial progenitor cells via CXCR4/PI3K/AKT pathway. J. Mol. Histol. 2021, 52, 1155–1164. [Google Scholar] [CrossRef]
- Gong, J.; Meng, H.B.; Hua, J.; Song, Z.S.; He, Z.G.; Zhou, B.; Qian, M.P. The SDF-1/CXCR4 axis regulates migration of transplanted bone marrow mesenchymal stem cells towards the pancreas in rats with acute pancreatitis. Mol. Med. Rep. 2014, 9, 1575–1582. [Google Scholar] [CrossRef]
- Rempel, S.A.; Dudas, S.; Ge, S.; Gutiérrez, J.A. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin. Cancer Res. 2000, 6, 102–111. [Google Scholar] [PubMed]
- Sehgal, A.; Keener, C.; Boynton, A.L.; Warrick, J.; Murphy, G.P. CXCR-4, a chemokine receptor, is overexpressed in and required for proliferation of glioblastoma tumor cells. J. Surg. Oncol. 1998, 69, 99–104. [Google Scholar] [CrossRef]
- Ehtesham, M.; Min, E.; Issar, N.M.; Kasl, R.A.; Khan, I.S.; Thompson, R.C. The role of the CXCR4 cell surface chemokine receptor in glioma biology. J. Neurooncol. 2013, 113, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.B.; Kung, A.L.; Klein, R.S.; Chan, J.A.; Sun, Y.; Schmidt, K.; Kieran, M.W.; Luster, A.D.; Segal, R.A. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13513–13518. [Google Scholar] [CrossRef] [PubMed]
- Nowak, B.; Rogujski, P.; Janowski, M.; Lukomska, B.; Andrzejewska, A. Mesenchymal stem cells in glioblastoma therapy and progression: How one cell does it all. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188582. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.D.; Vieira de Castro, J.; Costa, B.M.; Salgado, A.J. The impact of Mesenchymal Stem Cells and their secretome as a treatment for gliomas. Biochimie 2018, 155, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Vieira de Castro, J.; Gomes, E.D.; Granja, S.; Anjo, S.I.; Baltazar, F.; Manadas, B.; Salgado, A.J.; Costa, B.M. Impact of mesenchymal stem cells’ secretome on glioblastoma pathophysiology. J. Transl. Med. 2017, 15, 200. [Google Scholar] [CrossRef]
- Marofi, F.; Vahedi, G.; Biglari, A.; Esmaeilzadeh, A.; Athari, S.S. Mesenchymal Stromal/Stem Cells: A New Era in the Cell-Based Targeted Gene Therapy of Cancer. Front. Immunol. 2017, 8, 1770. [Google Scholar] [CrossRef] [PubMed]
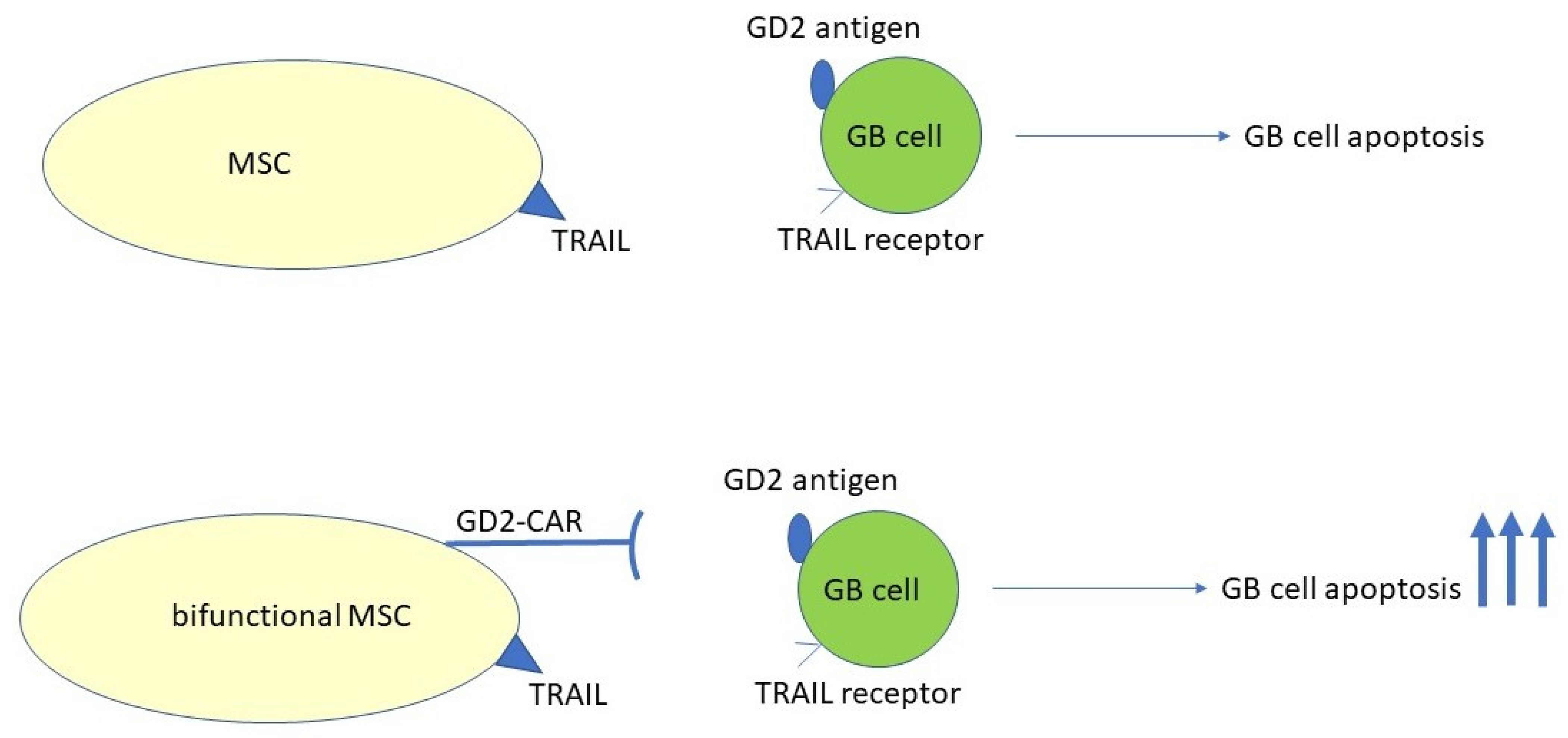
- Fakiruddin, K.S.; Ghazalli, N.; Lim, M.N.; Zakaria, Z.; Abdullah, S. Mesenchymal Stem Cell Expressing TRAIL as Targeted Therapy against Sensitised Tumour. Int. J. Mol. Sci. 2018, 19, 2188. [Google Scholar] [CrossRef]
- Loebinger, M.R.; Eddaoudi, A.; Davies, D.; Janes, S.M. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009, 69, 4134–4142. [Google Scholar] [CrossRef]
- Loebinger, M.R.; Sage, E.K.; Davies, D.; Janes, S.M. TRAIL-expressing mesenchymal stem cells kill the putative cancer stem cell population. Br. J. Cancer 2010, 103, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Cafforio, P.; Viggiano, L.; Mannavola, F.; Pellè, E.; Caporusso, C.; Maiorano, E.; Felici, C.; Silvestris, F. pIL6-TRAIL-engineered umbilical cord mesenchymal/stromal stem cells are highly cytotoxic for myeloma cells both in vitro and in vivo. Stem Cell Res. Ther. 2017, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [PubMed]
- Amano, S.; Li, S.; Gu, C.; Gao, Y.; Koizumi, S.; Yamamoto, S.; Terakawa, S.; Namba, H. Use of genetically engineered bone marrow-derived mesenchymal stem cells for glioma gene therapy. Int. J. Oncol. 2009, 35, 1265–1270. [Google Scholar] [CrossRef]
- Kenarkoohi, A.; Bamdad, T.; Soleimani, M.; Soleimanjahi, H.; Fallah, A.; Falahi, S. HSV-TK Expressing Mesenchymal Stem Cells Exert Inhibitory Effect on Cervical Cancer Model. Int. J. Mol. Cell. Med. 2020, 9, 146–154. [Google Scholar] [CrossRef]
- Bashyal, N.; Lee, T.Y.; Chang, D.Y.; Jung, J.H.; Kim, M.G.; Acharya, R.; Kim, S.S.; Oh, I.H.; Suh-Kim, H. Improving the Safety of Mesenchymal Stem Cell-Based Ex Vivo Therapy Using Herpes Simplex Virus Thymidine Kinase. Mol. Cells 2022, 45, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Dührsen, L.; Hartfuß, S.; Hirsch, D.; Geiger, S.; Maire, C.L.; Sedlacik, J.; Guenther, C.; Westphal, M.; Lamszus, K.; Hermann, F.G.; et al. Preclinical analysis of human mesenchymal stem cells: Tumor tropism and therapeutic efficiency of local HSV-TK suicide gene therapy in glioblastoma. Oncotarget 2019, 10, 6049–6061. [Google Scholar] [CrossRef]
- Oishi, T.; Ito, M.; Koizumi, S.; Horikawa, M.; Yamamoto, T.; Yamagishi, S.; Yamasaki, T.; Sameshima, T.; Suzuki, T.; Sugimura, H.; et al. Efficacy of HSV-TK/GCV system suicide gene therapy using SHED expressing modified HSV-TK against lung cancer brain metastases. Mol. Ther. Methods Clin. Dev. 2022, 26, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Ghaleh, H.E.G.; Vakilzadeh, G.; Zahiri, A.; Farzanehpour, M. Investigating the potential of oncolytic viruses for cancer treatment via MSC delivery. Cell Commun. Signal. 2023, 21, 228. [Google Scholar] [CrossRef]
- Shah, S. Novel Therapies in Glioblastoma Treatment: Review of Glioblastoma; Current Treatment Options; and Novel Oncolytic Viral Therapies. Med. Sci. 2023, 12, 1. [Google Scholar] [CrossRef]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol. Ther. 2014, 22, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Opyrchal, M.; Aderca, I.; Schroeder, M.A.; Sarkaria, J.N.; Domingo, E.; Federspiel, M.J.; Galanis, E. Oncolytic measles virus strains have significant antitumor activity against glioma stem cells. Gene Ther. 2013, 20, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef]
- Ali, S.; Xia, Q.; Muhammad, T.; Liu, L.; Meng, X.; Bars-Cortina, D.; Khan, A.A.; Huang, Y.; Dong, L. Glioblastoma Therapy: Rationale for a Mesenchymal Stem Cell-based Vehicle to Carry Recombinant Viruses. Stem Cell Rev. Rep. 2022, 18, 523–543. [Google Scholar] [CrossRef]
- Ramírez, M.; García-Castro, J.; Melen, G.J.; González-Murillo, Á.; Franco-Luzón, L. Patient-derived mesenchymal stem cells as delivery vehicles for oncolytic virotherapy: Novel state-of-the-art technology. Oncolytic Virother. 2015, 4, 149–155. [Google Scholar] [CrossRef]
- Ghasemi Darestani, N.; Gilmanova, A.I.; Al-Gazally, M.E.; Zekiy, A.O.; Ansari, M.J.; Zabibah, R.S.; Jawad, M.A.; Al-Shalah, S.A.J.; Rizaev, J.A.; Alnassar, Y.S.; et al. Mesenchymal stem cell-released oncolytic virus: An innovative strategy for cancer treatment. Cell Commun. Signal. 2023, 21, 43. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.P.; Castresana, J.S.; Shahi, M.H. Glioblastoma and MiRNAs. Cancers 2021, 13, 1581. [Google Scholar] [CrossRef]
- Hasan, H.; Afzal, M.; Castresana, J.S.; Shahi, M.H. A Comprehensive Review of miRNAs and Their Epigenetic Effects in Glioblastoma. Cells 2023, 12, 1578. [Google Scholar] [CrossRef]
- Jegathesan, Y.; Stephen, P.P.; Sati, I.; Narayanan, P.; Monif, M.; Kamarudin, M.N.A. MicroRNAs in adult high-grade gliomas: Mechanisms of chemotherapeutic resistance and their clinical relevance. Biomed. Pharmacother. 2024, 172, 116277. [Google Scholar] [CrossRef]
- Nikolova, E.; Laleva, L.; Milev, M.; Spiriev, T.; Stoyanov, S.; Ferdinandov, D.; Mitev, V.; Todorova, A. miRNAs and related genetic biomarkers according to the WHO glioma classification: From diagnosis to future therapeutic targets. Noncoding RNA Res. 2024, 9, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.A.J.; Altamimi, A.S.A.; Afzal, M.; Gupta, G.; Singla, N.; Gilhotra, R.; Almalki, W.H.; Kazmi, I.; Alzarea, S.I.; Prasher, P.; et al. Unraveling the impact of miR-21 on apoptosis regulation in glioblastoma. Pathol. Res. Pract. 2024, 254, 155121. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xue, Z.; Wang, Y.; Imran, M.; Assiri, M.; Fahad, S. Insights into the roles of non-coding RNAs and angiogenesis in glioblastoma: An overview of current research and future perspectives. Biochim. Biophys. Acta Gen. Subj. 2024, 1868, 130567. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Zhu, Z.; Lin, M.; Xu, D.; Liang, Y.; Wang, Y.; Qiao, Z.; Cao, T.; Yang, D.; Gao, L.; et al. MiR-9 Promotes Apoptosis Via Suppressing SMC1A Expression in GBM Cell Lines. Curr. Chem. Genom. Transl. Med. 2017, 11, 31–40. [Google Scholar] [CrossRef]
- Ngadiono, E.; Hardiany, N.S. Advancing towards Effective Glioma Therapy: MicroRNA Derived from Umbilical Cord Mesenchymal Stem Cells’ Extracellular Vesicles. Malays J. Med. Sci. 2019, 26, 5–16. [Google Scholar] [CrossRef]
- Qin, F.; Tang, H.; Zhang, Y.; Zhang, Z.; Huang, P.; Zhu, J. Bone marrow-derived mesenchymal stem cell-derived exosomal microRNA-208a promotes osteosarcoma cell proliferation, migration, and invasion. J. Cell. Physiol. 2020, 235, 4734–4745. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Wang, J.; Xu, S.; Leleu, X.; Himpe, E.; Maes, K.; De Bruyne, E.; Van Valckenborgh, E.; Vanderkerken, K.; Menu, E.; et al. Induction of miR-146a by multiple myeloma cells in mesenchymal stromal cells stimulates their pro-tumoral activity. Cancer Lett. 2016, 377, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Chen, S.; Liu, Z.; Xie, H.; Deng, H.; Shang, S.; Wang, X.; Xia, M.; Zuo, C. miRNA-221 of exosomes originating from bone marrow mesenchymal stem cells promotes oncogenic activity in gastric cancer. Onco Targets Ther. 2017, 10, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.A.; Fueyo, J.; Walters, E.T.; et al. Exosomes from Glioma-Associated Mesenchymal Stem Cells Increase the Tumorigenicity of Glioma Stem-like Cells via Transfer of miR-1587. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Mu, X.; Liu, L.; Wu, H.; Hu, X.; Chen, L.; Liu, J.; Mu, Y.; Yuan, F.; Liu, W.; et al. Bone marrow mesenchymal stem cells-derived exosomal microRNA-193a reduces cisplatin resistance of non-small cell lung cancer cells via targeting LRRC1. Cell Death Dis. 2020, 11, 801. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Wang, J.; Chen, S.; Tian, R.; Zeng, H.; Wang, L.; Xia, M.; Zhu, H.; Zuo, C. Exosomal miRNA-1231 derived from bone marrow mesenchymal stem cells inhibits the activity of pancreatic cancer. Cancer Med. 2019, 8, 7728–7740. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, G.; Zhang, Y.; Jiang, H.; Wang, W.; Zhao, D.; Hong, J.; Yu, H.; Qi, L. Mesenchymal stem cell-derived exosomal microRNA-133b suppresses glioma progression via Wnt/β-catenin signaling pathway by targeting EZH2. Stem Cell Res. Ther. 2019, 10, 381. [Google Scholar] [CrossRef]
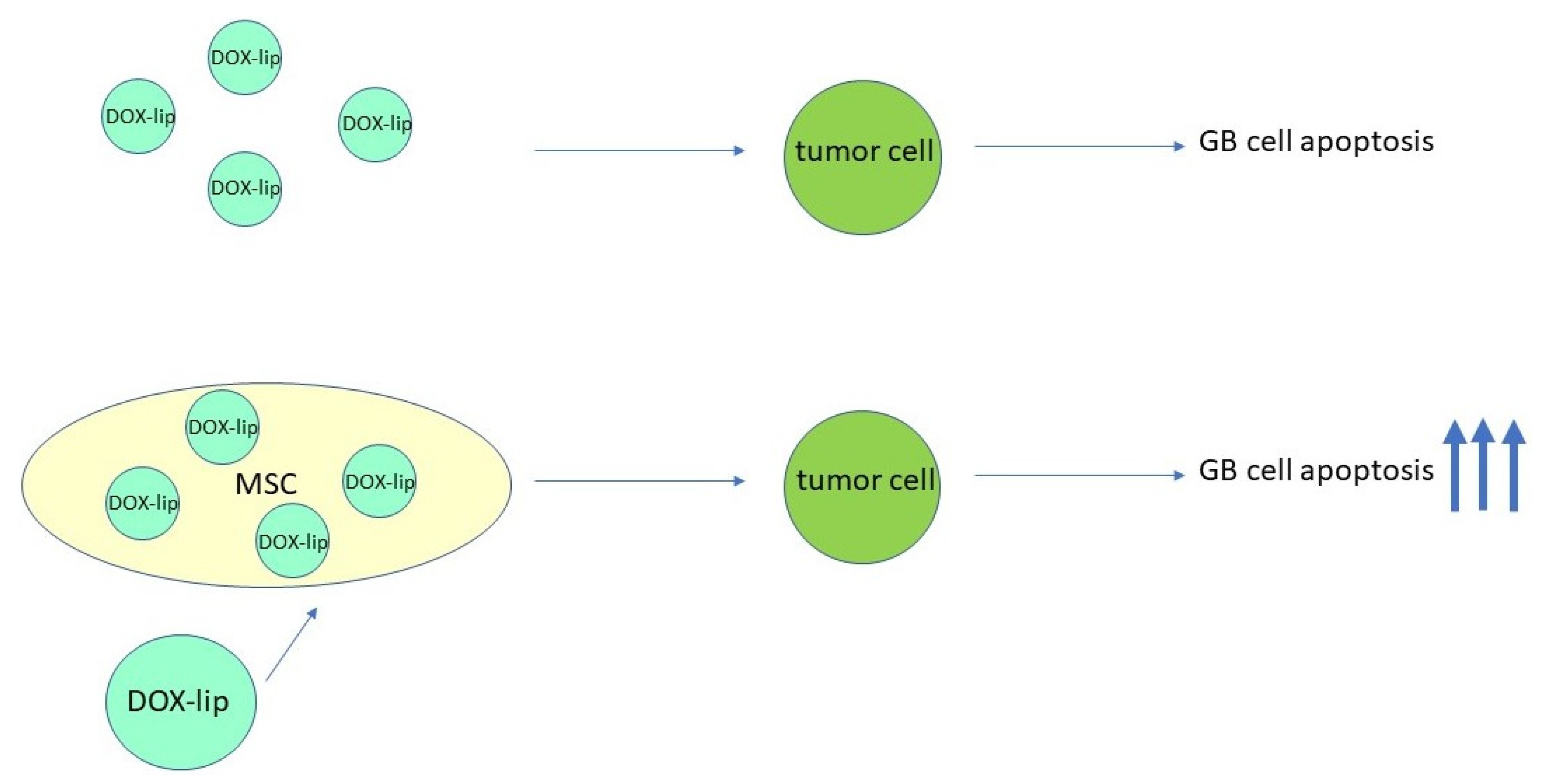
- Takayama, Y.; Kusamori, K.; Tsukimori, C.; Shimizu, Y.; Hayashi, M.; Kiyama, I.; Katsumi, H.; Sakane, T.; Yamamoto, A.; Nishikawa, M. Anticancer drug-loaded mesenchymal stem cells for targeted cancer therapy. J. Control. Release 2021, 329, 1090–1101. [Google Scholar] [CrossRef]
- Yan, L.; Li, J.; Zhang, C. The role of MSCs and CAR-MSCs in cellular immunotherapy. Cell Commun. Signal. 2023, 21, 187. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Geumann, U.; Günther, C.; Hermann, F.G.; Abken, H. IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells. Cells 2020, 9, 873. [Google Scholar] [CrossRef]
- Chan, L.Y.; Dass, S.A.; Tye, G.J.; Imran, S.A.M.; Wan Kamarul Zaman, W.S.; Nordin, F. CAR-T Cells/-NK Cells in Cancer Immunotherapy and the Potential of MSC to Enhance Its Efficacy: A Review. Biomedicines 2022, 10, 804. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Liu, X.; He, W.; Huang, Q.; Feng, Q. Nanoparticles and their effects on differentiation of mesenchymal stem cells. Biomater. Transl. 2020, 1, 58–68. [Google Scholar] [CrossRef]
- Raghav, P.K.; Mann, Z.; Ahlawat, S.; Mohanty, S. Mesenchymal stem cell-based nanoparticles and scaffolds in regenerative medicine. Eur. J. Pharmacol. 2022, 918, 174657. [Google Scholar] [CrossRef]
- Merino, J.J.; Cabaña-Muñoz, M.E. Nanoparticles and Mesenchymal Stem Cell (MSC) Therapy for Cancer Treatment: Focus on Nanocarriers and a si-RNA CXCR4 Chemokine Blocker as Strategies for Tumor Eradication In Vitro and In Vivo. Micromachines 2023, 14, 2068. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xin, Y.; Cao, H.; Li, W.; Hua, Y.; Webster, T.J.; Zhang, C.; Tang, W.; Liu, Z. Recent advances in mesenchymal stem cell membrane-coated nanoparticles for enhanced drug delivery. Biomater. Sci. 2021, 9, 1088–1103. [Google Scholar] [CrossRef]
- Ren, N.; Feng, Z.; Liang, N.; Xie, J.; Wang, A.; Sun, C.; Yu, X. NaGdF(4):Yb/Er nanoparticles of different sizes for tracking mesenchymal stem cells and their effects on cell differentiation. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 111, 110827. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Maus, M.V.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T-cell Strategies. Clin. Cancer Res. 2019, 25, 2042–2048. [Google Scholar] [CrossRef]
- Golinelli, G.; Grisendi, G.; Prapa, M.; Bestagno, M.; Spano, C.; Rossignoli, F.; Bambi, F.; Sardi, I.; Cellini, M.; Horwitz, E.M.; et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Ther. 2020, 27, 558–570. [Google Scholar] [CrossRef]
- Zottel, A.; Videtič Paska, A.; Jovčevska, I. Nanotechnology Meets Oncology: Nanomaterials in Brain Cancer Research, Diagnosis and Therapy. Materials 2019, 12, 1588. [Google Scholar] [CrossRef] [PubMed]
- Farhat, W.; Yeung, V.; Kahale, F.; Parekh, M.; Cortinas, J.; Chen, L.; Ross, A.E.; Ciolino, J.B. Doxorubicin-Loaded Extracellular Vesicles Enhance Tumor Cell Death in Retinoblastoma. Bioengineering 2022, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Xunian, Z.; Kalluri, R. Biology and therapeutic potential of mesenchymal stem cell-derived exosomes. Cancer Sci. 2020, 111, 3100–3110. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.; Zheng, C.X.; Sui, B.D.; Liu, W.J.; Jin, Y. Mesenchymal stem cell-derived exosomes: A novel and potential remedy for cutaneous wound healing and regeneration. World J. Stem Cells 2022, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, B.; Dayeri, B.; Zahedi, E.; Khoshbakht, S.; Nezamabadi Pour, N.; Ranjbar, H.; Davari Nejad, A.; Noureddini, M.; Alani, B. Mesenchymal stem cell (MSC)-derived exosomes as novel vehicles for delivery of miRNAs in cancer therapy. Cancer Gene Ther. 2022, 29, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Nikfarjam, S.; Rezaie, J.; Zolbanin, N.M.; Jafari, R. Mesenchymal stem cell derived-exosomes: A modern approach in translational medicine. J. Transl. Med. 2020, 18, 449. [Google Scholar] [CrossRef] [PubMed]
- Lotfy, A.; AboQuella, N.M.; Wang, H. Mesenchymal stromal/stem cell (MSC)-derived exosomes in clinical trials. Stem Cell Res. Ther. 2023, 14, 66. [Google Scholar] [CrossRef] [PubMed]
- Panda, B.; Sharma, Y.; Gupta, S.; Mohanty, S. Mesenchymal Stem Cell-Derived Exosomes as an Emerging Paradigm for Regenerative Therapy and Nano-Medicine: A Comprehensive Review. Life 2021, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Kou, M.; Huang, L.; Yang, J.; Chiang, Z.; Chen, S.; Liu, J.; Guo, L.; Zhang, X.; Zhou, X.; Xu, X.; et al. Mesenchymal stem cell-derived extracellular vesicles for immunomodulation and regeneration: A next generation therapeutic tool? Cell Death Dis. 2022, 13, 580. [Google Scholar] [CrossRef]
- Ghasempour, E.; Hesami, S.; Movahed, E.; Keshel, S.H.; Doroudian, M. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy in the brain tumors. Stem Cell Res. Ther. 2022, 13, 527. [Google Scholar] [CrossRef]
- Rahmani, R.; Kiani, J.; Tong, W.Y.; Soleimani, M.; Voelcker, N.H.; Arefian, E. Engineered anti-EGFRvIII targeted exosomes induce apoptosis in glioblastoma multiforme. J. Drug Target 2023, 31, 310–319. [Google Scholar] [CrossRef]
- Rodini, C.O.; Gonçalves da Silva, P.B.; Assoni, A.F.; Carvalho, V.M.; Okamoto, O.K. Mesenchymal stem cells enhance tumorigenic properties of human glioblastoma through independent cell-cell communication mechanisms. Oncotarget 2018, 9, 24766–24777. [Google Scholar] [CrossRef] [PubMed]
- Onzi, G.R.; Faccioni, J.L.; Pereira, L.C.; Thomé, M.P.; Bertoni, A.P.S.; Buss, J.H.; Fazolo, T.; Filippi-Chiela, E.; Wink, M.R.; Lenz, G. Adipose-derived stromal cell secretome disrupts autophagy in glioblastoma. J. Mol. Med. 2019, 97, 1491–1506. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, D.; Giacomelli, C.; Marchetti, L.; Martini, C.; Trincavelli, M.L. High Adenosine Extracellular Levels Induce Glioblastoma Aggressive Traits Modulating the Mesenchymal Stromal Cell Secretome. Int. J. Mol. Sci. 2020, 21, 7706. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.T.; Fu, P.; Juerchott, K.; Motaln, H.; Selbig, J.; Lah, T.; Tonn, J.C.; Schichor, C. The expression of Wnt-inhibitor DKK1 (Dickkopf 1) is determined by intercellular crosstalk and hypoxia in human malignant gliomas. J. Cancer Res. Clin. Oncol. 2014, 140, 1261–1270. [Google Scholar] [CrossRef]
- Zhang, Q.; Yi, D.Y.; Xue, B.Z.; Wen, W.W.; Lu, Y.P.; Abdelmaksou, A.; Sun, M.X.; Yuan, D.T.; Zhao, H.Y.; Xiong, N.X.; et al. CD90 determined two subpopulations of glioma-associated mesenchymal stem cells with different roles in tumour progression. Cell Death Dis. 2018, 9, 1101. [Google Scholar] [CrossRef]
- Svensson, A.; Ramos-Moreno, T.; Eberstål, S.; Scheding, S.; Bengzon, J. Identification of two distinct mesenchymal stromal cell populations in human malignant glioma. J. Neurooncol. 2017, 131, 245–254. [Google Scholar] [CrossRef]
- Shahar, T.; Rozovski, U.; Hess, K.R.; Hossain, A.; Gumin, J.; Gao, F.; Fuller, G.N.; Goodman, L.; Sulman, E.P.; Lang, F.F. Percentage of mesenchymal stem cells in high-grade glioma tumor samples correlates with patient survival. Neuro Oncol. 2017, 19, 660–668. [Google Scholar] [CrossRef]
- Motaln, H.; Gruden, K.; Hren, M.; Schichor, C.; Primon, M.; Rotter, A.; Lah, T.T. Human mesenchymal stem cells exploit the immune response mediating chemokines to impact the phenotype of glioblastoma. Cell Transplant. 2012, 21, 1529–1545. [Google Scholar] [CrossRef]
- Oliveira, M.N.; Pillat, M.M.; Motaln, H.; Ulrich, H.; Lah, T.T. Kinin-B1 Receptor Stimulation Promotes Invasion and is Involved in Cell-Cell Interaction of Co-Cultured Glioblastoma and Mesenchymal Stem Cells. Sci. Rep. 2018, 8, 1299. [Google Scholar] [CrossRef]
- Li, S.; Xiang, W.; Tian, J.; Wang, H.; Hu, S.; Wang, K.; Chen, L.; Huang, C.; Zhou, J. Bone Marrow-Derived Mesenchymal Stem Cells Differentially Affect Glioblastoma Cell Proliferation, Migration, and Invasion: A 2D-DIGE Proteomic Analysis. Biomed. Res. Int. 2021, 2021, 4952876. [Google Scholar] [CrossRef] [PubMed]
- Dasari, V.R.; Velpula, K.K.; Kaur, K.; Fassett, D.; Klopfenstein, J.D.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Cord blood stem cell-mediated induction of apoptosis in glioma downregulates X-linked inhibitor of apoptosis protein (XIAP). PLoS ONE 2010, 5, e11813. [Google Scholar] [CrossRef] [PubMed]
- Kucerova, L.; Matuskova, M.; Hlubinova, K.; Altanerova, V.; Altaner, C. Tumor cell behaviour modulation by mesenchymal stromal cells. Mol. Cancer 2010, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Castro-Oropeza, R.; Vazquez-Santillan, K.; Díaz-Gastelum, C.; Melendez-Zajgla, J.; Zampedri, C.; Ferat-Osorio, E.; Rodríguez-González, A.; Arriaga-Pizano, L.; Maldonado, V. Adipose-derived mesenchymal stem cells promote the malignant phenotype of cervical cancer. Sci. Rep. 2020, 10, 14205. [Google Scholar] [CrossRef]
- Chen, Y.; He, Y.; Wang, X.; Lu, F.; Gao, J. Adipose-derived mesenchymal stem cells exhibit tumor tropism and promote tumorsphere formation of breast cancer cells. Oncol. Rep. 2019, 41, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Gao, Y.; Li, X.; Zhang, M.; Yang, Y.; Han, Q.; Sun, Z.; Bai, C.; Zhao, R.C. Mesenchymal stem cells derived from adipose accelerate the progression of colon cancer by inducing a MT-CAFs phenotype via TRPC3/NF-KB axis. Stem Cell Res. Ther. 2022, 13, 335. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, G.; Huo, X.; Wang, Y.; Tigyi, G.; Zhu, B.M.; Yue, J.; Zhang, W. Adipose-Derived Stem Cells Facilitate Ovarian Tumor Growth and Metastasis by Promoting Epithelial to Mesenchymal Transition Through Activating the TGF-β Pathway. Front. Oncol. 2021, 11, 756011. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Jiang, Q.; Deng, J.; Xu, F.; Chen, X.; Cheng, F.; Zhang, Y.; Yao, Y.; Xia, Z.; et al. Human Adipose-Derived Mesenchymal Stem Cell-Secreted CXCL1 and CXCL8 Facilitate Breast Tumor Growth By Promoting Angiogenesis. Stem Cells 2017, 35, 2060–2070. [Google Scholar] [CrossRef] [PubMed]
- Kucerova, L.; Altanerova, V.; Matuskova, M.; Tyciakova, S.; Altaner, C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007, 67, 6304–6313. [Google Scholar] [CrossRef]
- Szyposzynska, A.; Bielawska-Pohl, A.; Murawski, M.; Sozanski, R.; Chodaczek, G.; Klimczak, A. Mesenchymal Stem Cell Microvesicles from Adipose Tissue: Unraveling Their Impact on Primary Ovarian Cancer Cells and Their Therapeutic Opportunities. Int. J. Mol. Sci. 2023, 24, 15862. [Google Scholar] [CrossRef]
- Jung, P.Y.; Ryu, H.; Rhee, K.J.; Hwang, S.; Lee, C.G.; Gwon, S.Y.; Kim, J.; Kim, J.; Yoo, B.S.; Baik, S.K.; et al. Adipose tissue-derived mesenchymal stem cells cultured at high density express IFN-β and TRAIL and suppress the growth of H460 human lung cancer cells. Cancer Lett. 2019, 440–441, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Tang, Q.; Yin, X.; Yan, D.; Tang, M.; Xin, J.; Pan, Q.; Ma, C.; Yan, S. The Therapeutic Potential of Adipose Tissue-Derived Mesenchymal Stem Cells to Enhance Radiotherapy Effects on Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2019, 7, 267. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.A.; Lee, J.Y.; Wang, K.C.; Phi, J.H.; Song, S.H.; Song, J.; Kim, S.K. Human adipose tissue-derived mesenchymal stem cells: Characteristics and therapeutic potential as cellular vehicles for prodrug gene therapy against brainstem gliomas. Eur. J. Cancer 2012, 48, 129–137. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santillán-Guaján, S.M.; Shahi, M.H.; Castresana, J.S. Mesenchymal-Stem-Cell-Based Therapy against Gliomas. Cells 2024, 13, 617. https://doi.org/10.3390/cells13070617
Santillán-Guaján SM, Shahi MH, Castresana JS. Mesenchymal-Stem-Cell-Based Therapy against Gliomas. Cells. 2024; 13(7):617. https://doi.org/10.3390/cells13070617
Chicago/Turabian StyleSantillán-Guaján, Sisa M., Mehdi H. Shahi, and Javier S. Castresana. 2024. "Mesenchymal-Stem-Cell-Based Therapy against Gliomas" Cells 13, no. 7: 617. https://doi.org/10.3390/cells13070617